

Abstract
The discovery that gene expression can be controlled by the Watson–Crick base-pairing of small RNAs with messenger RNAs containing complementary sequence — a process known as RNA interference — has markedly advanced our understanding of eukaryotic gene regulation and function. The ability of short RNA sequences to modulate gene expression has provided a powerful tool with which to study gene function and is set to revolutionize the treatment of disease. Remarkably, despite being just one decade from its discovery, the phenomenon is already being used therapeutically in human clinical trials, and biotechnology companies that focus on RNA-interference-based therapeutics are already publicly traded.
Before 1980, RNA was generally considered to be no more than a passive intermediate carrying information between DNA and protein synthesis. The discovery of catalytic RNAs in the early 1980s merited a shared Nobel prize to Tom Cech and Sidney Altman, and in 1986 the concept of ‘the RNA world’, an idiom created by Walter Gilbert, was proposed. Today, this is a common expression, and RNA has claimed a pivotal place in cellular biology.
Just ten years ago, RNA’s functional repertoire was expanded further with the discovery in the nematode Caenorhabditis elegans1 that double-stranded RNAs (dsRNAs) can trigger silencing of complementary messenger RNA sequences, and the term ‘RNA interference’ (RNAi) was born. Shortly thereafter, short dsRNAs — or short interfering RNAs (siRNAs) (reviewed in ref. 1) — were generated artificially and used to demonstrate that this process also occurs in mammalian cells, usually, but not always, without triggering the innate immune sys tem, which normally recognizes RNAs as part of an antiviral defence mechanism (see page 421). The knowledge that small RNAs can affect gene expression has had a tremendous impact on basic and applied research, and RNAi is currently one of the most promising new approaches for disease therapy.
That RNAi could be triggered in vivo in mammals was first shown in animals infected with hepatitis B virus2. This was followed by the first therapeutic application of siRNAs: siRNAs were targeted to Fas mRNA in a mouse model of autoimmune hepatitis, resulting in protection of the treated animals against liver fibrosis3. In 2004, only six years after the discovery of RNAi, the first siRNA-based human therapeutics — developed as treatments for wet age-related macular degeneration — entered phase I clinical trials. RNAi is one of the fastest advancing fields in biology, and the flow of discoveries gives true meaning to the expression ‘from the bench to the bedside’.
Although much is known about the mechanisms of RNAi, there are a number of challenges that applications of this gene-silencing technology need to overcome. For one, RNAi is a fundamentally important regulatory mechanism in the cell, and tapping into it in the interests of therapeutic benefit could result in side effects. Exogenously introduced dsRNA sequences can sequester components that make up the cellular machinery involved in gene silencing (see page 396), thereby reducing the accessibility of the machinery to a class of small RNAs known as microRNAs (miRNAs) that are entering the natural cellular pathway4,5. In addition, some synthetic siRNAs contain sequence motifs that can induce type I interferon responses and stimulate the production of pro-inflammatory cytokines6-8.
During the past few years, many scientists have searched for solutions to overcome these limitations and to increase the safety of potential RNAi-based therapeutics. This article explores recent strategies to minimize undesirable secondary effects, describes new approaches to delivery and discusses RNAi therapies that are being tested. As it is anticipated that this technology will be applied to an increasing range of diseases, the potential problems and solutions that could one day transform RNAi into a conventional treatment for human diseases warrant careful attention.
Endogenous gene silencing
The effector RNA molecules of RNAi consist of ~20–30 nucleotides9. They are complexed with the protein components of the RNA-induced silencing complex (RISC). Its catalytic core in plants and animals (with the exception of single-celled organisms) is AGO2, a member of the highly conserved Argonaute protein family10. These small RNAs can silence gene expression by two mechanisms: post-transcriptional gene silencing (PTGS)11, and transcriptional gene silencing (TGS)12,13 (Fig. 1). PTGS can, in turn, be divided into two main mechanisms: direct sequence-specific cleavage, and translational repression and RNA degradation. Direct sequence-specific cleavage occurs when the targeted mRNA is perfectly complementary to the siRNA and is degraded after site-specific cleavage by the RISC. Translational repression and RNA degradation occur when the small RNA guide sequence has only limited complementarity to the target in the ‘seed’ region (nucleotides 2 to 8 from the 5′ end of the guide strand), with base-pairing usually occurring in the 3′ untranslated region (UTR). The latter mechanism is used by miRNAs.
Figure 1. Mechanisms of cellular gene silencing.
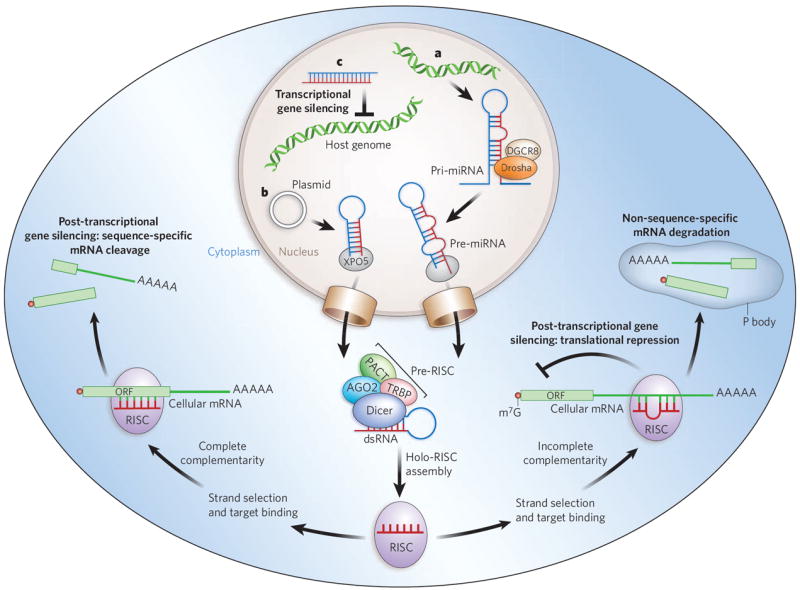
a, Primary microRNAs (pri-miRNAs) are, in plants and animals, processed by Drosha and its partner DGCR8 into precursor miRNAs (pre-miRNAs) and then transported to the cytoplasm by exportin 5 (XPO5). In the cytoplasm, they are bound by a Dicer-containing pre-RISC and processed to yield the guide sequence that is loaded into the holo-RISC, which contains all the components required for gene silencing. AGO2 is the catalytic core of the RISC (present but not shown in the schematically drawn holo-RISC). The guide sequence binds to the corresponding target sequences in the 3′ UTRs of cellular mRNAs. If the miRNA guide sequence is fully complementary to its target site (left pathway), it triggers site-specific cleavage and degradation of the mRNA through the catalytic domain of AGO2. If the base-pairing is incomplete (right pathway) but includes pairing of the seed region (nucleotides 2–8 of the miRNA) with the target, translational inhibition occurs, and this can be accompanied by non-sequence-specific degradation of the mRNA in P bodies. b, Similarly to miRNAs, artificially transcribed shRNAs (in this case from a plasmid) are transported to the cytoplasm by XPO5. The dsRNA in the cytoplasm is recognized and processed by Dicer into ~21–25-nucleotide siRNA fragments that are loaded into the RISC. The siRNAs can target complementary sequences of cellular mRNAs and trigger their degradation through AGO2-mediated cleavage. c, When siRNAs are present in the nucleus and are complementary to promoter regions, they can trigger chromatin remodelling and histone modifications that result in transcriptional gene silencing. In mammalian cells, the details of this mechanism are still under investigation but are known to include Argonaute-family proteins. Accessory proteins indicated in the figure are TRBP (HIV tar-RNA-binding protein; also known as TRBP2P) and PACT (activator of protein kinase PKR; also known as PRKRA). m7G, 7-methylguanosine.
