

Abstract
Oxidative stress plays an important role in neurodegenerative disorders such as Parkinson's Disease and Alzheimer's Disease. Methamphetamine (METH) is an amphetamine analog that causes degeneration of the dopaminergic system in mammals and subsequent oxidative stress. In our present study, we have used immortalized human brain microvascular endothelial (HBMVEC) cells to test whether N-Acetylcysteineamide (NACA), a novel antioxidant, prevents METH-induced oxidative stress in vitro. Our studies showed that NACA protects against METH-induced oxidative stress in HBMVEC cells. NACA significantly protected the integrity of our blood brain barrier (BBB) model, as shown by permeability and trans-endothelial electrical resistance (TEER) studies. NACA also significantly increased the levels of intracellular glutathione (GSH) and glutathione peroxidase (GPx). Malondialdehyde (MDA) levels increased dramatically after METH exposure, but this increase was almost completely prevented when the cells were treated with NACA. Generation of reactive oxygen species (ROS) also increased after METH exposure, but was reduced to control levels with NACA treatment, as measured by dichlorofluorescin (DCF). These results suggest that NACA protects the BBB integrity in vitro, which could prevent oxidative stress-induced damage; therefore, the effectiveness of this antioxidant should be evaluated for the treatment of neurodegenerative diseases in the future.
Keywords: methamphetamine, oxidative stress, antioxidant, reactive oxygen species, N-acetyl cysteine amide, blood brain barrier, trans-endothelial electric resistance
1. Introduction
Methamphetamine (METH) is a highly addictive abused psycho stimulant whose use has widely increased in popularity (Barr et al., 2006). In 1996, the World Health Organization (WHO) reported METH as the second most popular illicit drug in the world, with 35 million users worldwide (WHO, 1997). Generally, METH causes a euphoric effect accompanied with decreased appetite, hypothermia, paranoia, aggression, and a heightened sense of pleasure. Magnetic resonance imaging of the brain has shown that chronic use of METH causes neuronal damage (Ernst et al., 2000) and produces numerous adverse effects on the central nervous system (Scott et al., 2000). Further, damage to dopaminergic nerve terminals has also been reported in the brains of METH-treated animals (Frey et al., 1997; Villemagne et al., 1998; Axt et al., 1991).
Numerous mechanisms for METH-induced neurotoxicity have been proposed, of which the release of dopamine (DA) in the nucleus accumbens (Cho et al., 1990; Wise et al., 1992) and the successive selective degeneration of striatal DA terminals (Seiden et al., 1987) are the most commonly known. Recently, however, the focus has been on generation of METH-induced oxidative stress. In the brain, DA oxidizes under physiological conditions to form short-lived, toxic reactive oxygen species (ROS) (Davidson et al., 2001). These ROS overwhelm the antioxidant defense mechanisms and lead to a condition known as oxidative stress. DA-dependent oxidative stress has been observed after pharmacologically relevant levels of METH have been administered (Cubells et al., 1994). Additionally, extracellular concentrations of oxidized glutathione also increased after METH administration (Acikgoz et al., 2001), suggesting either another mechanism of toxicity or an effect of oxidative stress. Impairment of the mitochondrial function has also been linked to pathways involving ROS, leading to a further oxidized environment and a decrease in ATP production (Moszczynska et al., 1998). These findings indicate that oxidative stress may play a pivotal role in the neurotoxic damage caused by METH abuse. Although the initiating cause of toxicity is not known, it seems that oxidative stress acts as a propagating force in spreading the toxicity to mitochondria and other systems that are susceptible to oxidation, as observed in various neurodegenerative diseases like Parkinson's and Alzheimer's diseases (Andersen et al., 2004). Indeed, evidence of increased oxidative stress has been observed both in vitro (SH-SY5Y neuroblastoma cells) and in vivo (rat brain tissues), following METH abuse (Yamamoto et al., 1998; Fitzmaurice et al., 2006; Wu et al., 2007).
METH abuse has also been reported to cause degeneration in the various regions of the brain (Park et al., 2006). In the literature, brain degeneration has been associated with modifications of the BBB (Dallasta et al., 1999; Fiala et al., 2002; Bar-Or et al., 2003). The vascular BBB is the capillary bed of the brain that is modified by having the adjacent cerebrovascular endothelial cells joined by intercellular tight junctions. These junctions and other endothelial cell modifications eliminate endothelial pores, resulting in a barrier between blood and the brain interstitial fluid that tightly regulates the exchange of substances between the brain and blood. The BBB helps to maintain the homeostatic environment of the brain, supplies the brain's nutritional needs, and plays a role in communication between the brain and the peripheral tissues (Banks., 1999; Davson and Segal., 1996). Under physiological conditions, the integrity of the BBB is protected from oxidative stress because the BBB has high levels of antioxidant enzymes (Plateel et al., 1995). Oxidative stress is one of the important mechanisms responsible for the disruption of the BBB. This disruption allows the passage of toxic substances into the brain, leading to development and progression of various neurological diseases (Dallasta et al., 1999; Fiala et al., 2002; Bar-Or et al., 2003). Thiol antioxidants have been shown to be effective in treating various neurological disorders. N-acetylcysteine amide (NACA), a novel thiol antioxidant, has been shown to be effective in various oxidative stress-related diseases (Penugonda et al., 2005; Wu et al., 2008; Price et al., 2006; Amer et al., 2008; Grinberg et al., 2005). It has also been shown to be more effective in neurotoxic cases than the parent compound N-acetylcysteine (NAC) because the neutral carboxyl group of NACA increases its ability to permeate cell membranes and the BBB. This allows NACA to be administered at a lower dose than NAC and prevents many side effects generally associated with NAC toxicity.
