

Abstract
Here we review how GFAP mutations cause Alexander disease. The current data suggest that a combination of events cause the disease. These include: i) the accumulation of GFAP and the formation of characteristic aggregates, called Rosenthal fibres, ii) the sequestration of the protein chaperones αB-crystallin and HSP27 into Rosenthal fibres, and iii) the activation of both Jnk and the stress response. These then set in motion events that lead to Alexander disease. We discuss parallels with other intermediate filament diseases and assess potential therapies as part of this review as well as emerging trends in disease diagnosis and other aspects concerning GFAP.
Keywords: Alexander disease, GFAP, chaperone, alphaB-crystallin, Jnk, MLK
Alexander Disease – A brief overview
Alexanders disease (OMIM #203450) was first described by W. S. Alexander [1]. It is a rare, but often fatal neurological disorder that has been divided into three subtypes based on the age of onset (Table 1): the infantile, juvenile and adult forms [2]. The infantile form, with onset between birth and about two years of age, is currently the most common form of the disease [3-5]. The characteristic neuropathological feature of all forms of Alexander's disease is the presence of Rosenthal fibres (Figure 1). These unique cytoplasmic inclusions are found within astrocytes and contain GFAP [6, 7], the major astrocytic intermediate filament protein, that like other cytoplasmic intermediate filament proteins is found in individual filaments and bundles within the cytoplasm, passing from the perinuclear region to the cell periphery of the cell. Although GFAP can coassemble with vimentin in such cells [8], neither nestin nor vimentin are required for GFAP to form intermediate filaments [9].
Table 1.
Age of onset classification of Alexander disease
| ALEXANDER DISEASE | ||
|---|---|---|
| Category | Age of Onset (years) |
Associated Phenotypes |
| Infantile | 0-2 | The clinical and pathological features associated with Alexander disease are related to the age of onset. Alexander disease is a progressive neurological disorder. Whilst in infantile cases, macrocephaly, seizures, and impaired cognitive function are relatively common, such phenotypes become progressively less common for the juvenile and adult cases. Rosenthal fibres are the classical histopathological feature of the disease and MRI is an extremely important diagnostic tool, but genotyping is the most secure method for confirming Alexander disease. |
| Juvenile | 2-12 | |
| Adult | >12 | |
Figure 1.
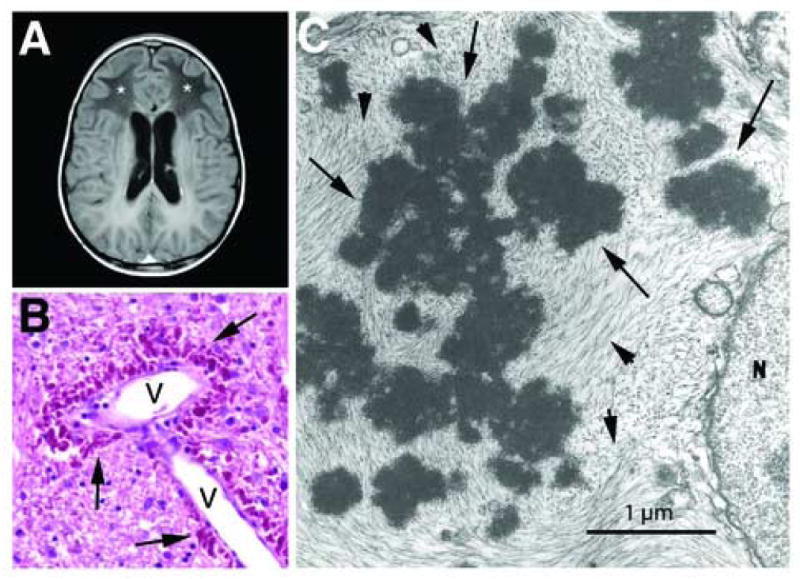
Features of Alexander disease. A. MR image of a 10-year-old boy with Alexander disease, showing extensive abnormalities (*) of the cerebral white matter with frontal predominance. (generously provided by Dr. Marjo van der Knaap [17]). Reprinted with permission from Elsevier.
B. Rosenthal fibres (arrows) concentrated in the astrocytic endfeet surrounding a blood vessel (V) in the brain stem of a 1-year-old child with Alexander disease. Hematoxylin & eosin stain, paraffin section. Reprinted with permission from Elsevier [81].
C. Rosenthal fibres (arrows) surrounded by intermediate filaments (arrowheads) in an astrocyte cell body from a 17-month old child with Alexander disease, viewed by transmission electron microscopy (reprinted from [82], with permission of Wiley-Liss, Inc., a subsidiary of John Wiley & Sons, Inc.).
GFAP is a type III intermediate filament protein transcribed from a 10kb region located on chromosome 17q21 (chromosome 11 in the mouse). The gene is conserved, existing as a single copy in the human [10] and rodent genomes [11] producing a major protein product α-GFAP that is 91% identical (95% similarity) between humans and mice at the amino acid level [12]. Although GFAP is alternatively spliced (reviewed in [13]), the major splice variant termed α-GFAP, is expressed at highest levels in astrocytes of the central nervous system.
The genetic basis of Alexander's disease was recently defined [14] when de novo mutations in four different GFAP residues, R79, R239, R258, and R416 were identified in 12 unrelated individuals. The decision to search for mutations in the GFAP gene of these individuals was in part due to the observation that Rosenthal fibre formation could be induced by the overexpression of human GFAP in transgenic mice in a dose dependent manner [15]. Subsequent work by a number of investigators supports the idea that Alexander disease is a genetically homogeneous disorder, with >95% of patients who meet recognized MRI criteria for diagnosis having GFAP mutations [16]. In addition, two obvious hot-spots for mutations exist, with approximately half of all patients affected at either the R79 or R239. Many other mutations, however, are distributed throughout the other protein domains (Figure 2).
Figure 2.

Location of Alexander disease-associated mutations in GFAP in relation to protein domain structure of intermediate filaments. The red boxes indicate the four α-helical sub-domains within the central rod domain, separated by non-helical linkers. The grey boxes at the ends of helix 1A and 2B reflect the location of the highly conserved LNDR- and TYRKLLEGE-motifs and correspond to residues L76-R79 and T365-E373 respectively in GFAP. Each letter (on the right) represents an individual patient, using the single letter code for the mutated amino acid. Multiple letters adjacent to numbered residues indicate the number of independent mutations linked to that residue. Only one letter is shown for familial patients or identical twins. Symbols are color coded for clinical category based on age of onset (infantile, juvenile, or adult). A boxed letter indicates that this mutation was inherited and found in multiple family members. If a particular family with an inherited mutation contained affected individuals with variable phenotypes (for instance, some juvenile, and some adult), the classification for the proband only is shown. A circle around a mutation indicates that the patient was asymptomatic at the time of diagnosis. Mutations or polymorphisms shown on the left are presently considered innocuous and not responsible for disease (although there is one report of putative Alexander disease in a patient with E223Q). The diagram includes all patients published as of 1/07, as well as some unpublished patients. A continually updated list of mutations, along with links to the relevant publications, is available at the Alexander disease web site maintained at the University of Wisconsin-Madison (www.waisman.wisc.edu/alexander).