TGS has been demonstrated in Schizosaccharomyces pombe (fission yeast), plants and, most recently, mammalian cells14-17. In S. pombe, the process is mediated by the RNA-induced transcriptional silencing complex (RITS), which contains Ago1, the chromodomain protein Chp1 and the glycine and tryptophan (GW)-repeat-containing protein Tas3 (ref. 18) (see page 413). Although in mammalian cells the mechanism by which small-RNA-directed silencing occurs is still hotly debated, both AGO1 and AGO2 have been shown to be integral to the overall process19,20. Most recently, a miRNA (miR-320) has been shown to regulate transcription of the POLR3D subunit of RNA polymerase III (Pol III)21 in human cell culture.
Endogenous small RNAs have been found in various organisms, including humans, mice, the fruitfly Drosophila melanogaster and C. elegans. Many of these originate from transposons, viruses and repetitive sequences and are characterized by their interactions with the PIWI subfamily (or PIWI clade) of Argonaute proteins22-25 — these are thus named PIWI-interacting RNAs (piRNAs). The identification of piRNAs has been restricted to germline cells. Recently, a new class of endogenous siRNAs (endo-siRNAs or esiRNAs) has been identified in the gonads and somatic tissues of D. melanogaster26-29 and in mouse oocytes30,31. In mice, endo-siRNAs have been proposed to regulate retrotransposon movement30,31. Several families of small RNAs, including repeat-associated siRNAs (ra-siRNAs), tiny non-coding RNAs (tncRNAs), trans-acting siRNAs (ta-siRNAs) and scan RNAs (scnRNAs) (Table 1) are found in fungi, plants and animals, but so far none of these has been observed in mammals. The evidence suggests that piRNAs act through different cellular pathways from siRNAs and miRNAs and so could offer alternative targeting strategies for therapeutic targets.
Table 1.
Cellular small RNAs involved in gene silencing
| Class | Size (nucleotides) | Functions | Mechanisms | Origin | Organisms found in |
|---|---|---|---|---|---|
| siRNAs | 21–25 | Regulating gene expression, providing antiviral response, restricting transposons | RNA degradation, transposon restriction | Intergenic regions, exons, introns | Caenorhabditis elegans, Drosophila melanogaster, Schizosaccharomyces pombe, Arabidopsis thaliana, Oryza sativa (rice) |
| endo-siRNAs | 21–25 | Restricting transposons, regulating mRNAs and heterochromatin | RNA degradation | Transposable elements, pseudogenes | D. melanogaster, mammals |
| miRNAs | 21–25 | Regulating gene expression at the post-transcriptional level | Blocking translation, RNA degradation | Intergenic regions, introns | C. elegans, D. melanogaster, S. pombe, A. thaliana, O. sativa, mammals |
| piRNAs | 24–31* | Regulating germline development and integrity, silencing selfish DNA | Unknown | Defective transposon sequences and other repeats | C. elegans, D. melanogaster, Danio rerio, mammals |
| ra-siRNAs | 23–28 | Remodelling chromatin, transcriptional gene silencing | Unknown | Repeated sequence elements (subset of piRNAs) | C. elegans, D. melanogaster, S. pombe, Trypanosoma brucei, D. rerio, A. thaliana |
| ta-siRNAs | 21–22 | Trans-acting cleavage of endogenous mRNAs | RNA degradation | Non-coding endogenous transcripts | D. melanogaster, S. pombe, A. thaliana, O. sativa |
| natRNAs | 21–22 | Regulating gene expression at the post-transcriptional level | RNA degradation | Convergent partly overlapping transcripts | A. thaliana |
| scnRNAs | 26–30 | Regulating chromatin structure | DNA elimination | Meiotic micronuclei | Tetrahymena thermophila, Paramecium tetraurelia |
| tncRNAs | 22 | Unknown | Unknown | Non-coding regions | C. elegans |
C. elegans piRNAs are 21 nucleotides. endo-siRNAs, endogenous siRNAs; miRNAs, microRNAs; natRNAs, natural antisense transcript siRNAs; piRNAs, PIWI-interacting RNAs; ra-siRNAs, repeat-associated siRNAs; scnRNAs, scan RNAs; siRNAs, short interfering RNAs; ta-siRNAs, trans-acting siRNAs; tncRNAs, tiny non-coding RNAs.
Superior designs for small molecules
Cellular genes can be targeted by exogenous introduction of siRNAs, which then take advantage of the endogenous PTGS mechanism. The siRNAs can be either transfected into cells, where they enter the RISC directly, or generated within cells through gene expression by the use of vectors containing Pol II or Pol III promoters. These RNAi triggers can be expressed in animals and plants, but not in S. pombe, in the form of miRNAs or as short hairpin RNAs (shRNAs), which are cleaved into small (~21–25-nucleotide) RNAs by the enzymes Drosha and/or Dicer. In both cases, if the two strands of the RNA trigger are completely complementary, the passenger strand is cleaved by AGO2 (refs 32, 33), leaving behind a single-stranded guide sequence, which acts as the template for recognition of the targeted gene sequence by the RISC (Fig. 1).
Most of the impending therapeutic applications based on RNAi propose using direct introduction of synthetic siRNAs. The advantage of using a chemically synthesized molecule is that chemical modifications can be introduced to increase stability, promote efficacy, block binding to unintended targets that contain sequence mismatches (specific off-target effects), and reduce or abrogate potential immunostimulatory effects (general off-target effects). However, the effects of these molecules are transient, whereas the promoter-expressed shRNAs or miRNAs can potentially mediate long-term silencing with a single application.
Conventional siRNAs are ~22 nucleotides and have 3′ dinucleotide overhangs that mimic Dicer cleavage products. Because not all siRNAs achieve equivalent levels of target knockdown, large-scale siRNA screening is often performed for any given target to find the most potent inhibitors. These have yielded some rules for siRNA design. For example, to facilitate incorporation into the RISC, the 5′ end of the antisense (guide) strand should be designed to have a lower thermodynamic stability than the 5′ end of the sense strand. The proportion of the nucleotides guanosine and cytidine should be around 50% or lower, and targeting of known protein-binding sites in mRNA regulatory regions should be avoided because binding of regulatory proteins may block siRNA–target pairing. For the same reason, intramolecular structures in the target should be avoided. Statistical analyses have also found a preference for certain nucleotides at specific positions within the siRNA34. Many computer programs are available for identifying the optimal target sequences for a given gene34,35. One of these, an artificial neural network, has been used to develop a genome-wide siRNA library for humans and to identify effective siRNAs for 34 targets36.