The objective of this study was to investigate the possible role of NACA in protecting the human microvascular endothelial cells (HBMVEC, an in vitro model of BBB) from METH-induced oxidative stress. Initial experiments were performed to determine the suitable dosage for METH and NACA. Following this, cells were treated with NACA 30 min prior to METH challenge. The protective effects of NACA were assessed by measurement of parameters like reduced glutathione (GSH), malondialdehyde (MDA), ROS levels, and glutathione peroxidase activity (GPx) in both the treatment and the control groups. Functional endpoint assays, like dextran permeability and trans endothelial electric resistance (TEER), were also performed in both groups to assess the antioxidant capability of NACA in protecting the BBB from METH-induced toxicity.
2. Results
2.1 Protection from METH Toxicity
In order to find the optimum concentration of NACA administration in the HBMVEC cells, a NACA dose-response curve was created and 1 mM of NACA was found to be the optimum dose, which was used in all of the experiments. The METH concentration used in this study was based on previous studies by Choi et al., (2002) and Wu et al., (2007). A METH dose-response curve was created, and 2.5 mM was found to be the minimum toxic dose (20% of cell death). Figures 1 and 2 represent these findings. To determine whether NACA can protect cells from METH toxicity, HBMVEC cells were exposed to 2.5 mM METH for 24 h, with pretreatment of 1 mM of NACA for 2 h. Cell viability was measured by a Calcein AM cell viability assay kit. As shown in Figure 3, 1 mM NACA completely protected the cells from METH- induced cell death. The cells in the NACA-treated group had similar cell viability as that of the controls.
Fig. 1.
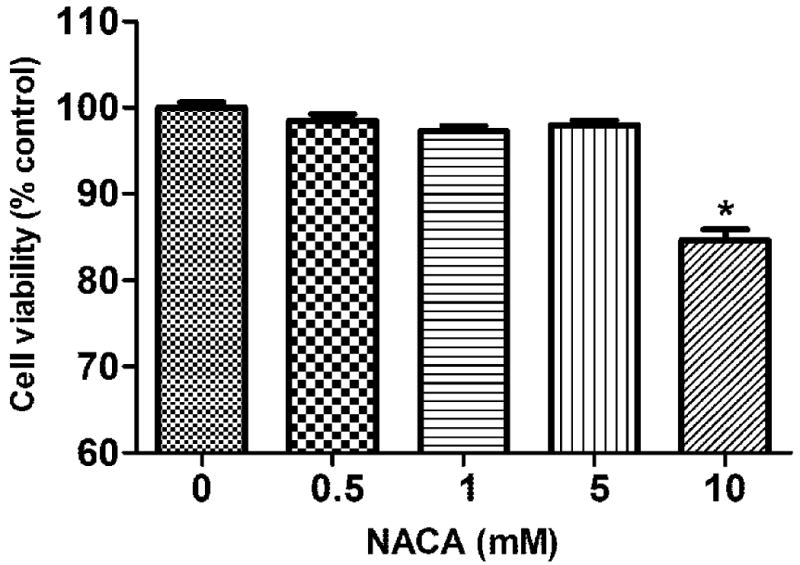
Cytotoxicity of NACA on HBMVEC cells. Cells were treated with various concentrations of NACA (500 μM, 1 mM, 5 mM, and 10 mM). After 24 h of treatment, the cell viability was quantified by a Calcein AM assay. Values represent mean ± SD. (*) refers to significant differences from the control with p < 0.05. The graph is representative of three independent experiments.
Fig. 2.

Cytotoxicity of METH on HBMVEC cells. Cells were treated with various concentrations of METH (100 μM, 500 μM, 1 mM, 2.5mM, 5 mM, and 10mM)). After 24 h of treatment, the cell viability was quantified by a Calcein AM assay. Values represent mean ± SD. (*) refers to significant differences from the control with p < 0.05. The graph is representative of three independent experiments.
Fig. 3.
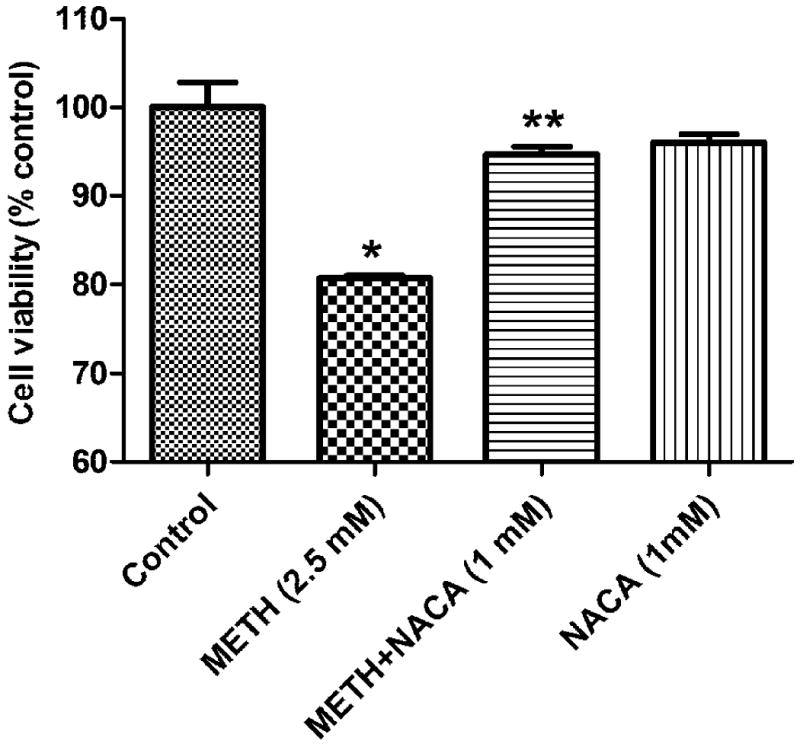
Effect of NACA on METH-induced cytotoxicity in HBMVEC cells. Cells were pretreated with NACA for 2 h followed by treatment with METH (2.5 mM) or serum free media for 24 h. The cell viability was quantified by a Calcein AM assay. Values represent mean ± SD. (*) refers to significant differences from the control with p<0.05. (**) refers to significant difference from METH-treated group, with p<0.05. The graph is representative of three independent experiments.