Early clinical descriptions of Alexander disease emphasized macrocephaly, frontal leukodystrophy, and a variety of developmental delays, but based on reliable genetic diagnoses and larger numbers of patients, it is now clear that these features are not always present [16, 17]. Macrocephaly is frequent in the R239 patients, but only found half the time in the others. Leukodystrophy is common in the infantile group, but progressively less so in the older patients, and sometimes not found at all [18, 19]. Seizures, cognitive delays and/or regression, are common symptoms, but again these are not found in all patients. In the adult onset cases, there are a number of clinical features that frequently coincide with mutations in GFAP and these include palatal tremors, dysphagia, and other bulbar and pseudobulbar signs.
While the genetic basis for Alexander disease is now firmly established, little is known regarding the mechanisms by which GFAP mutations lead to disease [20]. In this review we concentrate on recent studies that have appeared during the past two years that bear on these questions, emphasizing model systems such as in vitro assembly, cell culture, and mouse models.
Update on GFAP mutations and their origins
A recent study [16] significantly expanded the number of Alexander disease mutations that have been published for GFAP, and other isolated case reports or small groups of patients continue to appear in the literature. The current status of GFAP mutations is shown in Fig. 1. The mutation hot spots in GFAP contain CpG dinucleotides which are substrates for methylation-induced deamination [21] and includes the R79 position, which is the same arginine in the LNDR-motif that is frequently mutated in other intermediate filament genes that cause disease. Mutations have now been reported that alter 36 of the 432 GFAP amino acids, converting them to 50 different substitutions (Figure 2). The R79 and R239 positions hot spots have produced the greatest variety of substitutions: C, H, L, G and S for R79 (only P is missing among possible single nucleotide mutations), and C, H, L and P for R239 (only G and S are missing). New mutations continue to be reported at a high rate. For example, in an analysis of 43 putative Alexander disease cases, Li et al. [16] found 27 to carry novel changes. In the past year (2006), 5 additional novel amino substitutions were reported, as well as 2 mutations involving insertions [19, 22-26]. Nearly all of the Alexander disease mutations involve amino acid substitutions, but several insertion or deletion/insertion alterations have also been reported [16, 19]. Several instances of vertical transmission of GFAP mutations have been reported, but the large majority arise de novo (reviewed in [16]). Some GFAP amino acid substitutions, however, do not produce disease. D157N is found in normal controls at a frequency of about 4%, and it is uncertain whether P47L, V115I and E223Q are disease causing or innocuous polymorphisms [16]. All the mutations are heterozygous, and none results in premature termination, including the insertion/deletions. These observations are consistent with the mutations producing disease by a dominant gain of function. The distribution of the mutations throughout the GFAP sequence, and possible genotype/phenotype correlations, have been recently discussed [16].
In order for the heterozygous mutations to be detected over background noise by the sequencing procedures used, they must be present in at least 50% of the cells in the analyzed sample. This suggests that the de novo mutations occur either in the germ line of one of the parents or in the first stages of development. This question was recently addressed by determining whether the chromosome bearing the mutation had been inherited from the father or mother [27]. Point mutations arise in the paternal germline much more frequently than in the maternal (reviewed in [28]). Thus if the GFAP mutations were occurring in a parental germ cell rather than early in the affected individual's development, the mutation would more often be found on the paternal chromosome. This proved to be the case; in 24 of the 28 cases analyzed, the paternal chromosome carried the GFAP mutation (P<0.001). The ratio obtained of 6:1 for paternal to maternal mutations is consistent with indirect estimates of mutation rate based on the rate of evolution of the sex chromosome relative to the autosomic chromosomes, and thus strongly argues that Alexander disease GFAP mutations arise in the parental germline [27]. Exceptions are possible and an infantile case is currently being examined in which the GFAP mutation is mosaic, strongly suggesting a somatic origin rather than originating in a parental germ line.
Model systems
The discovery of the GFAP mutations paved the way to develop model systems in tissue culture cells and transgenic mice to study Alexander Disease. Previous observations about the disease, such as the accumulation of GFAP in Rosenthal fibres along with several stress proteins, including αB-crystallin and HSP27, and the phosphorylation and ubiquitination of the Rosenthal fibre components, are all clues about the disease mechanism. Several appealing hypotheses have been suggested concerning the sequence of events following the expression of the mutant GFAPs. These include the sequestration of protein chaperones, the hyperactivation of the stress response and the altered assembly and subsequent accumulation of GFAP in aggregates. The combination of these downstream consequences then ultimately compromises astrocyte function to impact other cells of the CNS. There is a growing realization that astrocytes, have much more than a support role in the brain [29] as they help form the blood brain barrier, protect neurons against neurotransmitter excesses, promote synaptic plasticity and coordinate neuronal activity via direct communication with neurons (reviewed in [30]).
Over-expression of wild type GFAP in mice
The idea that elevated levels of GFAP contribute to astrocyte dysfunction in Alexander disease began with the initial studies of over-expressing wild type GFAP in transgenic mice, which resulted in the formation of Rosenthal fibres indistinguishable from those found in Alexander disease patients [15]. Recently the effects of wild type GFAP over-expression was studied in these same mice by microarray analysis. Focusing on olfactory bulbs from the Tg73.7 line, expression profiles at two different ages (3 wks, 4 mo) were compared to understand how the pathology progresses in these mice [31]. This revealed a stress response that included genes involved in glutathione metabolism, peroxide detoxification, and iron homeostasis. Many of these genes are regulated by the transcription factor Nrf2 (also known as Nfe2l2) via a short anti-oxidant response element (ARE) in their promoters; NRF2 was also increased. Utilizing a transgenic line that places the human placental alkaline phosphatase reporter gene (hPAP) under the control of the ARE, the same studies revealed dramatic induction of the ARE-driven reporter throughout the brain and spinal cord. An immune-related response occurs as evidenced by activation of cytokine and cytokine receptor genes, complement components, and acute phase response genes. These transcripts are further elevated with age, with additional induction of macrophage-specific markers such as Mac1 and CD68, suggesting activation of microglia. At 4 months, decreased expression of genes for microtubule associated proteins, vesicular trafficking proteins, and neurotransmitter receptors becomes apparent. Interneuron-specific transcription factors including Dlx family members and Pax6 are down-regulated as well as Gad1 and Gad2, suggesting impairment of GABAergic granule cells. Together these data implicate an initial stress response by astrocytes that results in the activation of microglia and compromised neuronal function. These studies highlight the dramatic up-regulation of microglial genes as well as impairment of neuronal function that likely takes place in Alexander disease.
These studies also provided the first direct demonstration that GFAP expression is increased in Alexander disease brain, at both the mRNA [31] and protein levels [32]. Previously this finding had been generally assumed based on the known histopathology where gliosis was a common feature. Other key genes identified through the microarray analysis of the mice were subsequently evaluated in brain samples from Alexander disease patients, documenting elevations of NQO1 (a stress protein oxidoreductase that is regulated by Nrf2), ITGAM (a microglial marker), and ceruloplasmin (a copper-binding ferroxidase) [31].