Chemical modifications are often included in the design of synthetic siRNAs. Selective addition of phosphorothioate linkages or substitution of 2′ fluoropyrimidines or a 2′-O-methyl for the 2′ ribose at certain positions does not compromise siRNA activity and concomitantly increases resistance to ribonucleases37, which is important for in vivo applications. A single 2′-O-methyl group on the passenger strand of an siRNA duplex can abrogate activation of the Toll-like receptors38 and prevent tox icities due to the activation of type I interferon pathway gene expression. It has recently been demonstrated that fluoro-β-d-arabinonucleic acid (FANA39 or as 4′-S-FANA40) or arabinonucleic acid (ANA41) modifications can increase both the serum stability and the potency of siRNAs. Some chemical modifications also have the important advantage of decreasing or blocking the activity of the siRNA’s sense (passenger) strand, thereby reducing specific off-target effects. Other modifications, such as the addition of lauric acid, lithocholic acids and cholesterol derivatives, can be made to increase cellular uptake42, which is currently one of the main hurdles of RNAi therapy.
Breaking and entering
Therapeutic applications of siRNAs require effective delivery to the target cells and tissues. The two main strategies are delivery of chemically synthesized siRNAs (non-viral delivery), or delivery of shRNA-encoding genes by engineered viruses that will ultimately generate siRNAs by transcription in the target cells.
Non-viral delivery
Because of their size and negative charge, siRNAs cannot easily cross cell membranes. Delivery has therefore been one of the major challenges for RNAi technology. Various means of delivery have been developed and tested in murine and non-human primate models, ranging from the injection of naked RNAs into a target organ such as the lung or eye to systemic delivery of the RNA in nanoparticles, complexed with polycations, attached to cholesterol groups or conjugated with cell-surface receptors. Some delivery approaches are detailed in Fig. 2.
Figure 2. In vivo delivery strategies for therapeutic siRNAs.
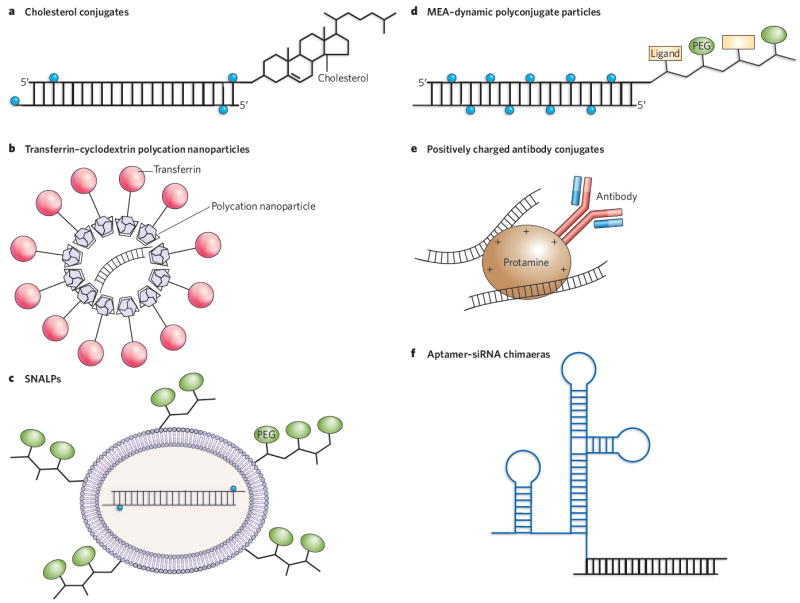
a, Cholesterol groups can be linked to modified siRNAs to enhance their stability before systemic delivery. The most common siRNA modifications are 2′-O-methyluridine or 2′-fluorouridine substitutions (blue circles) combined with phosphorothioate linkages. b, Polycation nanoparticles can direct delivery of the siRNAs to specific cells through the use of surface ligands (such as transferrin) that bind to receptors on target cells. c, SNALPs encapsulate modified siRNAs into cationic or neutral lipid bilayers coated with diffusible PEG–lipid conjugates. SNALPs allow siRNAs to be taken up by cells and released by endosomes. d, Masked endosomolytic agent (MEA)–Dynamic PolyConjugates (DPCs) are similar to SNALPS but smaller, and contain a ligand that allows targeted cell delivery. The release of the siRNA from the endosome is also improved by the inclusion of a pH-labile bond in the MEA–DPC particles. e, Tagging specific antibodies with protamine or other positive charges allows the delivery of siRNAs to specific cell types via receptor-mediated uptake. f, Chemically linking or co-transcribing siRNAs with RNA aptamers allows the targeted delivery of the siRNAs to cells expressing the appropriate receptor.
Two polymers that have been examined for their delivery properties are atelocollagen and chitosan. Chitosans have mucoadhesive properties and have been used for intranasal delivery to bronchiolar epithelial cells43. Intranasal delivery has proved an effective means of delivering siRNA in mice44 and in non-human primates45 to block respiratory syncytial virus infection of the upper respiratory tract. In fact, the delivery of siRNAs to mucosal membranes seems to be an effective approach in general. For example, intravaginal delivery of lipid-encapsulated siRNAs targeting herpes simplex virus 2 (HSV-2) provided protection against lethal viral infection in more than two-thirds of the siRNA-treated mice46.
Targeting of anti-apolipoprotein B (APOB) and peroxisome proliferator-activated receptor-α (PPAR-α) siRNAs to the liver has been achieved by means of a ‘membrane-active’ polymer, which can mask its activity until it reaches the endosome, resulting in the delivery of siRNAs to hepatocytes after a simple intravenous injection47. A different siRNA delivery approach used transferrin conjugated to a cyclodextrin-polycation polymer to deliver siRNAs targeting the Ewing’s sarcoma Ews–Fli1 fusion mRNA by means of the transferrin receptor in mice48, resulting in inhibition of tumour progression. And conjugation of an siRNA to a cholesterol group permitted its delivery to the liver and the jejunum, where it downregulated its target, APOB, leading to consequent lowering of blood cholesterol levels in a murine model system49.
An important advance for siRNA delivery was the successful application of stable nucleic-acid lipid particles decorated with poly ethylene glycol (PEG) polymer chains (termed SNALPs) for the delivery of siRNAs directed against APOB mRNA (APOB-targeted siRNAs) to the livers of non-human primates50. In this case, the siRNA effect of a single intravenous injection lasted for more than 11 days and resulted in greater than 90% target knockdown and no toxicity50. These exciting results have increased confidence in the potential of therapeutic siRNAs for treating liver diseases.
Until recently, most approaches to in vivo delivery have targeted a particular organ, primarily the eye, the lungs or the liver. A significant advance in targeting siRNAs to a specific class of leukocytes involved in gut inflammation has now been reported51. In this study, a cyclin D1 (Cyd1)-targeted siRNA was loaded into stabilized nanoparticles, the surfaces of which incorporated an antibody specific for a receptor expressed by the leukocytes. The targeted siRNA-containing nanoparticles down-regulated the cyclin D1 target, suppressed leukocyte proliferation and reversed experimentally induced colitis in mice51.