2.2 Intracellular GSH and GPx Activity
Table 1 depicts the protective effect of NACA on the intracellular concentration of GSH. Treatment with 2.5 mM METH significantly altered the GSH level to 70% of that of the control. A 2.5 mM treatment of METH, with a 1 mM pretreatment of NACA, was significantly different from that of the METH group alone, with p≤ 0.05 and a GSH concentration of 85% of that of the control group. The NACA alone treated group was found to have no significant changes in the GSH levels as compared to that of the control group. Table 1 shows GPx levels in the cells. METH administration markedly decreased the levels of GPx to 50% of that of the control. Pretreatment with NACA significantly attenuated this decrease to 80% of the GPx level of the control. In both of these experiments, NACA alone did not significantly alter the results from those of the control group; all the experiments were conducted in triplicate.
Table 1.
Effects of NACA one the levels of GSH, GPx and MDA
| Groups | GSH (nM/mg protein) |
GPx (mU/mg protein) |
MDA (nM/100mg protein) |
|---|---|---|---|
| Control | 42.10 ± 3.44 | 43.50 ± 0.08 | 86.10 ± 3.89 |
| METH (2.5 mM) | 33.09 ± 2.61* | 26.80 ± 0.09* | 193.16 ± 5.62* |
| METH+NACA (1mM) | 40.54 ± 0.69** | 37.20 ± 0.19** | 88.24 ± 2.71** |
| NACA (1 mM) | 44.32 ± 2.34 | 39.35 ± 0.36 | 84.78 ± 34.38 |
HBMVEC cells were pretreated with either NACA or serum free media for 2 h, followed by exposure to 2.5 mM of METH for 24 h. The control groups were exposed to serum free media. The NACA-only group showed similar result to those of the control group. All the experiments were performed in triplicate, and the values reported are mean ± SD.
p < 0.05 compared to that of control group,
p < 0.05 compared to METH-only group.
2.3 Lipid Peroxidation Byproduct MDA
MDA levels were determined in cells pretreated with NACA for 2 h, followed by exposure to METH for 24 h. Cells treated with 2.5 mM METH had increased MDA levels, that were nearly 200% of that of the control (Table 1). Pretreatment with 1 mM NACA completely reduced this increase, with MDA levels becoming nearly the same as those of the control, and with a p value of P<0.05, as compared to that of the control. The NACA and METH group had a p value of P<0.05, as compared to that of the METH-only group. The NACA-only treated group showed no significant difference when compared to the control.
2.4 Intracellular ROS Measurements
After treatment with METH (2.5 mM), ROS production was found to have increased. METH (2.5 mM) induced an increase in the DCF fluorescence of approximately 40%, as compared to that of the control (Figure 4), while pretreatment with 1 mM NACA (2 h prior to exposure) completely erased this increase. NACA alone did not significantly alter DCF fluorescence compared to that of the control.
Fig. 4.
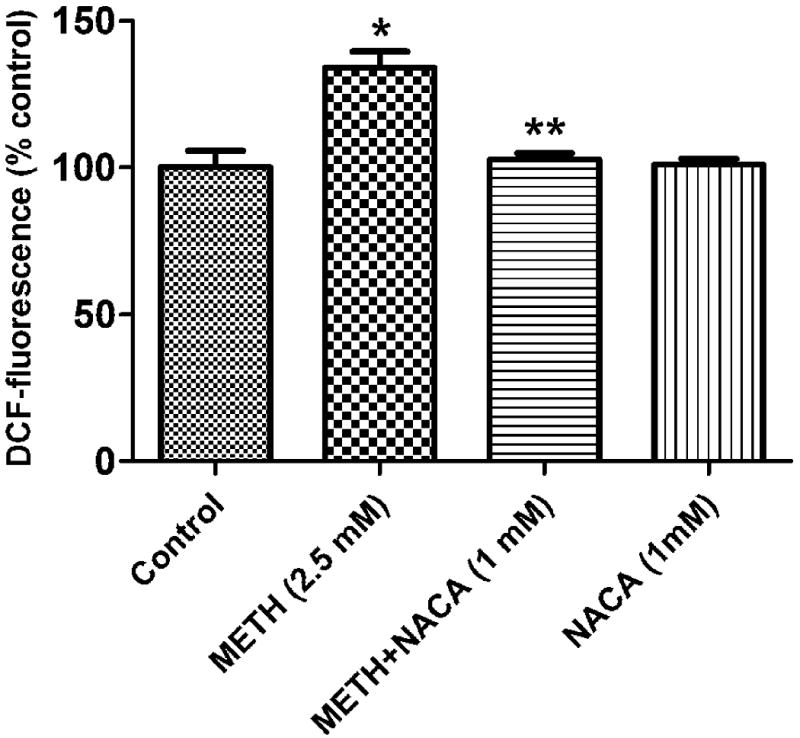
Effect of NACA on METH-induced intracellular ROS levels. After treatment with 2.5 mM of METH for 24 h, the intracellular ROS level increased, as determined by the evaluation of the DCF fluorescence. The increase of ROS was inhibited in the presence of NACA (1 mM). The DCF fluorescence of the NACA-only group was similar to that of the control group. Values represent mean ± SD. (*) refers to significant differences from the control with p<0.05. (**) refers to the significant difference from the METH group, with p<0.05. The graph is representative of three independent experiments.
2.5 TEER and Cell Permeability Assay
The cell permeability assay and TEER are especially important in this study because they simulated the integrity of the BBB. Permeability studies showed that 5 mM METH treatment increased permeability by approximately 25%, as compared to that of the control (Figure 5), while pretreatment with NACA resulted in a significant change of permeability to only 10% of that of the control. As in the permeability study, TEER results (as shown in Figure 6) also indicate the protective effect of NACA. Exposure to 5 mM METH resulted in a decrease in TEER by about 60%, as compared to that of the control, while pretreatment with 1 mM NACA reduced the resistance by only 30%. Treatment with NACA alone did not significantly alter TEER, as compared to that of the control. All experiments were conducted in triplicate.