Expression of Alexander disease causing GFAP point mutations in mice
More recently, knock-in mice with missense mutations homologous to two common mutations found in Alexander disease patients, R79H and R239H, have been generated that mimic the actual genetic defect present in the human disease [33]. Because mouse GFAP has three fewer amino acids in the head domain, the corresponding numbers for these mouse mutations are R76H and R236H. Mice with these mutations spontaneously develop Rosenthal fibres, in multiple sites throughout the CNS. Astrocytes in these areas appear reactive and total GFAP expression is elevated. Although general white matter architecture and myelination appear normal, when crossed with an anti-oxidant response element reporter line, the mutant mice show a distinct pattern of reporter gene induction that is especially prominent in the hippocampus, corpus callosum, and sub-pial regions. Histochemical staining also reveals an accumulation of iron in these same regions. The mutant mice have a normal lifespan and show no overt behavioral defects, but are smaller than wild type littermates and show increased susceptibility to kainate-induced seizures. While the mutant mice have higher than normal GFAP expression by themselves, further elevation of GFAP via crosses to GFAP transgenic animals leads to a shift in GFAP solubility, an increased stress response, and ultimately death by 35 days. Video recordings of these mice suggest that prolonged seizures are the proximate cause of death. The knock-in mice do not display the full spectrum of pathology observed in human infantile Alexander disease, but more closely resemble the juvenile or adult forms of the disease. Thus the knock-in mice provide formal proof linking GFAP mutations with Rosenthal fibres and oxidative stress. In addition, conditional lethality induced by increasing GFAP expression highlights the potential significance of GFAP levels per se, independent of mutant protein.
A similar increased susceptibility to kainate-induce seizures has been observed in mice expressing mutant human GFAP as a transgene [34]. Since seizures are one of the most common symptoms associated with the human disease these two mouse models may help elucidate the physiological link between aberrant intermediate filament expression in astrocytes and the resulting hyperexcitability in neurons.
Cell models – changes in protein properties trigger GFAP accumulation
To determine the initiating events that accompany the expression of Alexander disease causing mutations in GFAP, a number of cell studies have revealed that the presence of the mutant GFAPs in filaments causes a variety of effects that include altered filament stability [35], increased GFAP levels [36], increased association with plectin [32], and the accumulation of αB-crystallin and HSP27 into aggregates that resemble Rosenthal fibres [37]. These events trigger the activation of Jnk [36]. There are therefore important parallels with other intermediate filament based diseases, for instance in the activation of stress pathways [38, 39] and the problem created by excess protein [40].
Of course, such studies assume that patients with Alexander disease express and accumulate the mutant protein in the Rosenthal fibres. This was formally proven by the generation of antibodies specific to the mutant R416W GFAP [37]. These antibodies were then used to label sections from Alexander disease brain and show that the mutant GFAP was found in both the Rosenthal fibres and also in filamentous patterns of astrocytes [37]. In fact, this was the first demonstration for any intermediate filament disease that the mutant protein was a component of the histopathological features that most typified the disease [37].
GFAP mutants alter filament properties
When R239C GFAP was expressed into SW13Vim- cells, the protein tended not to form filaments, but instead gave diffuse staining patterns comprising many distinct, variable foci [35]. This GFAP material was more resistant to extraction with Triton containing buffers. The results of the in vitro assembly revealed that whilst the GFAP still retained the ability to assemble, the filaments appeared less uniform and also had the tendency to pellet more efficiently in sedimentation assays. When transfected into primary rat astrocytes, the R239C mutant could induce intermediate filament aggregation, but also incorporate successfully into the endogenous GFAP networks. These were the first data to suggest that the mutations didn't necessarily prevent GFAP assembly. The transfection into SW13Vim- cells, however, showed that the mutation clearly compromised its assembly ability and by transfection into the primary astrocytes the endogenous filament network provided the filament scaffold into which the R239 GFAP could assemble [35]. This observation has been made for other GFAP mutations that cause Alexander disease (K63Q; E210K, A253G; [16]), but some mutants retain the ability to assemble in the absence of other partners (A244V; [16]). The R416W GFAP has the most dramatic effects upon in vitro assembly of any of the mutants tested thus far, but this mutant was still capable of incorporation into filament networks in transfected cells when assembly partners, such as wild type GFAP or vimentin, were available [37]. The parallel with Alexander disease is that Rosenthal fibres and 10nm filaments are always present and therefore the disease is probably not the result of the prevention of GFAP assembly, but rather of changed filament properties.
These observations also suggest two further possibilities. First, the presence of the mutant protein would increase protein stability and, second, there was perhaps altered association with other proteins that would lead to the formation of protein aggregates. In the latter, plectin appears to be a very important associated protein and it is certainly required for the subcellular distribution of R239C GFAP [32]. The expression of R239C GFAP in plectin-/- fibroblasts produced a preponderance of aggregates and restoring plectin by cotransfection prevented GFAP aggregate formation [32]. Thus this cytoskeletal cross-linker [41] could play a role in the developing pathology, especially if its levels are altered as part of the disease.
GFAP mutants trigger the stress response
One of the downstream consequences of the expression of R239C GFAP is the activation of the stress kinases Jnk and the upstream mixed lineage kinases (MLK) MLK2 and 3 and ASK1 [36]. The coexpression of a dominant negative Jnk along with both wild type and R239 GFAP tagged constructs restored the extraction of GFAP into low ionic strength, Triton-containing buffers. Some of the downstream consequences of GFAP overexpression included a compromised ubiquitin-proteosome system and the accumulation of the small heat shock protein αB-crystallin [36]. GFAP overexpression made the transfected cells more sensitive to the apoptogen, camptothecin, demonstrating that there is indeed a functional consequence to increased GFAP expression as reported in an Alexander disease mouse model [31] and in samples from Alexander disease brain tissue [36]. As activated Jnk is found in Alexander disease samples both by immunoblotting and immunohistochemistry [36], collectively these data suggest that the Jnk pathway is important in propagating the deleterious effects of R239C GFAP.
Overexpression of FLAG-tagged wild type human GFAP also induced similar responses in Jnk, MLK and ASK activation and also caused GFAP aggregation and the loss of proteosome activity [36]. Despite these similarities, cell lines overexpressing FLAG tagged wild type GFAP were not as sensitive to the apoptogen, camptothecin, as those cell lines expressing FLAG-tagged R239C GFAP and therefore, something in addition to the Jnk pathway is needed to mediate the different responses to camptothecin by cells transfected with wild type and R239C GFAP. Although MLKs and associated pathways are targets for the treatment of other neurodegenerative diseases [42], further work will be required to finalise such targets in Alexander disease.