Delivery of siRNAs to the nervous system has been particularly challenging. The brain is notoriously refractory to targeting because of difficulties in crossing the blood–brain barrier. However, delivery of siRNAs to the peripheral nervous system by direct infusion into the brain for the relief of chronic pain52-54 or anxiety55 has been demonstrated in rats. Conjugates of liposomes and antibodies or neuropeptides have also been used to deliver siRNAs into the murine brain56. Nevertheless, these methods do not target neurons, and a less invasive alternative to direct cranial injection is required to make such therapies more palatable.
A recent study unlocked the possibility of selective delivery of siRNAs to the central nervous system by systemic intravenous injection57. The siRNA involved — designed to target Japanese encephalitis virus — was conjugated with a short peptide derived from the rabies virus glycoprotein, which binds to the neuronal cell acetylcholine receptor. After transvascular delivery, 80% of the mice treated with the therapeutic siRNA survived infection with Japanese encephalitis virus, whereas 100% of the untreated controls died from complications of the infection57.
Another interesting approach that allows systemic and targeted siRNA delivery uses a protamine–antibody fusion protein58. The protamine moiety is linked to the heavy-chain antigen-binding region (Fab) of an antibody to the human immunodeficiency virus 1 (HIV-1) envelope protein gp160. The positively charged protamine binds the negatively charged siRNAs — which are targeted against the HIV gene gag — allowing selective delivery to cells expressing the gp160 envelope protein on their surfaces58. This results in internalization of the antibody–siRNA complex, release of the siRNAs and downregulation of the HIV Gag-encoding transcripts in a murine model in vivo. In this same study, fusion to protamine of an antibody specific for the hormone receptor ERBB2 allowed siRNA targeting of cancer cells expressing that receptor58.
A similar technology for specific targeted delivery is based on aptamer–RNAi chimaeras59. Aptamers are in vitro-evolved, synthetically prepared nucleic acids that selectively bind specific ligands. An RNA aptamer designed to bind prostate-specific membrane antigen (PSMA; also known as FOLH1) was linked to a PLK1-targeted siRNA, and binding of the aptamer to the PSMA receptor resulted in the selective delivery into prostate cancer cells of siRNAs that target pro-survival genes59,60. Intratumoral injection of the PSMA–Plk1-targeted siRNA or PSMA–Bcl2-targeted siRNA conjugates into a mouse xenograft model resulted in triggering of apoptosis, growth inhibition and tumour regression59.
A different conjugation of an siRNA to vitamin-A-coupled liposomes succeeded in delivering antifibrotic siRNAs to hepatic stellate cells, which are produced in response to liver damage61. In this study, multiple siRNA treatments targeting collagen chaperone-encoding genes reversed liver fibrosis by preventing collagen deposition and increased survival in rats, providing a potential therapeutic approach to treating liver cirrhosis.
Also noteworthy is the recent report of libraries of lipid-like molecules (lipidoids) that can be selected for siRNA delivery to various tissues62.
Viral delivery
An alternative means of triggering RNAi is through promoter-expressed siRNA sequences processed from shRNAs or miRNA mimics. The genes encoding these hairpin structures are most commonly inserted into the backbones of viral vectors under the control of Pol II or Pol III promoters. A potential advantage of vector delivery is that a single administration triggers long-term expression of the therapeutic RNAi. This is particularly appropriate for chronic viral diseases such as HIV and viral hepatitis.
Lentiviral vectors have been used successfully to deliver shRNA constructs in various mammalian systems. For example, it was shown that downregulation of an activated Ras oncogene by a lentiviral-delivered shRNA resulted in suppression of tumour growth in mice63. And down-regulation of the expression of a mutant form of superoxide dismutase 1 (SOD1) in mouse models of amyotrophic lateral sclerosis delayed the onset of disease64,65. More recently, a lentiviral vector was used to deliver a Smad3-targeted shRNA for regeneration of satellite cells and repair of old tissue in aged and injured muscle66. Viral-vector expression of shRNAs has also been explored in mouse models of neurodegenerative disorders such as Huntington’s disease and Alzheimer’s disease67.
To deliver genes to the central nervous system, adenoviral vectors have proved very useful. For instance, direct brain injection of an adenoviral vector expressing a shRNA directed against the mRNA encoding the polyQ-harbouring SCA1-encoding transcript of spinocerebellar ataxia type 1 was shown to be an effective treatment in a mouse model of this disorder68.
Despite the successes of viral delivery, it is important to bear in mind that although some viruses are non-pathogenic, they are still potentially immunogenic. Another major concern with this technique is the risk of incurring mutations in viral sequences, causing insertional mutagenesis or triggering aberrant gene expression. However, viral vectors can transduce both dividing and non-dividing cells, yield a prolonged expression of the therapeutic gene and need not be delivered in large doses. Ultimately, any therapeutic gene when expressed in large quantities has the potential to cause toxicity and immunogenicity. Critical parameters such as tolerability, long-term expression, efficacy and the ability to regulate expression and targeting should be taken into consideration when choosing a delivery method. There is no ideal delivery system for every application; rather, the delivery method needs to be tailored to the application.
Clinical trials using RNAi to treat human diseases
For a new technology, siRNAs have moved into the clinic at an unprecedented pace. Some examples of the diseases and siRNA-targeting strategies that are currently under investigation are described below.
The first siRNA protocol granted investigational new drug (IND) status and tested in a human clinical trial is the vascular endothelial growth factor (VEGF)-targeted siRNA Bevasiranib (Acuity Pharmaceuticals, Philadelphia, Pennsylvania) for the treatment of wet age-related macular degeneration (see Table 2 for a summary of ongoing siRNA clinical trials). This involves the overgrowth of blood vessels behind the retina, and causes severe and irreversible loss of vision; it affects 1.6 million people in the United States alone, and it is predicted that 11 million individuals worldwide will have the disease by 2013. Preclinical studies of Bevasiranib in mice showed reduced neovascularization resulting from downregulation of Vegf expression after direct ocular injection of the siRNA69. This siRNA, which is now in a phase III trial, is also in a phase II clinical trial for the treatment of diabetic macular oedema. By the conclusion of these trials, several hundred patients will have received the siRNA treatments.
Table 2.
Current clinical trials of RNAi-based therapeutics
| siRNA | Company | Disease | Stage |
|---|---|---|---|
| Bevasiranib | Acuity Pharmaceuticals | Wet age-related macular degeneration | Phase III |
| Diabetic macular oedema | Phase II | ||
| Sirna-027 | Merck–Sirna Therapeutics | Wet age-related macular degeneration | Phase II |
| RTP801i-14 | Quark Pharmaceuticals, and Silence Therapeutics | Wet age-related macular degeneration | Phase I/IIA |
| ALN-RSV01 | Alnylam Pharmaceuticals | Respiratory syncytial virus infection | Phase II |
| NUC B1000 | Nucleonics | Hepatitis B | Phase I |
| Anti-tat/rev shRNA | City of Hope National Medical Center, and Benitec | AIDS | Pilot feasibility study |
| CALAA-01 | Calando Pharmaceuticals | Solid tumours | Phase I |
| TD101 | Trans Derm, and the International Pachyonychia Congenita Consortium | Pachyonychia congenita | Phase I |
Two other companies are also focusing on siRNA-based treatments against macular degeneration: Merck’s Sirna Therapeutics (San Francisco, California) with an siRNA (Sirna-027) that targets the VEGF receptor VEGFR1, and Quark Pharmaceuticals (Fremont, California) in collaboration with Silence Therapeutics (London and Berlin; previously SR Pharma), with one targeted against a hypoxia-inducible gene, RTP801 (also known as DDIT4), that is known to be involved in disease progression. This siRNA, RTP801i-14, has been licensed to Pfizer, UK, which is now running a phase I/IIA clinical trial. Quark Pharmaceuticals has also received IND status for another preclinical trial, in which it is currently enrolling patients. This trial is for an siRNA targeting TP53 mRNA (which encodes the protein p53), inhibition of which delays the induction of cell-death pathways and thereby reduces acute kidney injury after surgery.