Fig. 5.
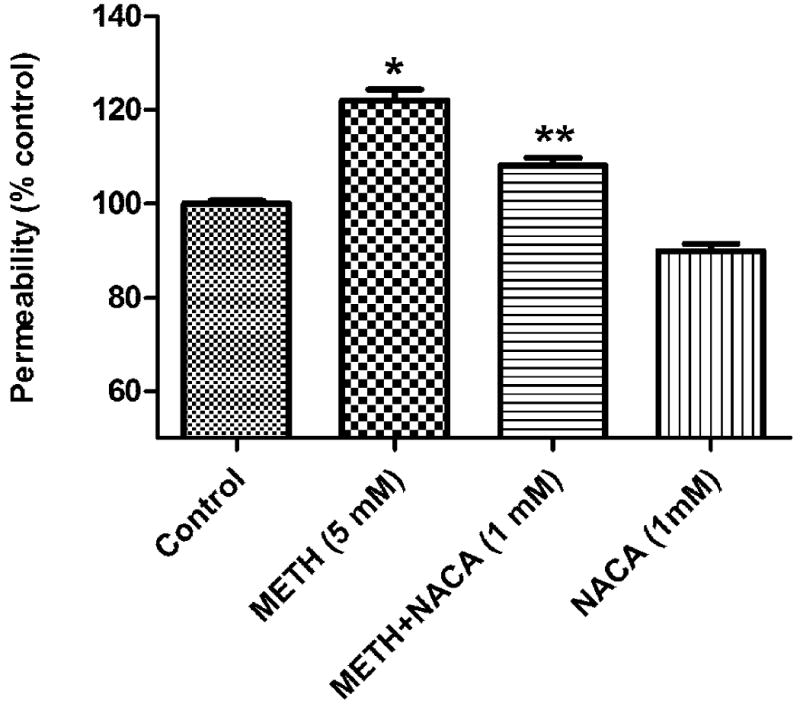
Effects of NACA on FITC-Dextran permeability in HBMVEC cells. HBMVEC cells were seeded onto a collagen-coated insert with a pore size of 0.4 μm at a density of 15×103 cells/ well and allowed to grow until a monolayer was formed. The cell monolayer was then treated with 1 mM NACA or serum free media for 2 h, followed by 5 mM of METH or serum free media for 24 h. Fluorescence was read with a 485 nm and 530 nm filter set. The NACA-only group showed similar results to those of the control group. *P<0.05 compared to the control group, and **P<0.05 compared to the corresponding value of the METH group. Three independent experiments were performed.
Fig. 6.
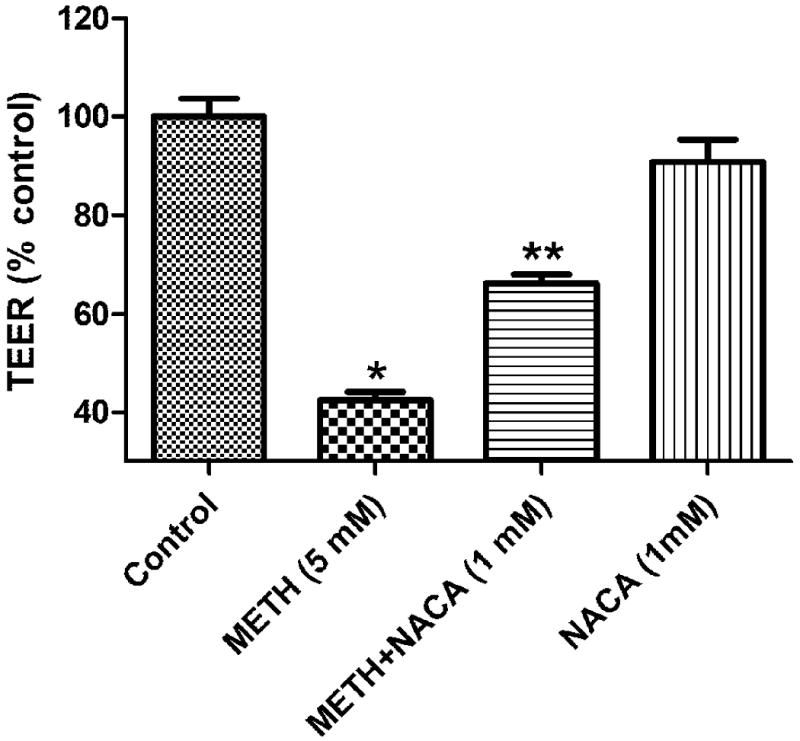
Effects of NACA on TEER in HBMVEC cells. HBMVEC cells were seeded onto a collagen-coated insert with a pore size of 0.4μm at a density of 15×103/well, and allowed to culture until a monolayer formed. The cell monolayer was then treated with 5 mM of METH with or without NACA (1 mM), for 24 h. Cells treated with NACA (1 mM) and METH (5 mM, 24 h) had increased TEER values as compared to the METH-alone treated group. * P<0.05 compared to the control, and **P<0.05 compared to the METH group.
3. Discussion
Abuse of metaphetamine (METH), an amphetamine analog, is a serious problem worldwide. The neurotoxic effect of METH has been reported to be caused by an increase in dopamine and glutamate formation in the brain that, in turn, mediates damage to the dopamine neurons through the formation of toxic ROS (Cadet et.al., 1994; Yamamoto et al., 1998; LaVoie and Hastings, 1999; Davidson et al., 2001). Although the role of oxidative stress in METH-induced toxicity is well-known, little is known about how METH-induced oxidative stress affects the BBB. In this study, we determined the oxidative stress parameters in the HBMVEC cells exposed to METH, and measured GSH, lipid peroxidation byproduct (MDA), levels of ROS formation, and the activity of the antioxidant enzyme GPx. We also investigated the role of the antioxidant NACA in abrogating oxidative stress and maintaining the permeability of the BBB in vitro.