Chaperone sequestration – a feature of Rosenthal fibres and a factor in disease progression
One of the consistent features of the disease is the overexpression of αB-crystallin [15, 43, 44]. This is also a consistent feature of recent animal [31] and cell models [36, 37] that suggests another facet to Alexander disease development. The sequestration of both HSP27 and αB-crystallin into the aggregates of GFAP will likely attenuate the ability of the astrocytes to resist stress [45-47] and prevent apoptosis [48]. In a recent cell model that investigated the consequences of the expression of R416W GFAP in human astrocyte cell lines, this was precisely one of the consequences. Both the small heat shock proteins HSP27 and αB-crystallin accumulated in the filament aggregates whereas the other protein chaperone HSP70 did not [37] and both sHSPs can prevent filament associations in vitro, whereas HSP70 is unable to do so [47]. A shift of αB-crystallin to the cytoskeletal fraction has been reported before as a response to stress in GFAP expressing glioma cells [49]. These observations and the fact that αB-crystallin can regulate GFAP filament interactions in vitro [47] help explain why the overexpression of αB-crystallin is capable of disaggregating GFAP accumulations [50]. The association of this chaperone with GFAP filaments is therefore of functional relevance.
Future directions
Alternative spliced forms of GFAP and their function
Most intermediate filament genes are not alternatively spliced, but there are notable exceptions and these include LAMA [51-54], peripherin [55, 56] and DNM (synemin; [57]). GFAP is also alternatively spliced ([58-63]; Table 2), involving the 5′ UTR (β-GFAP), exon 6 (Δ6, Δ135, Δ164; [62]) and exon 7 (δ-/ε-GFAP [59, 60] and κ-GFAP [61]). The relative abundance of these GFAP transcripts is often low [61] and can be dependent upon astrocyte location [64], whilst the expression of others (Δ6, Δ135, Δ164) is induced by specific diseases and then in neurons rather than astrocytes [62]). Many of the GFAP products from these transcripts will also be assembly incompetent and therefore the function of these various GFAP splice variants has to be appreciated within the context of α-GFAP [65], but not exclusively with respect to astrocytes [13].
Table 2.
Review of the alternatively spliced forms of human GFAP.
A review of all reported transcripts and their products from the human GFAP gene.
| Isoforms | Detected by | Transcript change | Affected protein feature relative to αGFAP | Human tissue expression pattern | Ref | ||
|---|---|---|---|---|---|---|---|
| cDNA/RNA | IH | IB | |||||
| α | + | + | + | 5′-UTR | Protein product of 432 residues | Astrocytes, enteric glia, nonmyelinating Schwann cells, liver stellate cells, breast myoepithelial cells, lymophycytes and respiratory tract chondrocytes – reviewed in [13) | 10 |
| β | + | + | + | 5′-UTR | Product same as α-GFAP | Schwann cells | 58, 68 |
| γ | + | - | - | 5′-UTR and exon 1 | Coding region predicted to start at residue 275, removing N-terminus and helix 1 | Corpus callosum | 63 |
| δ | + | + | - | Exon 7+ added | # Novel C-terminal sequence, replacing the exons 8 and 9 of αGFAP to give a 431 residue product | Astrocytes of subpial border and subventricular zone | 59 |
| ε | + | + | - | Exon 7+ added, different polyA to δ | ## Same C-terminal sequence as δ-form | 60, 64 | |
| κ | + | - | - | Both 7 and 7+ spliced together | ⊗ Novel C-terminal tail sequence to give a 438 residue product | Frontal cortex | 61 |
| +1 | + | + | - | Molecular Misreading in Exon 6 | Reading frame shifted from residue 307 to generate a unique 113 residue C-terminus and a predicted product 420 residues long* | Immunoreactivity in hippocampal neurones from (12/16) Alzheimer disease, (4/4) Down syndrome, (4/12) epilepsy and (4/20) non-demented control patient brain tissue | 62 |
| Δ6 | + | - | - | Exon 6 deleted | Exon 6 missing causing a frameshift with some sequences in common with the +1 form forming a predicted product 347 residues long ** | Transcript found in 1 out of 2 controls and 1 out 2 Alzheimer brain samples | 62 |
| Δ164 | + | - | - | Exon 6 and new acceptor site in exon 7 | Deletion of 164bps spanning the end of exon 6 and the start of exon 7 to produce a predicted 366 product with a novel C-terminal sequence*** | Transcript found in both Down Syndrome samples tested, but neither the two controls nor the two Alzheimer samples | 62 |
| Δ135 | + | - | - | Exon 6 | Removal of 135bp, but results in a predicted “in frame” deletion$ | Transcript found in both Down syndrome samples and one of two Alzheimer brain sample | 62 |
Novel C-terminal sequence GKSTKDGENHKVTRYLKSLTIRVIPIQAHQIVNGTPPARG generated from an additional exon (7+), located in intron 6, which incorporates an in frame termination codon and replaces exons 8 and 9 of αGFAP.
This transcript utilises a polyA signal in exon 7+ to the δGFAP, but coding sequence is identical to δGFAP.
Uses both exon 7 and 7+ to generate a novel 3′ sequence.
Novel C-terminal sequence predicted to correspond to: ADARAGGAARAGGGQLSGGAGAAGGRGAEPQGRDGPPLAGVPGPAQCQAGPGHRDRHLQEAARGRGEPDHHSRADLLQPADSRNQPGH QVCVRRPPQEEHRGEDRGDAGWRGH
C-terminal sequence similar to +1 GFAP predicted to be: DHHSRADLLQPADSRNQPGHQVCVRRPPQEEHRGEDRGDAGWRGH
C-terminal sequence similar to +1 GFAP predicted to be: SRADLLQPADSRNQPGHQVCVRRPPQEEHRGEDRGDAGWRGH
Predicted deleted sequences are: NESLERQMREQEERHVREAASYQEALARLEEEGQSLKDEMARHLQ
cDNA/RNA = RT-PCR, qPCR or Northern blotting; IH = immunohistochemistry; IB = immunblotting
GFAP is expressed in a range of other cells besides astrocytes, including enteric glia, nonmyelinating Schwann cells, liver stellate cells, breast myoepithelial cells, lymophycytes and respiratory tract chondrocytes (reviewed recently in [13]). The levels of GFAP are low in these other cells eg [66], suggesting that its primary role is not to form a filament network, a conclusion that also has application to the role of the different GFAP splice variants. Indeed, in Schwann cells, GFAP is required for cell proliferation in a pathway involving the αvβ8 integrin complex [67].
Schwann cells express the β-transcript of GFAP [58, 66, 68] and this differs only in the 5′ UTR to α-GFAP. The protein product from this splice variant therefore has the same predicted protein sequence as α-GFAP. The δ-/ε-GFAP [59, 60] and κ-GFAP [61] have quite different C-terminal domains, although the α-helical rod domain in not affected [61]. In contrast, the Δ6- and Δ164-GFAP transcripts [62]) introduce a frameshift that alters not only the sequence of the C-terminal tail but also part of the helix IIB sequence including the TYRKLLEGE helix-termination sequence. These transcripts are predicted to share the same C-terminal epitope as the +1-GFAP product, although the +1 GFAP product is thought to arise by molecular misreading of the α-GFAP transcript [62]. The Δ135-GFAP transcript lacks exon 6, but retains the reading frame. In all cases except β-GFAP, the identification of these alternatively spliced forms of GFAP is restricted to immunohistochemical techniques and has not been confirmed by immunoblotting because all such products are likely to be minor components of any cytoskeletal preparation on the basis of the low relative abundance of their transcripts [61].