Calando Pharmaceuticals (Pasadena, California), meanwhile, has initiated a phase I clinical trial for solid tumours using an siRNA that targets a subunit of ribonucleotide reductase (RRM2), an enzyme required for the synthesis of DNA building blocks. Importantly, this trial is the first to utilize receptor-mediated delivery of siRNAs, which are encapsulated in cyclodextrin particles decorated with transferrin. This results in uptake by cells expressing the transferrin receptor, which is highly expressed on cancer cell surfaces.
The clinical trials performed by Acuity Pharmaceuticals and Merck’s Sirna Therapeutics successfully stabilized patients’ conditions against further degeneration and improved their vision without adverse effects. These results engendered great optimism for intravitreal injection of siRNAs, but in a stunning turn of events a report by Kleinman et al. demonstrated that the observed decrease in vascularization could be a consequence not of an siRNA-specific effect on angiogenesis, but rather a nonspecific activation of Toll-like receptor 3 (TLR3) and subsequent activation of interferon-γ and interleukin 12, which, in turn, downregulate VEGF70. In other words, both the targeted and the control siRNAs mediated nonspecific inhibition of angiogenesis through a direct interaction of the siRNAs with TLR3. Cellular uptake is not necessary for this effect, and because TLR3 is involved in several other cellular pathways the finding has highlighted another level of concern for safe clinical use of siRNAs.
Alnylam Pharmaceuticals (Cambridge, Massachusetts) is a well-established siRNA-therapeutics company whose leading candidate siRNA, ALN-RSV01, is now in a phase II clinical trial. This siRNA targets respiratory syncytial virus — which affects almost 300,000 people every year in the United States alone — by silencing the virus’s nucleocapsid ‘N’ gene, a gene essential to viral replication. ALN-RSV01 was the first antiviral siRNA to enter clinical trials, and trials will soon be expanded to paediatric patients. Thus far it has been shown to be effective and well tolerated. Recently, Alnylam Pharmaceuticals formed an exclusive alliance with Kyowa Hakko Kogyo to develop and commercialize ALN-RSV01 in Japan and other Asian countries.
Also in development at Alnylam Pharmaceuticals are siRNAs directed against genes implicated in hypercholesterolaemia, Huntington’s disease (in a joint venture with Medtronic of Minneapolis, Minnesota), hepatitis C (in a joint venture with Isis Pharmaceuticals in Carlsbad, California), progressive multifocal leukoencephalopathy (in a joint venture with Biogen Idec of Cambridge, Massachusetts) and pandemic flu (in a joint venture with the Swiss company Novartis).
The International Pachyonychia Congenita Consortium (IPCC), in collaboration with TransDerm (Santa Cruz, California), has developed an siRNA to allow the correct production of keratin as a treatment for a rare skin disorder called pachyonychia congenita.
The City of Hope National Medical Center in Duarte, California, in collaboration with Benitec (Melbourne, Australia), has started a phase I trial for the treatment of AIDS lymphoma. This trial uses a Pol III promoter-expressed shRNA targeting the HIV tat and rev shared exons. The shRNA has been incorporated into an HIV-based lentiviral vector, which in turn has been used to insert the shRNA gene (along with two other RNA-based anti-HIV genes) into blood stem cells71. The gene-modified stem cells have been infused into HIV-positive patients in a trial that uses autologous bone marrow transplantation to treat AIDS-related lymphomas. Four patients have now been treated in this trial.
As indicated above, partnerships have become quite accepted in the field of siRNA biotechnology. These consortia are considerably increasing the capital available for these efforts and are shortening the time involved in commercializing siRNA-based drugs.
Some companies, such as Regulus Therapeutics (Carlsbad, California), have chosen to focus on miRNAs as therapeutic targets. Santaris Pharma in Hørsholm, Denmark, has recently started the first phase I trial to target a human miRNA (miR-122). In this trial, miR-122 is being targeted for downregulation with a locked nucleic acid (LNA) anti-miRNA (SPC3649). LNA is a backbone modification that enhances the hybridization of the oligonucleotide with its target and protects it from nuclease degradation. The approach is intended to treat hepatitis C virus infection because miR-122 facilitates replication of this virus in the liver72,73. Downregulation of miR-122 is also potentially useful in the treatment of hypercholesterolaemia. Targeting miRNAs expressed in the heart, such as miR-208, which regulates cardiac hypertrophy and fibrosis74, may have an advantage, because in the medical field there is a considerable experience in delivering drugs directly into this organ.
Gain or loss of miRNA function has been linked to the onset and progression of various diseases75-77. Protein function can be regulated either directly or indirectly by miRNAs, and alterations in miRNA expression can have profound effects on gene regulation. In instances in which disease results from altered miRNA expression, it is conceivable that normal levels could be achieved, either by targeting the specific miRNA if expression is too high or by delivering a miRNA mimic if expression is too low. However, the specificity and efficacy of delivery systems would need to be improved for this goal to be accomplished. Moreover, correct modulation of the targeted miRNA’s expression is not an easy task, and it is not clear whether one miRNA can be specifically targeted without affecting other miRNAs of the same family.
The regulatory complexities of miRNAs should also be taken into consideration when either ablation or restoration of miRNA function is being considered in a therapeutic setting. A single miRNA can regulate the levels of hundreds of proteins78,79, raising cautionary flags about the consequences of downregulating or ectopically expressing even a single miRNA species.
The safety issue
The application of siRNAs to therapeutics has raised a number of concerns about their safety. After the initial excitement, a number of reports underscored potential drawbacks to this promising technology. The first warning came from a study that recorded the deaths of mice after Pol III promoter-driven expression of shRNAs in the liver4. The exact mechanisms leading to mortality are still under investigation, but seem to be due at least in part to saturation of the transport factor, exportin 5, that ferries miRNAs from the nucleus to the cytoplasm. There are now indications that other factors involved in the RNAi process can also be saturated by high-level expression of exogenous siRNAs, which can sequester them from their cognate cellular miRNAs. Because each cellular miRNA can potentially modulate the expression of several hundred genes78,79, minor alterations in the miRNA pathway can have major consequences.
One strategy to mitigate this problem is to use the lowest possible concentration of siRNAs that provides therapeutic efficacy by designing the exogenous siRNAs to be Dicer substrates (by increasing their length). These RNAs enter the RNAi pathway upstream of the RISC at the step of Dicer cleavage, which facilitates passing the siRNA to AGO2 for selection of the guide strand, often resulting in enhanced RNAi at lower concentrations than can be achieved with the exogenous delivery of cognate 21-base siRNAs80-83. Although small amounts of siRNAs are not expected to saturate the RNAi machinery, they can compete with miRNAs for selective incorporation into the RISC5. The long-term consequences of such competition are poorly understood.