The BBB is the major regulatory interface between the central nervous system (CNS) and the blood, which controls the passage of nutrients, xenobiotics, cytokines, and immune cells in and out of the CNS (Banks, 1999); hence it is important for maintaining the integrity of this membrane. Glutathione (GSH, γ-glutamyl-cysteinyl-glycine), an intracellular thiol, is one of the important factors that is critical for maintaining the integrity of the BBB (Agarwal and Shukla, 1999). Even though the mechanism by which GSH depletion leads to the dysfunction of the BBB is not known, GSH has been reported to be a direct scavenger of ROS, leading to reduction in oxidative stress (Yamamoto et al., 1998). In addition, GSH depletion also leads to a loss in protein sulfhydryls, which are believed to be essential for membrane functions such as enzyme activities and transport systems (Agarwal and Shukla, 1999). Results from our study show that HBMVEC cells treated with METH experience a significant decrease in cell viability and GSH levels, indicating that the increases in oxidative stress due to decreases in GSH levels were responsible for reduced cell viability. Pretreatment of the METH-treated cells with NACA (an antioxidant) increased the GSH levels and cell viability in this group, indicating that NACA had replenished the GSH levels in these cells. Further, depletion of intracellular GSH has been reported to increase production of ROS in the cells (Penugonda et al., 2005). ROS levels were measured using a peroxide-sensitive dye, DCF. METH-treated cells were found to have significant increases in ROS accumulation, as compared to that of the controls, but pretreatment with NACA significantly reduced the ROS accumulation in these cells.
The endothelial cells of the BBB are an important target of oxidative stress because they are rich in polyunsaturated fatty acids (PUFA) (Tayarani et al., 1987). The double bond in these fatty acids undergoes lipid peroxidation in the presence of free radicals and forms stable byproducts, such as malondialdehyde (MDA), which are used as markers of lipid peroxidation (Belghmi et al., 1988; Janero, 1990). HBMVEC cells that were exposed to METH had increased levels of MDA, compared to those of the controls, indicating increased lipid peroxidation in the BBB cells. However, pretreatment of cells in the METH-treated group with NACA significantly decreased their MDA levels.
Oxidative stress is a condition in which the pro-oxidants outweigh the antioxidant levels (including the enzymes) in the cells. The antioxidant enzymes are involved in the major detoxification pathways for peroxides in the cells. The antioxidant enzyme, GPx, is responsible for the detoxification of hydrogen peroxide by using glutathione as its substrate. Decreased activity of GPx was noted in METH-treated cells, as compared to activity in the controls, indicating that the GPx levels in the BBB cells were inadequate for combating the overwhelming oxidative stress induced by METH treatment. However, pretreatment of these cells with NACA resulted in increased levels of GPx in the METH-treated group, indicating that NACA was acting as a potent antioxidant. This is in accordance with previous studies, where NACA has been reported to inhibit radiation-induced cytotoxicity (Wu et al., 2008), HIV protein induced oxidative stress in the brain endothelial cells (Price et al., 2006) and Ab (1-42)-induced oxidative stress in neuronal cell lines (Bartov et al., 2006).
The brain microvascular endothelial cells, situated at the interface of the blood and the brain, perform the essential functions of shielding the brain from toxic substances in the blood stream, transporting micronutrients and macronutrients, leukocyte trafficking, and osmoregulation (Banks et al., 2006; Persidsky et al., 1997). Due to the unique regulatory function of the BBB, these endothelial cells possess several modifications (tight junctions), which are important for maintaining the integrity of these membranes. Loss of BBB integrity has been reported to be critical in the development and progression of neurological diseases like Alzheimers's (Fiala et al., 2002), human immunodeficiency virus-1 (HIV-1) encephalitis (Dallasta et al., 1999), multiple sclerosis (Bar-Or et al., 2003), stroke (Ilzecka, 1996), and traumatic brain injury (Morganti-Kossmann et al., 2002). Further, recent studies by Mahajan et al., (2008) and Bowyer and Ali (2006) have shown that METH causes disruption of the BBB by modulation of the tight junction expression and Rho-A activation. Our studies are in accordance with these, showing a decrease in permeability of the BBB (as evidenced by measurements of TEER and a dextran permeability assay) in HBMVEC cells treated with METH. Further, studies by Haorah et al., 2005, have shown that an increase in oxidative stress activates myosin light chain kinase, resulting in the enhanced phosphorylation of Rho in brain microvascular endothelial cells, leading to the loss of tight junction integrity. In our studies, the loss of cell permeability, due to METH treatment, decreased after pretreatment with NACA, indicating that the antioxidant was able to protect the BBB from oxidative stress-induced damage. However, a complete reversal of permeability of the BBB was not evident by pretreatment of the cells with NACA. This could be attributed to METH-induced cell death or apoptosis. Previous studies have reported that cells treated with METH experience a decrease in mitochondrial membrane potential which, in turn, leads to increased apoptosis (Wu et al., 2007). Further, METH treatment has also been reported to increase the calcium levels that activate a variety of proteases and kinases, resulting in the breakdown of cytoskeletal proteins (Sattler and Tymianski, 2000). Increased proteolysis of the cytoskeletal membrane protein Spectrin, and the microtubule-binding protein, Tau, has also been reported when a high dose of METH is administered, and may be responsible for the loss of permeability of the BBB (Warren et al., 2005; Staszewski and Yamamoto, 2006).