The expression of some of the GFAP transcripts is influenced by disease [61]. The +1 GFAP is induced by Alzheimer's disease and Down syndrome [62], and in these situations is expressed mainly in neurons. Peripheral nerve injury also increases GFAP levels in Schwann cells [67] and of course increased GFAP expression is one of the hallmarks of astrogliosis [29, 65]. Interestingly Alzheimers disease induced astrogliosis does not uniformly increase all GFAP transcripts [64] and this makes the association of +1 GFAP with Alzheimer's disease and Down Syndrome rather unusual. This is, however, not unique amongst type III intermediate filament genes as motor neuron disease stabilises the expression of a particular peripherin splice variant, PE-61, but motor neuron disease does not appear to induce the expression of PE61 in other cell types [69]. These reports indicate that there is still much to learn about GFAP function, not least the impact of δ/ε- and κ-GFAP upon α-GFAP function and their role in Alexander disease.
GFAP mutations, Alexander disease and associated phenotypes
Recently, adult and inherited forms of Alexander disease have been identified and this has suggested several clinical symptoms that could help in the detection of more GFAP mutations in the population. In a recent review of adult cases (disease onset at 21, 24 and 45), ataxia and bulbar/pseudobulbar symptoms were found in all three cases available at that time [16], whereas cognitive function defects, macrocephaly and seizure, which typify infantile Alexander disease, were not seen. For instance the first inherited adult-onset cases of Alexander disease were found in a family with familial palatal myoclonus, which was caused by the V83G mutation in GFAP [70]. Subsequently two other adult onset cases were identified, both with R416W GFAP mutations and once again symptomatic palatal tremors was a clinical symptom [71, 72]. Whilst palatal tremors are also rare, they are distinctive and GFAP mutations have now been shown to be a genetic basis for their appearance in patients.
Analysis of various inherited cases of Alexander disease leads to the conclusion that there is significant phenotypic variability and age of onset for the same mutation eg R416W [16, 72]. Therefore like other diseases caused by intermediate filament mutations in keratin 17 [73], desmin [74, 75] and CP49 [76-78] for instance, it is clear that epigenetic and environmental factors influence the appearance and timing of disease symptoms [16]. This could be a reason why the incidence of reported GFAP mutations linked to human disease is still very rare.
Therapeutics and drug screening
With GFAP mutations providing a firm genetic foundation for Alexander disease, attention is naturally turning to therapeutics. The GFAP mutations are genetically dominant and appear to act in a gain-of-function fashion. No recessive mutations have ever been identified, nor have any mutations been found that are null or truncations. In addition, mouse knock-outs of GFAP have only subtle phenotypes that do not resemble Alexander disease. Hence the strategy of simply providing additional wild type GFAP to astrocytes is unlikely to be useful in Alexander disease. One publication has recently claimed modest success in treating a juvenile Alexander disease patient with thyrotropin releasing hormone (TRH), but the gains were small, the rationale for TRH was not strong, and the study itself had several shortcomings that limit the ability to interpret the significance of this result [24]. The rationale for TRH was based on the success that TRH had in treating the ataxic gait of cerebellar mutant mice [24]. An alternative approach to therapy that is currently under investigation is to screen drug libraries that are already approved by the FDA for the ability to reduce expression of GFAP. Gorospe et al [79] suggested that quercetin or other compounds known to reduce GFAP expression in cultured astrocytes might be useful as therapeutics. The activity of the GFAP promoter is controlled, in part, by NFκB, and even common agents such as aspirin may be capable of suppressing its activity [80]. Whether the high concentrations used in this study will be useful will require further tests using suitable in vivo models. Since astrocyte dysfunction appears to depend in part upon elevation of GFAP above a toxic threshold, any damping of the elevation that occurs during the natural course of disease is likely to be beneficial.
Abbreviations
- GFAP
glial fibrillary acidic protein
- HSP
heat shock protein
- Jnk
Jun N-terminal kinase
- MLK
mixed linage kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alexander WS. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain. 1949;72:373–81. doi: 10.1093/brain/72.3.373. 3 pl. [DOI] [PubMed] [Google Scholar]
- 2.Russo LS, Jr, Aron A, Anderson PJ. Alexander's disease: a report and reappraisal. Neurology. 1976;26:607–14. doi: 10.1212/wnl.26.7.607. [DOI] [PubMed] [Google Scholar]
- 3.Rodriguez D, Gauthier F, Bertini E, Bugiani M, Brenner M, N'Guyen S, Goizet C, Gelot A, Surtees R, Pedespan JM, Hernandorena X, Troncoso M, Uziel G, Messing A, Ponsot G, Pham-Dinh D, Dautigny A, Boespflug-Tanguy O. Infantile Alexander disease: spectrum of GFAP mutations and genotype-phenotype correlation. Am J Hum Genet. 2001;69:1134–40. doi: 10.1086/323799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neal JW, Cave EM, Singhrao SK, Cole G, Wallace SJ. Alexander's disease in infancy and childhood: a report of two cases. Acta Neuropathol (Berl) 1992;84:322–7. doi: 10.1007/BF00227826. [DOI] [PubMed] [Google Scholar]
- 5.Deprez M, D'Hooghe M, Misson JP, de Leval L, Ceuterick C, Reznik M, Martin JJ, D'Hooge M. Infantile and juvenile presentations of Alexander's disease: a report of two cases. Acta Neurol Scand. 1999;99:158–65. doi: 10.1111/j.1600-0404.1999.tb07338.x. [DOI] [PubMed] [Google Scholar]
- 6.Tomokane N, Iwaki T, Tateishi J, Iwaki A, Goldman JE. Rosenthal fibers share epitopes with alpha B-crystallin, glial fibrillary acidic protein, and ubiquitin, but not with vimentin. Immunoelectron microscopy with colloidal gold. Am J Pathol. 1991;138:875–85. [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson AB, Bettica A. On-grid immunogold labeling of glial intermediate filaments in epoxy-embedded tissue. Am J Anat. 1989;185:335–41. doi: 10.1002/aja.1001850228. [DOI] [PubMed] [Google Scholar]