With the use of microarrays, it has become increasingly obvious that introducing foreign siRNAs into the cell alters the expression of non-target genes, as well as target genes84,85; as few as six or seven nucleotides complementary to the seed region could result in a specific off-target effect86 through a miRNA-like mechanism. Because microarrays only reflect mRNA levels, they do not take into account any genes affected at the translational level, and so at present it is not clear how extensive the problem of off-target effects really is. Given that the application of synthetic siRNAs results in transient inhibition of gene expression, specific off-targeting may not be a major concern for many clinical applications. Nevertheless, appropriate toxicity testing should take into account the potential for a particular siRNA to target 3′ UTRs in non-target genes.
Some strategies can be used in siRNA design to minimize the problem of off-targeting. For instance, it has been shown that 2′-O-Me modifications87 or DNA substitutions88 in siRNA duplexes can significantly reduce off-target effects. It would also be valuable to improve antisense-strand selectivity by taking into account thermodynamic stability (see ‘Superior designs for small molecules’) or by blocking the 5′ phosphorylation of the sense strand89.
RNAi is a widely conserved mechanism that may originally have evolved to combat viral infections. As such, it is perhaps not surprising that in some cases siRNAs can act as agonists of Toll-like receptors90 and that specific sequence motifs, such as uridine-rich regions and guano sine- and uridine-rich regions, can induce cellular immune responses6,7. The ability of an siRNA to stimulate cellular immune responses is based not only on specific sequences but also on structure, the type of delivery system used and the cell type7,91. Although the immunostimulatory potential of siRNAs could be advantageous in certain circumstances92, it is usually an unwanted outcome. The above-mentioned finding of the TLR3 response to non-sequence-specific modulation of VEGF or the VEGF receptor70, as well as a separate report showing that a macrophage migration inhibitory factor (Mif)-targeted siRNA (in a murine model) and a nonspecific control siRNA increased the proliferation of breast cancer cells through activation of dsRNA-activated protein kinase (PKR)93, raise serious concerns in interpreting the results of in vivo siRNA applications.
Although we have yet to reach a universal solution for avoiding all off-target effects, it is foreseeable that these problems will be overcome by the use of appropriate backbone modifications, as well as delivery systems that can mask RNAs from the receptors of the innate immune system94.
Gazing ahead
Despite the technique’s youth, the list of diseases for which RNAi is being tested as a therapeutic agent is extensive, and includes Parkinson’s disease, Lou Gehrig’s disease, HIV infection, wet age-related macular degeneration, type 2 diabetes, obesity, hypercholesterolaemia, rheumatoid arthritis, respiratory diseases and cancers. It is already a multimillion dollar business, projected to reach US$1 billion by 2010, and intellectual property rights will become an increasingly important concern in the coming years.
However, although much has been accomplished, obstacles remain that will hamper the race to the clinic. The ultimate goal of achieving RNAi-based therapies for life-threatening or debilitating diseases cannot be attained without improving the safety, effectiveness and reliability of RNAi-trigger delivery systems. The use of targeted delivery strategies that permit systemic delivery will be a big step towards fulfilling this difficult task. The development of new, noninvasive imaging methods to monitor the in vivo delivery of siRNAs, such as labelling with near-infrared dyes95, will aid studies of tissue uptake and biodistribution.
Although RNAi is not yet an accepted therapeutic modality, the enormous interest in this phenomenon ensures that we will soon witness fast advances and new applications for RNAi-based therapies. Given the way that RNAi has transformed basic research and the unprecedented speed with which it has reached the clinic, the coming years promise to be exciting.
Acknowledgments
I thank the National Institutes of Health for grant assistance.
Footnotes
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at www.nature.com/nature.
References
- 1.Zamore PD. RNA interference: big applause for silencing in Stockholm. Cell. 2006;127:1083–1086. doi: 10.1016/j.cell.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 2.McCaffrey AP, et al. RNA interference in adult mice. Nature. 2002;418:38–39. doi: 10.1038/418038a.. This study was the first to show siRNA activity in vivo in mammals.
- 3.Song E, et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nature Med. 2003;9:347–351. doi: 10.1038/nm828.. This paper provided the first therapeutic RNAi demonstration in animals.
- 4.Grimm D, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791.. This article raised cautionary concerns about the danger of high-level shRNA expression in animals.
- 5.Castanotto D, et al. Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC. Nucleic Acids Res. 2007;35:5154–5164. doi: 10.1093/nar/gkm543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hornung V, et al. Sequence-specific potent induction of IFN-α by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nature Med. 2005;11:263–270. doi: 10.1038/nm1191. [DOI] [PubMed] [Google Scholar]
- 7.Judge AD, et al. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nature Biotechnol. 2005;23:457–462. doi: 10.1038/nbt1081. [DOI] [PubMed] [Google Scholar]
- 8.Gantier MP, Williams BR. The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev. 2007;18:363–371. doi: 10.1016/j.cytogfr.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elbashir SM, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107.. This study was the first to show that RNAi triggers can work in mammalian cells without stimulating interferon pathways.