In conclusion, data from the present study indicate that METH causes oxidative stress to BBB cells, as demonstrated by decreased intracellular GSH, increased MDA levels and intracellular ROS production, and decreased GPx activity. In addition, METH treatment also altered the integrity of the BBB by increasing the permeability of the cells. These toxic effects of METH were reversed, however, by pretreatment of the cells with NACA. This antioxidant restored the levels of GSH, and scavenged the ROS produced by treatment with METH, thereby maintaining the permeability of the BBB. Considering the ability of NACA to protect BBB cells from METH-induced oxidative stress, the effectiveness of this antioxidant should be evaluated for the treatment of neurodegenerative diseases in the future.
4. Experimental Procedure
4.1 Reagents and Chemicals
N-(1-pyrenyl)-maleimide (NPM) was purchased from Sigma (St. Louis, MO). NACA was provided by Dr. Glenn Goldstein (David Pharmaceuticals, New York, NY 10021, USA). High-performance liquid chromatography (HPLC) grade solvents were purchased from Fisher Scientific (Fair Lawn, NJ). All other chemicals were purchased from Sigma (St. Louis, MO, USA). Methamphetamine was obtained from the National Institute on Drug Abuse (NIDA).
4.2 HPLC System
The HPLC system (Thermo Electron Corporation) consists of a Finnigan TM Spectra SYSTEM SCM1000 Vacuum Membrane Degasser, Finnigan TM SpectraSYSTEM P2000 Gradient Pump, Finnigan™ SpectraSYSTEM AS3000 Autosampler, and Finnigan™ SpectraSYSTEM FL3000 Fluorescence Detector (λex = 330 nm and λem = 376 nm). The HPLC column was a Reliasil ODS-1 C18 column (Column Engineering, Ontario, CA, USA). The mobile phase was 70% acetonitrile and 30% water and was adjusted to a pH 2.5 through the addition of 1 ml/L of both acetic and o-phosphoric acids. The NPM derivatives were eluted from the column isocratically at a flow rate of 1 ml/min.
4.3 Culture of Human Brain Microvascular Endothelail Cells (HBMVEC)
As an in vitro BBB model, immortalized human brain endothelial cells, HBMVEC (a gift from Dr. Pierre Couraud), were plated on 25 cm2 tissue culture flasks coated with type 1 rat tail collagen (Sigma, St.Louis, MO) and maintained in EBM-2 medium in humidified 5% CO2/95% air at 37°C. Culture medium was changed twice a week and endothelial cells at passages 28-34 were used in this study. A serum-free and growth-factor-free medium was used in the experiments. All assays were performed in triplicate and each experiment was repeated three times. EBM-2 medium (Lonza, Walkersville, MD), was supplemented with VEGF, IGF-1, EGF, basic FGF, hydrocortisone, ascorbate, gentamycin and 2.5% fetal bovine serum (FBS), as recommended by the manufacturer; this fully supplemented medium is designated as Microvascular Endothelial Cell Medium-2 (EGM-2 MV, herein referred to as EGM-2 medium).
4.4 Cytotoxicity Measurement
HBMVEC cells were treated with METH for 24 h, after which, the medium was discarded and a Calcein AM assay KIT (Biotium, Inc. CA 94545) was used to determine cell viability relative to the control group. Briefly, the cells were seeded in a 96-well plate, at densities of approximately 5,000 cells per well, for a day, after which they were treated with NACA or serum-free media for 2 h. The media was then discarded, and the cells were treated with METH (2.5mM) in serum-free media for 24 h. The cells were then washed three times with PBS, and 100 μl of 2M Calcein AM in PBS were added to each well for 30 min at 37°C. The fluorescence was measured with an excitation wavelength at 485 nm and an emission wavelength of 530 nm, using a microplate reader (FLOURstar, BMG Labtechnologies, Durham, NC, USA).
4.5 Oxidative Stress Studies
HBMVEC cells were plated in culture flasks and incubated to facilitate attachment. Cells (5 × 106/ flask) in the culture flasks were then treated with 1 mM NACA (2 h), followed by 2.5mM METH (24 h), as indicated before. The following groups were used for the experiment: (1) control (no METH or NACA); (2) NACA only (1 mM NACA for 2 h followed by fresh medium for 24 h); (3) METH only (2.5 mM METH for 24 h); (4) METH + NACA (1 mM NACA for 2 h, followed by 2.5 mM METH for 24 h). At the end of the incubation period, the cells were collected by using a cell scraper and re-suspended in fresh media. After being centrifugated, the cell pellet was re-suspended in a serine–borate buffer (SBB: 100 mM Tris–HCl, 10 mM borate, 5 mM serine, 1 mM diethyllene triaminepenta-acetic acid, pH 7.0) to prevent artifactual oxidation. Samples were immediately homogenized with Tissue-Tearor (Model 985-370, Biospec Products, Inc.) and analyzed for oxidative stress parameters. A portion of this preparation was used for the determination of reduced GSH, MDA, and protein levels, and the remainder was centrifuged at 700 × g for 10 min to yield a supernatant for enzyme assays (GPx).
4.6 Determination of GSH
Intracellular endothelial cell GSH content was determined by reverse phase HPLC, according to the method developed in our laboratory (Ridnour et al., 1999). HBMVEC cell samples were homogenized in SBB. Twenty microliters of this homogenate were added to 230 μl of HPLC grade water and 750 μl of NPM (1 mM in acetonitrile). The resulting solutions were incubated at room temperature for 5 min. The reaction was stopped by addition of 5 μl of 2N HCl. The samples were then filtered through a 0.45 μm filter (Advantec MFS, Inc. Dulin, CA USA) and injected onto the HPLC system. An aliquot of 5 μl sample was injected for analysis using a Thermo Finnigan TM Spectra SYSTEM SCM1000 Vacuum Membrane Degasser, Finnigan TM SpectraSYSTEM P2000 Gradient Pump, Finnigan™ SpectraSYSTEM AS3000 Autosampler, and Finnigan™ SpectraSYSTEM FL3000 Fluorescence Detector (λex = 330 nm and λem = 376 nm). The HPLC column was a Reliasil ODS-1 C18 column (Column Engineering, Ontario, CA, USA). The mobile phase was 70% acetonitrile and 30% water and was adjusted to a pH of 2.5 through the addition of 1 ml/L of both acetic and o-phosphoric acids. The NPM derivatives were eluted from the column isocratically at a flow rate of 1 ml/min.