- 8.Quinlan RA, Franke WW. Molecular interactions in intermediate-sized filaments revealed by chemical cross-linking. Heteropolymers of vimentin and glial filament protein in cultured human glioma cells. Eur J Biochem. 1983;132:477–84. doi: 10.1111/j.1432-1033.1983.tb07386.x. [DOI] [PubMed] [Google Scholar]
- 9.Eliasson C, Sahlgren C, Berthold CH, Stakeberg J, Celis JE, Betsholtz C, Eriksson JE, Pekny M. Intermediate filament protein partnership in astrocytes. J Biol Chem. 1999;274:23996–4006. doi: 10.1074/jbc.274.34.23996. [DOI] [PubMed] [Google Scholar]
- 10.Reeves SA, Helman LJ, Allison A, Israel MA. Molecular cloning and primary structure of human glial fibrillary acidic protein. Proc Natl Acad Sci USA. 1989;86:5178–5182. doi: 10.1073/pnas.86.13.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balcarek JM, Cowan NJ. Structure of the mouse glial fibrillary acidic protein gene: Implications for the evolution of the intermediate filament multigene family. Nuc Acid Res. 1985;13:5527–5543. doi: 10.1093/nar/13.15.5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenner M, Lampel K, Nakatani Y, Mill J, Banner C, Mearow K, Dohadwala M, Lipsky R, Freese E. Characterization of human cDNA and genomic clones for glial fibrillary acidic protein. Brain Res Mol Brain Res. 1990;7:277–286. doi: 10.1016/0169-328x(90)90078-r. [DOI] [PubMed] [Google Scholar]
- 13.Su M, Hu H, Lee Y, d'Azzo A, Messing A, Brenner M. Expression specificity of GFAP transgenes. Neurochem Res. 2004;29:2075–93. doi: 10.1007/s11064-004-6881-1. [DOI] [PubMed] [Google Scholar]
- 14.Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet. 2001;27:117–20. doi: 10.1038/83679. [DOI] [PubMed] [Google Scholar]
- 15.Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M. Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Path. 1998;152:391–398. [PMC free article] [PubMed] [Google Scholar]
- 16.Li R, Johnson AB, Salomons G, Goldman JE, Naidu S, Quinlan R, Cree B, Ruyle SZ, Banwell B, D'Hooghe M, Siebert JR, Rolf CM, Cox H, Reddy A, Gutierrez-Solana LG, Collins A, Weller RO, Messing A, van der Knaap MS, Brenner M. Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann Neurol. 2005;57:310–26. doi: 10.1002/ana.20406. [DOI] [PubMed] [Google Scholar]
- 17.Messing A, Goldman JE. Alexander Disease. Elsevier; Amsterdam: 2004. [Google Scholar]
- 18.Barkovich AJ, Messing A. Alexander disease: not just a leukodystrophy anymore. Neurology. 2006;66:468–9. doi: 10.1212/01.wnl.0000200905.43191.4d. [DOI] [PubMed] [Google Scholar]
- 19.van der Knaap MS, Ramesh V, Schiffmann R, Blaser S, Kyllerman M, Gholkar A, Ellison DW, van der Voorn JP, van Dooren SJ, Jakobs C, Barkhof F, Salomons GS. Alexander disease: ventricular garlands and abnormalities of the medulla and spinal cord. Neurology. 2006;66:494–8. doi: 10.1212/01.wnl.0000198770.80743.37. [DOI] [PubMed] [Google Scholar]
- 20.Mignot C, Boespflug-Tanguy O, Gelot A, Dautigny A, Pham-Dinh D, Rodriguez D. Alexander disease: putative mechanisms of an astrocytic encephalopathy. Cell Mol Life Sci. 2004;61:369–85. doi: 10.1007/s00018-003-3143-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooper DN, Youssoufian H. The CpG dinucleotide and human genetic disease. Hum Genet. 1988;78:151–5. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- 22.Caceres-Marzal C, Vaquerizo J, Galan E, Fernandez S. Early mitochondrial dysfunction in an infant with Alexander disease. Pediatr Neurol. 2006;35:293–6. doi: 10.1016/j.pediatrneurol.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 23.Dinopoulos A, Gorospe JR, Egelhoff JC, Cecil KM, Nicolaidou P, Morehart P, DeGrauw T. Discrepancy between neuroimaging findings and clinical phenotype in Alexander disease. AJNR Am J Neuroradiol. 2006;27:2088–92. [PMC free article] [PubMed] [Google Scholar]
- 24.Ishigaki K, Ito Y, Sawaishi Y, Kodaira K, Funatsuka M, Hattori N, Nakano K, Saito K, Osawa M. TRH therapy in a patient with juvenile Alexander disease. Brain Dev. 2006;28:663–7. doi: 10.1016/j.braindev.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 25.Kawai M, Sakai N, Miyake S, Tsukamoto H, Akagi M, Inui K, Mushiake S, Taniike M, Ozono K. Novel mutation of gene coding for glial fibrillary acidic protein in a Japanese patient with Alexander disease. Brain Dev. 2006;28:60–2. doi: 10.1016/j.braindev.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 26.Lee JM, Kim AS, Lee SJ, Cho SM, Lee DS, Choi SM, Kim DK, Ki CS, Kim JW. A case of infantile Alexander disease accompanied by infantile spasms diagnosed by DNA analysis. J Korean Med Sci. 2006;21:954–7. doi: 10.3346/jkms.2006.21.5.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li R, Johnson AB, Salomons GS, van der Knaap MS, Rodriguez D, Boespflug-Tanguy O, Gorospe JR, Goldman JE, Messing A, Brenner M. Propensity for paternal inheritance of de novo mutations in Alexander disease. Hum Genet. 2006;119:137–44. doi: 10.1007/s00439-005-0116-7. [DOI] [PubMed] [Google Scholar]
- 28.Crow JF. The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet. 2000;1:40–7. doi: 10.1038/35049558. [DOI] [PubMed] [Google Scholar]
- 29.Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–34. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- 30.Seifert G, Schilling K, Steinhauser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7:194–206. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- 31.Hagemann TL, Gaeta SA, Smith MA, Johnson DA, Johnson JA, Messing A. Gene expression analysis in mice with elevated glial fibrillary acidic protein and Rosenthal fibers reveals a stress response followed by glial activation and neuronal dysfunction. Hum Mol Genet. 2005;14:2443–58. doi: 10.1093/hmg/ddi248. [DOI] [PubMed] [Google Scholar]
- 32.Tian R, Gregor M, Wiche G, Goldman JE. Plectin regulates the organization of glial fibrillary acidic protein in Alexander disease. Am J Pathol. 2006;168:888–97. doi: 10.2353/ajpath.2006.051028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hagemann TL, Connor JX, Messing A. Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress response. J Neurosci. 2006;26:11162–73. doi: 10.1523/JNEUROSCI.3260-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka KF, Takebayashi H, Yamazaki Y, Ono K, Naruse M, Iwasato T, Itohara S, Kato H, Ikenaka K. The murine model of Alexander disease: analysis of GFAP aggregate formation and its pathological significance. Glia. 2007 doi: 10.1002/glia.20486. in press. [DOI] [PubMed] [Google Scholar]