- 10.Tolia NH, Joshua-Tor L. Slicer and the argonautes. Nature Chem Biol. 2007;3:36–43. doi: 10.1038/nchembio848. [DOI] [PubMed] [Google Scholar]
- 11.Zamore PD, Tuschl T, Sharp PA, Bartel DP. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000;101:25–33. doi: 10.1016/S0092-8674(00)80620-0. [DOI] [PubMed] [Google Scholar]
- 12.Matzke MA, Birchler JA. RNAi-mediated pathways in the nucleus. Nature Rev Genet. 2005;6:24–35. doi: 10.1038/nrg1500. [DOI] [PubMed] [Google Scholar]
- 13.Wassenegger M. The role of the RNAi machinery in heterochromatin formation. Cell. 2005;122:13–16. doi: 10.1016/j.cell.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 14.Wassenegger M, Heimes S, Riedel L, Sanger HL. RNA-directed de novo methylation of genomic sequences in plants. Cell. 1994;76:567–576. doi: 10.1016/0092-8674(94)90119-8. [DOI] [PubMed] [Google Scholar]
- 15.Morris KV, Chan SW, Jacobsen SE, Looney DJ. Small interfering RNA-induced transcriptional gene silencing in human cells. Science. 2004;305:1289–1292. doi: 10.1126/science.1101372. [DOI] [PubMed] [Google Scholar]
- 16.Castanotto D, et al. Short hairpin RNA-directed cytosine (CpG) methylation of the RASSF1A gene promoter in HeLa cells. Mol Ther. 2005;12:179–183. doi: 10.1016/j.ymthe.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Ting AH, Schuebel KE, Herman JG, Baylin SB. Short double-stranded RNA induces transcriptional gene silencing in human cancer cells in the absence of DNA methylation. Nature Genet. 2005;37:906–910. doi: 10.1038/ng1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verdel A, et al. RNAi-mediated targeting of heterochromatin by the RITS complex. Science. 2004;303:672–676. doi: 10.1126/science.1093686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janowski BA, et al. Involvement of AGO1 and AGO2 in mammalian transcriptional silencing. Nature Struct Mol Biol. 2006;13:787–792. doi: 10.1038/nsmb1140. [DOI] [PubMed] [Google Scholar]
- 20.Kim DH, Villeneuve LM, Morris KV, Rossi JJ. Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nature Struct Mol Biol. 2006;13:793–797. doi: 10.1038/nsmb1142. [DOI] [PubMed] [Google Scholar]
- 21.Kim DH, Saetrom P, Snove O, Rossi JJ., Jr MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc Natl Acad Sci USA. 2008;105:16230–16235. doi: 10.1073/pnas.0808830105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aravin AA, Hannon GJ, Brennecke J. The Piwi–piRNA pathway provides an adaptive defense in the transposon arms race. Science. 2007;318:761–764. doi: 10.1126/science.1146484. [DOI] [PubMed] [Google Scholar]
- 23.Klattenhoff C, Theurkauf W. Biogenesis and germline functions of piRNAs. Development. 2008;135:3–9. doi: 10.1242/dev.006486. [DOI] [PubMed] [Google Scholar]
- 24.Batista PJ, et al. PRG-1 and 21U-RNAs interact to form the piRNA complex required for fertility in C. elegans. Mol Cell. 2008;31:67–78. doi: 10.1016/j.molcel.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Das PP, et al. Piwi and piRNAs act upstream of an endogenous siRNA pathway to suppress Tc3 transposon mobility in the Caenorhabditis elegans germline. Mol Cell. 2008;31:79–90. doi: 10.1016/j.molcel.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghildiyal M, et al. Endogenous siRNAs derived from transposons and mRNAs in Drosophila somatic cells. Science. 2008;320:1077–1081. doi: 10.1126/science.1157396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Czech B, et al. An endogenous small interfering RNA pathway in Drosophila. Nature. 2008;453:798–802. doi: 10.1038/nature07007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawamura Y, et al. Drosophila endogenous small RNAs bind to Argonaute 2 in somatic cells. Nature. 2008;453:793–797. doi: 10.1038/nature06938. [DOI] [PubMed] [Google Scholar]
- 29.Okamura K, et al. The Drosophila hairpin RNA pathway generates endogenous short interfering RNAs. Nature. 2008;453:803–806. doi: 10.1038/nature07015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tam OH, et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008;453:534–538. doi: 10.1038/nature06904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watanabe T, et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008;453:539–543. doi: 10.1038/nature06908. [DOI] [PubMed] [Google Scholar]
- 32.Matranga C, Tomari Y, Shin C, Bartel DP, Zamore PD. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell. 2005;123:607–620. doi: 10.1016/j.cell.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 33.Rand TA, Petersen S, Du F, Wang X. Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell. 2005;123:621–629. doi: 10.1016/j.cell.2005.10.020.. References 32 and 33 were the first studies to demonstrate the mechanism of guide-strand selection for siRNAs.
- 34.Li W, Cha L. Predicting siRNA efficiency. Cell Mol Life Sci. 2007;64:1785–1792. doi: 10.1007/s00018-007-7057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tafer H, et al. The impact of target site accessibility on the design of effective siRNAs. Nature Biotechnol. 2008;26:578–583. doi: 10.1038/nbt1404. [DOI] [PubMed] [Google Scholar]
- 36.Huesken D, et al. Design of a genome-wide siRNA library using an artificial neural network. Nature Biotechnol. 2005;23:995–1001. doi: 10.1038/nbt1118. [DOI] [PubMed] [Google Scholar]
- 37.Morrissey DV, et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nature Biotechnol. 2005;23:1002–1007. doi: 10.1038/nbt1122. [DOI] [PubMed] [Google Scholar]
- 38.Robbins M, et al. 2′-O-Methyl-modified RNAs act as TLR7 antagonists. Mol Ther. 2007;15:1663–1669. doi: 10.1038/sj.mt.6300240. [DOI] [PubMed] [Google Scholar]
- 39.Dowler T, et al. Improvements in siRNA properties mediated by 2′-deoxy-2′-fluoro-β-d-arabinonucleic acid (FANA) Nucleic Acids Res. 2006;34:1669–1675. doi: 10.1093/nar/gkl033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watts JK, et al. 2′-Fluoro-4′-thioarabino-modified oligonucleotides: conformational switches linked to siRNA activity. Nucleic Acids Res. 2007;35:1441–1451. doi: 10.1093/nar/gkl1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fisher M, et al. Inhibition of MDR1 expression with altritol-modified siRNAs. Nucleic Acids Res. 2007;35:1064–1074. doi: 10.1093/nar/gkl1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lorenz C, Hadwiger P, John M, Vornlocher H-P, Unverzagt C. Steroid and lipid conjugates of siRNAs to enhance cellular uptake and gene silencing in liver cells. Bioorg Med Chem Lett. 2004;14:4975–4977. doi: 10.1016/j.bmcl.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 43.Howard KA, et al. RNA interference in vitro and in vivo using a novel chitosan/siRNA nanoparticle system. Mol Ther. 2006;14:476–484. doi: 10.1016/j.ymthe.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 44.Bitko V, Musiyenko A, Shulyayeva O, Barik S. Inhibition of respiratory viruses by nasally administered siRNA. Nature Med. 2005;11:50–55. doi: 10.1038/nm1164. [DOI] [PubMed] [Google Scholar]
- 45.Li BJ, et al. Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in Rhesus macaque. Nature Med. 2005;11:944–951. doi: 10.1038/nm1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palliser D, et al. An siRNA-based microbicide protects mice from lethal herpes simplex virus 2 infection. Nature. 2006;439:89–94. doi: 10.1038/nature04263. [DOI] [PubMed] [Google Scholar]
- 47.Rozema DB, et al. Dynamic polyconjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc Natl Acad Sci USA. 2007;104:12982–12987. doi: 10.1073/pnas.0703778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci USA. 2007;104:15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soutschek J, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 50.Zimmermann TS, et al. RNAi-mediated gene silencing in non-human primates. Nature. 2006;441:111–114. doi: 10.1038/nature04688. [DOI] [PubMed] [Google Scholar]
- 51.Peer D, Park EJ, Morishita Y, Carman CV, Shimaoka M. Systemic leukocyte-directed siRNA delivery revealing cyclin D1 as an anti-inflammatory target. Science. 2008;319:627–630. doi: 10.1126/science.1149859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dorn G, et al. siRNA relieves chronic neuropathic pain. Nucleic Acids Res. 2004;32:e49. doi: 10.1093/nar/gnh044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawasaki Y, et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nature Med. 2008;14:331–336. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dore-Savard L, et al. Central delivery of Dicer-substrate siRNA: a direct application for pain research. Mol Ther. 2008;16:1331–1339. doi: 10.1038/mt.2008.98. [DOI] [PubMed] [Google Scholar]
- 55.Shishkina GT, Kalinina TS, Dygalo NN. Attenuation of α2A-adrenergic receptor expression in neonatal rat brain by RNA interference or antisense oligonucleotide reduced anxiety in adulthood. Neuroscience. 2004;129:521–528. doi: 10.1016/j.neuroscience.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 56.Pardridge WM. shRNA and siRNA delivery to the brain. Adv Drug Deliv Rev. 2007;59:141–152. doi: 10.1016/j.addr.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kumar P, et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901.. This paper demonstrated the important concept that an acetylcholine-receptor-binding peptide–polyarginine conjugate can deliver siRNAs across the blood–brain barrier.