4.7 Determination of Malondialdehyde (MDA)
MDA content was determined as described by Draper et al. (1993). Briefly, the cell pellets were homogenized in SBB. To 0.350 ml of cell homogenate, 0.550 ml of 5% trichloroacetic acid (TCA) and 0.100 ml of 500 ppm butylated hydroxytoluene (BHT) in methanol were added. The samples were then heated in a boiling water bath for 30 min. After cooling on ice, the samples were centrifuged. The supernatant fractions were mixed 1:1 with saturated thiobarbituric acid (TBA). The samples were again heated in a boiling water bath for 30 min. After cooling on ice, 0.50 ml of each sample was extracted with 1 ml of n-butanol and centrifuged to facilitate the separation phases. The resulting organic layers were first filtered through a 0.45 μm filter and then analyzed using the Shimadzu HPLC system with a fluorescence detector. Excitation wavelength and emission wavelength were set at 515 nm and 550 nm. The column was a 100 × 4.6 mm i.d C18 column (3 μm packing material, Astec, Bellefonte, PA). Twenty microliter samples were injected for analysis. The mobile phase consisted of 69.4% 50 mM sodium phosphate buffer (pH 7.0), 30% acetonitrile, and 0.6% THF. The flow rate of the mobile phase was 1.0 mL/min. The concentrations of the TBA-MDA complex in the mixture were determined by using the calibration curve obtained from a malondialdehyde bis (dimethyl acetal) standard solution.
4.8 Intracellular ROS Measurement
Intracellular ROS generation was measured using a well-characterized probe, 2′,7′-dichlorofluorescin diacetate (DCFH-DA) (Hong and Joseph, 1999). DCFH-DA is hydrolyzed by esterases to dichlorofluorescin (DCFH), which is trapped within the cell. This nonfluorescent molecule is then oxidized to fluorescent dichlorofluorescin (DCF) by the action of cellular oxidants. A DCFH-DA stock solution (in methanol) of 10 mM was diluted 500-fold in HBSS without serum or other additive to yield a 20 μM working solution. Cells were washed twice with HBSS and then incubated with a DCFH-DA working solution for 1 h in a dark environment (37 °C incubator). After exposure to 1 mM NACA or a serum-free media for 2 h, followed by 2.5 mM of METH or a serum-free media for 24 h, fluorescence was determined at 485 nm excitation and 520 nm emission, using a microplate reader (FLOURstar, BMG Labtechnologies, Durham, NC, USA).
4.9 Glutathione Peroxidase Activity Assay
Glutathione peroxidase (GPx) protects mammals against oxidative damage by catalyzing the reduction of a variety of ROOH, or H2O2, using GSH as the reducing substance. The GPx-340™ assay (a test kit from OxisResearch) is an indirect measure of the activity of GPx. Oxidized glutathione (GSSG), produced upon reduction of an organic peroxide by GPx, is recycled to its reduced state by the enzyme glutathione reductase (GR): The oxidation of NADPH to NADP + is accompanied by a decrease in absorbance at 340 nm (A340), providing a spectrophotometric means for monitoring GPx enzyme activity. The molar extinction coefficient for NADPH is 6220 M-1 cm-1 at 340 nm. To measure the activity of GPx, tissue homogenate is added to a solution containing glutathione, glutathione reductase, and NADPH. The enzyme reaction is initiated by adding the substrate, tert-butyl hydroperoxide, and the absorbance is recorded at A340. The rate of decrease in the A340 is directly proportional to the GPx activity in the sample.
4.10 Permeability Study
HBMVEC cells were seeded onto collagen-coated inserts with a pore size of 0.4 μm at densities of 15 × 103 / well, and allowed to culture until a monolayer formed. The cell monolayer was then treated with 1 mM NACA or saline-free media for 2 h, followed by 5 mM METH for 24 h. After this, the medium was replaced with 150 μl of FITC labeled dextran, and transferred the insert to a fresh plate well, containing 500 μl of serum-free medium. The plates were incubated for 30 min at room temperature, and 100 μl of the plate well solution were removed and transferred to a 96-well plate. Fluorescence was read with a 485 nm excitation and 530 nm emission, using a microplate reader (FLOURstar, BMG Labtechnologies, Durham, NC, USA).
4.11 Trans Endothelial Electric Resistance (TEER) Measurement
Trans endothelial electric resistance (TEER) measurement by EVOM voltohmmeter (World Precision Instrument, Sarasota, FL, USA) assessed the tightness of the HBMVEC monolayer. HBMVEC cells were seeded onto collagen-coated inserts with a pore size of 0.4 μm at densities of 15 × 103 / well, and allowed to culture until a monolayer formed (4-7 days). The cell monolayer was then treated with 1 mM NACA or serum-free media for 2 h, followed by 5 mM METH or serum-free media for 24 h. After this, the media was replaced with 150 μl of fresh medium. The insert containing the cell monolayer was then transferred in to a fresh plate containing 500 μl of serum-free medium. The TEER reading was recorded immediately. TEER values are calculated as: Resistance × 0.32 cm2 (insert surface area). Thus, resistance is proportional to the effective membrane.
4.12 Protein Determination
Protein levels were determined by the Bradford method with Coomassie blue (Bio-Rad) (Bradford, 1976). Concentrated Coomassie blue (Bio-Rad) was diluted 1:5 (v/v) with distilled water. Then 2.5 ml of this diluted dye was added to 50 μl of diluted cell homogenate. The mixture was incubated at room temperature for 10 min and the absorbance measurement was taken at 595 nm using a UV-VIS spectrophotometer. Bovine serum albumin (BSA) was used as a protein standard.