- 35.Hsiao VC, Tian R, Long H, Der Perng M, Brenner M, Quinlan RA, Goldman JE. Alexander-disease mutation of GFAP causes filament disorganization and decreased solubility of GFAP. J Cell Sci. 2005;118:2057–65. doi: 10.1242/jcs.02339. [DOI] [PubMed] [Google Scholar]
- 36.Tang G, Xu Z, Goldman JE. Synergistic effects of the SAPK/JNK and the proteasome pathway on glial fibrillary acidic protein (GFAP) accumulation in Alexander disease. J Biol Chem. 2006;281:38634–43. doi: 10.1074/jbc.M604942200. [DOI] [PubMed] [Google Scholar]
- 37.Der Perng M, Su M, Wen SF, Li R, Gibbon T, Prescott AR, Brenner M, Quinlan RA. The Alexander disease-causing glial fibrillary acidic protein mutant, R416W, accumulates into Rosenthal fibers by a pathway that involves filament aggregation and the association of alpha B-crystallin and HSP27. Am J Hum Genet. 2006;79:197–213. doi: 10.1086/504411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoneda K, Furukawa T, Zheng YJ, Momoi T, Izawa I, Inagaki M, Manabe M, Inagaki N. An autocrine/paracrine loop linking keratin 14 aggregates to tumor necrosis factor alpha-mediated cytotoxicity in a keratinocyte model of epidermolysis bullosa simplex. J Biol Chem. 2004;279:7296–303. doi: 10.1074/jbc.M307242200. [DOI] [PubMed] [Google Scholar]
- 39.D'Alessandro M, Russell D, Morley SM, Davies AM, Lane EB. Keratin mutations of epidermolysis bullosa simplex alter the kinetics of stress response to osmotic shock. J Cell Sci. 2002;115:4341–51. doi: 10.1242/jcs.00120. [DOI] [PubMed] [Google Scholar]
- 40.Nakamichi I, Toivola DM, Strnad P, Michie SA, Oshima RG, Baribault H, Omary MB. Keratin 8 overexpression promotes mouse Mallory body formation. J Cell Biol. 2005;171:931–7. doi: 10.1083/jcb.200507093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jefferson JJ, Leung CL, Liem RK. Plakins: goliaths that link cell junctions and the cytoskeleton. Nat Rev Mol Cell Biol. 2004;5:542–53. doi: 10.1038/nrm1425. [DOI] [PubMed] [Google Scholar]
- 42.Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–72. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- 43.Iwaki T, Kume-Iwaki A, Liem RKH, Goldman JE. αB-crystallin is expressed in non-lenticular tissues and accumulates in Alexander's disease brain. Cell. 1989;57:71–78. doi: 10.1016/0092-8674(89)90173-6. [DOI] [PubMed] [Google Scholar]
- 44.Head MW, Corbin E, Goldman JE. Overexpression and abnormal modification of the stress proteins alpha B-crystallin and HSP27 in Alexander disease. Am J Pathol. 1993;143:1743–53. [PMC free article] [PubMed] [Google Scholar]
- 45.Iwaki T, Iwaki A, Tateishi J, Goldman JE. Sense and antisense modification of glial alpha B-crystallin production results in alterations of stress fiber formation and thermoresistance. J Cell Biol. 1994;125:1385–93. doi: 10.1083/jcb.125.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Head MW, Corbin E, Goldman JE. Coordinate and independent regulation of alpha B-crystallin and hsp27 expression in response to physiological stress. J Cell Physiol. 1994;159:41–50. doi: 10.1002/jcp.1041590107. [DOI] [PubMed] [Google Scholar]
- 47.Perng MD, Cairns L, van den IP, Prescott A, Hutcheson AM, Quinlan RA. Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J Cell Sci. 1999;112(Pt 13):2099–112. doi: 10.1242/jcs.112.13.2099. [DOI] [PubMed] [Google Scholar]
- 48.Mehlen P, Preville X, Chareyron P, Briolay J, Klemenz R, Arrigo AP. Constitutive expression of human hsp27, Drosophila hsp27, or human alpha B-crystallin confers resistance to TNF- and oxidative stress-induced cytotoxicity in stably transfected murine L929 fibroblasts. J Immunol. 1995;154:363–74. [PubMed] [Google Scholar]
- 49.Inaguma Y, Shinohara H, Goto S, Kato K. Translocation and induction of alpha B crystallin by heat shock in rat glioma (GA-1) cells. Biochem Biophys Res Commun. 1992;182:844–850. doi: 10.1016/0006-291x(92)91809-5. [DOI] [PubMed] [Google Scholar]
- 50.Koyama Y, Goldman JE. Formation of GFAP cytoplasmic inclusions in astrocytes and their disaggregation by alphaB-crystallin. Am J Pathol. 1999;154:1563–72. doi: 10.1016/s0002-9440(10)65409-0. In Process Citation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Furukawa K, Inagaki H, Hotta Y. Identification and cloning of an mRNA coding for a germ cell-specific A-type lamin in mice. Exp Cell Res. 1994;212:426–30. doi: 10.1006/excr.1994.1164. [DOI] [PubMed] [Google Scholar]
- 52.Machiels BM, Zorenc AHG, Endert JM, Kuijpers HJH, Eys GJJMv, Ramaekers FCS, Broers JLV. An alternative splicing product of the lamin A/C gene lacks exon 10. J Biol Chem. 1996;271:9249–9253. doi: 10.1074/jbc.271.16.9249. [DOI] [PubMed] [Google Scholar]
- 53.McKeon FD, Kirschner MW, Caput D. Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature. 1986;319:463–468. doi: 10.1038/319463a0. [DOI] [PubMed] [Google Scholar]
- 54.Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem. 1993;268:16321–6. [PubMed] [Google Scholar]
- 55.Landon F, Lemonnier M, Benarous R, Huc C, Fiszman M, Gros F, Portier MM. Multiple mRNAs encode peripherin, a neuronal intermediate filament protein. Embo J. 1989;8:1719–1726. doi: 10.1002/j.1460-2075.1989.tb03564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Landon F, Wolff A, de Nechaud B. Mouse peripherin isoforms. Biol Cell. 2000;92:397–407. doi: 10.1016/s0248-4900(00)01099-6. [DOI] [PubMed] [Google Scholar]
- 57.Xue ZG, Cheraud Y, Brocheriou V, Izmiryan A, Titeux M, Paulin D, Li Z. The mouse synemin gene encodes three intermediate filament proteins generated by alternative exon usage and different open reading frames. Exp Cell Res. 2004;298:431–44. doi: 10.1016/j.yexcr.2004.04.023. [DOI] [PubMed] [Google Scholar]
- 58.Galea E, Dupouey P, Feinstein DL. Glial fibrillary acidic protein messenger-rna isotypes - expression in-vitro and in-vivo. Journal Of Neuroscience Research. 1995;41:452–461. doi: 10.1002/jnr.490410404. [DOI] [PubMed] [Google Scholar]
- 59.Condorelli DF, Nicoletti VG, Barresi V, Conticello SG, Caruso A, Tendi EA, Giuffrida Stella AM. Structural features of the rat GFAP gene and identification of a novel alternative transcript. J Neurosci Res. 1999;56:219–28. doi: 10.1002/(SICI)1097-4547(19990501)56:3<219::AID-JNR1>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 60.Nielsen AL, Holm IE, Johansen M, Bonven B, Jorgensen P, Jorgensen AL. A new splice variant of glial fibrillary acidic protein, GFAP epsilon, interacts with the presenilin proteins. J Biol Chem. 2002;277:29983–91. doi: 10.1074/jbc.M112121200. [DOI] [PubMed] [Google Scholar]