- 58.Song E, et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nature Biotechnol. 2005;23:709–717. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- 59.McNamara JO, et al. Cell type-specific delivery of siRNAs with aptamer–siRNA chimeras. Nature Biotechnol. 2006;24:1005–1015. doi: 10.1038/nbt1223. [DOI] [PubMed] [Google Scholar]
- 60.Chu TC, Twu KY, Ellington AD, Levy M. Aptamer mediated siRNA delivery. Nucleic Acids Res. 2006;34:e73. doi: 10.1093/nar/gkl388.. References 59 and 60 were the first to show aptamer-mediated delivery of siRNAs to a specific cellular receptor.
- 61.Sato Y, et al. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nature Biotechnol. 2008;26:431–442. doi: 10.1038/nbt1396. [DOI] [PubMed] [Google Scholar]
- 62.Akinc A, et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nature Biotechnol. 2008;26:561–569. doi: 10.1038/nbt1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell. 2002;2:243–247. doi: 10.1016/s1535-6108(02)00122-8. [DOI] [PubMed] [Google Scholar]
- 64.Raoul C, et al. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nature Med. 2005;11:423–428. doi: 10.1038/nm1207. [DOI] [PubMed] [Google Scholar]
- 65.Ralph GS, et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nature Med. 2005;11:429–433. doi: 10.1038/nm1205. [DOI] [PubMed] [Google Scholar]
- 66.Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008;454:528–532. doi: 10.1038/nature07034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Farah MH. RNAi silencing in mouse models of neurodegenerative diseases. Curr Drug Deliv. 2007;4:161–167. doi: 10.2174/156720107780362276. [DOI] [PubMed] [Google Scholar]
- 68.Xia H, et al. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nature Med. 2004;10:816–820. doi: 10.1038/nm1076. [DOI] [PubMed] [Google Scholar]
- 69.Shen J, et al. Suppression of ocular neovascularization with siRNA targeting VEGF receptor 1. Gene Ther. 2006;13:225–234. doi: 10.1038/sj.gt.3302641. [DOI] [PubMed] [Google Scholar]
- 70.Kleinman ME, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591–597. doi: 10.1038/nature06765.. This study found that macular vascularization could be inhibited in a non-sequence-specific manner by siRNA-mediated activation of TLR3.
- 71.Li MJ, et al. Inhibition of HIV-1 infection by lentiviral vectors expressing Pol III-promoted anti-HIV RNAs. Mol Ther. 2003;8:196–206. doi: 10.1016/s1525-0016(03)00165-5. [DOI] [PubMed] [Google Scholar]
- 72.Chang J, et al. Liver-specific microRNA miR-122 enhances the replication of hepatitis C virus in nonhepatic cells. J Virol. 2008;82:8215–8223. doi: 10.1128/JVI.02575-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Randall G, et al. Cellular cofactors affecting hepatitis C virus infection and replication. Proc Natl Acad Sci USA. 2007;104:12884–12889. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Rooij E, et al. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 75.Calin GA, Croce CM. MicroRNA–cancer connection: the beginning of a new tale. Cancer Res. 2006;66:7390–7394. doi: 10.1158/0008-5472.CAN-06-0800. [DOI] [PubMed] [Google Scholar]
- 76.Esau CC, Monia BP. Therapeutic potential for microRNAs. Adv Drug Deliv Rev. 2007;59:101–114. doi: 10.1016/j.addr.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 77.Soifer HS, Rossi JJ, Saetrom P. MicroRNAs in disease and potential therapeutic applications. Mol Ther. 2007;15:2070–2079. doi: 10.1038/sj.mt.6300311. [DOI] [PubMed] [Google Scholar]
- 78.Baek D, et al. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Selbach M, et al. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 80.Amarzguioui M, et al. Rational design and in vitro and in vivo delivery of Dicer substrate siRNA. Nature Protoc. 2006;1:508–517. doi: 10.1038/nprot.2006.72. [DOI] [PubMed] [Google Scholar]
- 81.Rose SD, et al. Functional polarity is introduced by Dicer processing of short substrate RNAs. Nucleic Acids Res. 2005;33:4140–4156. doi: 10.1093/nar/gki732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim DH, et al. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nature Biotechnol. 2005;23:222–226. doi: 10.1038/nbt1051. [DOI] [PubMed] [Google Scholar]
- 83.Siolas D, et al. Synthetic shRNAs as potent RNAi triggers. Nature Biotechnol. 2005;23:227–231. doi: 10.1038/nbt1052. [DOI] [PubMed] [Google Scholar]
- 84.Scacheri PC, et al. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc Natl Acad Sci USA. 2004;101:1892–1897. doi: 10.1073/pnas.0308698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jackson AL, et al. Expression profiling reveals off-target gene regulation by RNAi. Nature Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- 86.Birmingham A, et al. 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nature Methods. 2006;3:199–204. doi: 10.1038/nmeth854. [DOI] [PubMed] [Google Scholar]
- 87.Jackson AL, et al. Position-specific chemical modification of siRNAs reduces ‘off-target’ transcript silencing. RNA. 2006;12:1197–1205. doi: 10.1261/rna.30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ui-Tei K, et al. Functional dissection of siRNA sequence by systematic DNA substitution: modified siRNA with a DNA seed arm is a powerful tool for mammalian gene silencing with significantly reduced off-target effect. Nucleic Acids Res. 2008;36:2136–2151. doi: 10.1093/nar/gkn042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chiu YL, Rana TM. RNAi in human cells: basic structural and functional features of small interfering RNA. Mol Cell. 2002;10:549–561. doi: 10.1016/s1097-2765(02)00652-4. [DOI] [PubMed] [Google Scholar]
- 90.Agrawal S, Kandimalla ER. Role of Toll-like receptors in antisense and siRNA. Nature Biotechnol. 2004;22:1533–1537. doi: 10.1038/nbt1042. [DOI] [PubMed] [Google Scholar]
- 91.Judge AD, Bola G, Lee AC, MacLachlan I. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol Ther. 2006;13:494–505. doi: 10.1016/j.ymthe.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 92.Schlee M, Hornung V, Hartmann G. siRNA and isRNA: two edges of one sword. Mol Ther. 2006;14:463–470. doi: 10.1016/j.ymthe.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 93.Armstrong ME, et al. Small interfering RNAs induce macrophage migration inhibitory factor production and proliferation in breast cancer cells via a double-stranded RNA-dependent protein kinase-dependent mechanism. J Immunol. 2008;180:7125–7133. doi: 10.4049/jimmunol.180.11.7125. [DOI] [PubMed] [Google Scholar]
- 94.Sioud M. Does the understanding of immune activation by RNA predict the design of safe siRNAs? Front Biosci. 2008;13:4379–4392. doi: 10.2741/3011. [DOI] [PubMed] [Google Scholar]
- 95.Medarova Z, Pham W, Farrar C, Petkova V, Moore A. In vivo imaging of siRNA delivery and silencing in tumors. Nature Med. 2007;13:372–377. doi: 10.1038/nm1486. [DOI] [PubMed] [Google Scholar]