- 61.Blechingberg J, Holm IE, Nielsen KB, Jensen TH, Jorgensen AL, Nielsen AL. Identification and characterization of GFAPkappa, a novel glial fibrillary acidic protein isoform. Glia. 2007 doi: 10.1002/glia.20475. [DOI] [PubMed] [Google Scholar]
- 62.Hol EM, Roelofs RF, Moraal E, Sonnemans MA, Sluijs JA, Proper EA, de Graan PN, Fischer DF, van Leeuwen FW. Neuronal expression of GFAP in patients with Alzheimer pathology and identification of novel GFAP splice forms. Mol Psychiatry. 2003;8:786–96. doi: 10.1038/sj.mp.4001379. [DOI] [PubMed] [Google Scholar]
- 63.Zelenika D, Grima B, Brenner M, Pessac B. A novel glial fibrillary acidic protein messenger-rna lacking exon-1. Molecular Brain Research. 1995;30:251–258. doi: 10.1016/0169-328x(95)00010-p. [DOI] [PubMed] [Google Scholar]
- 64.Roelofs RF, Fischer DF, Houtman SH, Sluijs JA, Van Haren W, Van Leeuwen FW, Hol EM. Adult human subventricular, subgranular, and subpial zones contain astrocytes with a specialized intermediate filament cytoskeleton. Glia. 2005;52:289–300. doi: 10.1002/glia.20243. [DOI] [PubMed] [Google Scholar]
- 65.Pekny M, Pekna M. Astrocyte intermediate filaments in CNS pathologies and regeneration. J Pathol. 2004;204:428–37. doi: 10.1002/path.1645. [DOI] [PubMed] [Google Scholar]
- 66.Riol H, Tardy M, Rolland B, Levesque G, Murthy MR. Detection of the peripheral nervous system (PNS)-type glial fibrillary acidic protein (GFAP) and its mRNA in human ly mphocytes. J Neurosci Res. 1997;48:53–62. [PubMed] [Google Scholar]
- 67.Triolo D, Dina G, Lorenzetti I, Malaguti M, Morana P, Del Carro U, Comi G, Messing A, Quattrini A, Previtali SC. Loss of glial fibrillary acidic protein (GFAP) impairs Schwann cell proliferation and delays nerve regeneration after damage. J Cell Sci. 2006;119:3981–93. doi: 10.1242/jcs.03168. [DOI] [PubMed] [Google Scholar]
- 68.Jessen KR, Thorpe R, Mirsky R. Molecular identity, distribution and heterogeneity of glial fibrillary acidic protein: an immunoblotting and immunohistochemical study of Schwann cells, satellite cells, enteric glia and astrocytes. J Neurocytol. 1984;13:187–200. doi: 10.1007/BF01148114. [DOI] [PubMed] [Google Scholar]
- 69.Robertson J, Doroudchi MM, Nguyen MD, Durham HD, Strong MJ, Shaw G, Julien JP, Mushynski WE. A neurotoxic peripherin splice variant in a mouse model of ALS. J Cell Biol. 2003;160:939–49. doi: 10.1083/jcb.200205027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Okamoto Y, Mitsuyama H, Jonosono M, Hirata K, Arimura K, Osame M, Nakagawa M. Autosomal dominant palatal myoclonus and spinal cord atrophy. J Neurol Sci. 2002;195:71–6. doi: 10.1016/s0022-510x(01)00687-6. [DOI] [PubMed] [Google Scholar]
- 71.Kinoshita T, Imaizumi T, Miura Y, Fujimoto H, Ayabe M, Shoji H, Okamoto Y, Takashima H, Osame M, Nakagawa M. A case of adult-onset Alexander disease with Arg416Trp human glial fibrillary acidic protein gene mutation. Neurosci Lett. 2003;350:169–72. doi: 10.1016/s0304-3940(03)00900-5. [DOI] [PubMed] [Google Scholar]
- 72.Thyagarajan D, Chataway T, Li R, Gai WP, Brenner M. Dominantly-inherited adult-onset leukodystrophy with palatal tremor caused by a mutation in the glial fibrillary acidic protein gene. Mov Disord. 2004;19:1244–8. doi: 10.1002/mds.20161. [DOI] [PubMed] [Google Scholar]
- 73.Covello SP, Smith FJ, Sillevis Smitt JH, Paller AS, Munro CS, Jonkman MF, Uitto J, McLean WH. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol. 1998;139:475–80. doi: 10.1046/j.1365-2133.1998.02413.x. [DOI] [PubMed] [Google Scholar]
- 74.Li D, Tapscoft T, Gonzalez O, Burch PE, Quinones MA, Zoghbi WA, Hill R, Bachinski LL, Mann DL, Roberts R. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation. 1999;100:461–4. doi: 10.1161/01.cir.100.5.461. [DOI] [PubMed] [Google Scholar]
- 75.Dalakas MC, Dagvadorj A, Goudeau B, Park KY, Takeda K, Simon-Casteras M, Vasconcelos O, Sambuughin N, Shatunov A, Nagle JW, Sivakumar K, Vicart P, Goldfarb LG. Progressive skeletal myopathy, a phenotypic variant of desmin myopathy associated with desmin mutations. Neuromuscul Disord. 2003;13:252–8. doi: 10.1016/s0960-8966(02)00271-7. [DOI] [PubMed] [Google Scholar]
- 76.Conley YP, Erturk D, Keverline A, Mah TS, Keravala A, Barnes LR, Bruchis A, Hess JF, FitzGerald PG, Weeks DE, Ferrell RE, Gorin MB. A Juvenile-Onset, Progressive Cataract Locus on Chromosome 3q21-q22 Is Associated with a Missense Mutation in the Beaded Filament Structural Protein-2. Am J Hum Genet. 2000;66:1426–1431. doi: 10.1086/302871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang L, Gao L, Li Z, Qin W, Gao W, Cui X, Feng G, Fu S, He L, Liu P. Progressive sutural cataract associated with a BFSP2 mutation in a Chinese family. Mol Vis. 2006;12:1626–31. [PubMed] [Google Scholar]
- 78.Zhang Q, Guo X, Xiao X, Yi J, Jia X, Hejtmancik JF. Clinical description and genome wide linkage study of Y-sutural cataract and myopia in a Chinese family. Mol Vis. 2004;10:890–900. [PubMed] [Google Scholar]
- 79.Gorospe JR, Naidu S, Johnson AB, Puri V, Raymond GV, Jenkins SD, Pedersen RC, Lewis D, Knowles P, Fernandez R, De Vivo D, van der Knaap MS, Messing A, Brenner M, Hoffman EP. Molecular findings in symptomatic and pre-symptomatic Alexander disease patients. Neurology. 2002;58:1494–500. doi: 10.1212/wnl.58.10.1494. [DOI] [PubMed] [Google Scholar]
- 80.Bae MK, Kim SR, Lee HJ, Wee HJ, Yoo MA, Ock Oh S, Baek SY, Kim BS, Kim JB, Sik Y, Bae SK. Aspirin-induced blockade of NF-kappaB activity restrains up-regulation of glial fibrillary acidic protein in human astroglial cells. Biochim Biophys Acta. 2006;1763:282–9. doi: 10.1016/j.bbamcr.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 81.Messing A, Brenner M. Alexander disease: GFAP mutations unify young and old. Lancet Neurol. 2003;2:75. doi: 10.1016/s1474-4422(03)00301-6. [DOI] [PubMed] [Google Scholar]
- 82.Eng LF, Lee YL, Kwan H, Brenner M, Messing A. Astrocytes cultured from transgenic mice carrying the added human glial fibrillary acidic protein gene contain Rosenthal fibers. J Neurosci Res. 1998;53:353–60. doi: 10.1002/(SICI)1097-4547(19980801)53:3<353::AID-JNR9>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]