4.13 Statistical Analysis
Data was given as the mean ± SD. The one-way analysis of variance (ANOVA) and Student-Newman-Keuls multiple comparison tests were used to analyze the significance of the differences between the control and experimental groups. Values of p < 0.05 were considered significant.
Acknowledgments
Dr. Ercal was supported by 1 R15DA023409-01A2 from the NIDA, NIH. The contents of this paper are solely the responsibility of the authors and do not necessarily represent official views of the NIDA or NIH. Dr. Banks was supported by VA Merit Review and R01 AG029839. The authors appreciate the efforts of Charles Anderson and Barbara Harris for editing the manuscript.
Abbreviations
- METH
methamphetamine
- GSH
glutathione
- GSSG
glutathione disulfide
- GPx
glutathione peroxidase
- NACA
N-acetyl-cysteine-amide
- NAC
N-acetyl-cysteine
- MDA
Malondialdehyde
- ROS
Reactive oxygen species
- TEER
Trans-endothelial electric resistance
- BBB
blood brain barrier
- CNS
central nervous system
- HBMVEC
human brain microvascular endothelial cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acikgoz O, Gonenc S, Gezer S, Kayatekin BM, Uysal N, Semin I, Gure A. Methamphetamine causes depletion of glutathione and an increase in oxidized glutathione in the rat striatum and prefrontal cortex, Neurotox. Res. 2001;3:277–280. doi: 10.1007/BF03033266. [DOI] [PubMed] [Google Scholar]
- Agarwal R, Shukla GS. Potential role of cerebral glutathione in the maintenance of blood-brain barrier integrity in rat. Neurochemical Research. 1999;24:1507–1514. doi: 10.1023/a:1021191729865. [DOI] [PubMed] [Google Scholar]
- Amer J, Atlas D, Fibach E. N-acetylcysteine amide (AD4) attenuates oxidative stress in beta-thalassemia blood cells. Biochim Biophys Acta. 2008;1780:249–255. doi: 10.1016/j.bbagen.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Andersen J. Oxidative stress in neurodegeneration: cause or consequence? Nature Rev Neurosci. 2004;5:S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- Axt K, Molliver M. Immunocytochemical evidence for methamphetamine-induced serotonergic axon loss in the rat brain. Synapse. 1991;9:302–313. doi: 10.1002/syn.890090405. [DOI] [PubMed] [Google Scholar]
- Banks WA. Physiology and pathophysiology of the blood–brain barrier: implications for microbial pathogenesis, drug delivery and neurodegenerative disorders. J Neurovirol. 1999;5:538–555. doi: 10.3109/13550289909021284. [DOI] [PubMed] [Google Scholar]
- Banks WA, Ercal N, Price TO. The blood-brain barrier in neuroAIDS. Curr HIV Res. 2006;4:259–266. doi: 10.2174/157016206777709447. [DOI] [PubMed] [Google Scholar]
- Bar-Or A, Nuttall RK, Duddy M, Alter A, Kim HJ, Ifergan I, Pennington CJ, Bourgoin P, Edwards DR, Yong VW. Analyses of all matrix metalloproteinase members in leukocytes emphasize monocytes as major inflammatory mediators in multiple sclerosis. Brain. 2003;126:2738–2749. doi: 10.1093/brain/awg285. [DOI] [PubMed] [Google Scholar]
- Barr AM, Panenka WJ, MacEwan GW, Thorton AE, Lang DJ, Honer WG, Lecomte T. The need for speed: an update on methamphetamine addiction. J Psychiatry Neurosci. 2006;31:301–313. [PMC free article] [PubMed] [Google Scholar]
- Bartov O, Sultana R, Butterfield DA, Atlas D. Low molecular weight thiol amides attenuate MAPK activity and protect primary neurons from Ab (1-42) toxicity. Brain Res. 2006;1069:198–206. doi: 10.1016/j.brainres.2005.10.079. [DOI] [PubMed] [Google Scholar]
- Belghmi K, Nicolas JC, Crastes de Paulet A. Chemiluminescent assay of lipid hydroperoxides. J Biolumin Chemilumin. 1988;2:113–119. doi: 10.1002/bio.1170020302. [DOI] [PubMed] [Google Scholar]
- Bowyer JF, Ali S. High doses of methamphetamine that cause disruption of the blood-brain barrier in limbic regions produce extensive neuronal degeneration in mouse hippocampus. Synpase. 2006;60:521–532. doi: 10.1002/syn.20324. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding, Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Sheng P, Ali S, Rothman R, Carlson E, Epstein C. Attenuation of methamphetamine-induced neurotoxicity in copper/zinc superoxide dismutase transgenic mice. J Neurochem. 1994;62:380–383. doi: 10.1046/j.1471-4159.1994.62010380.x. [DOI] [PubMed] [Google Scholar]
- Cho A. Ice: a new dosage form of an old drug. Science. 1990;249:631–634. doi: 10.1126/science.249.4969.631. [DOI] [PubMed] [Google Scholar]
- Choi HJ, Yoo TM, Chung SY, Yang JS, Kim JI, Ha ES, Hwang O. Methamphetamine-induced apoptosis in a CNS-derived catecholaminergic cell line. Mol Cells. 2002;13:221–227. [PubMed] [Google Scholar]
- Cubells JF, Rayport S, Rajendran G, Sulzer D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J Neurosci. 1994;14:2260–2271. doi: 10.1523/JNEUROSCI.14-04-02260.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallasta LM, Pisarov LA, Esplen JE, Werley JV, Moses AV, Nelson JA, Achim CL. Blood-brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am J Pathol. 1999;155:1915–1927. doi: 10.1016/S0002-9440(10)65511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson C, Gow AJ, Lee TH, Ellinwood EH. Methamphetamine neurotoxicity: necrotic and apoptotic mechanisms and relevance to human abuse and treatment. Brain Res Brain Res Rev. 2001;36:1–22. doi: 10.1016/s0165-0173(01)00054-6. [DOI] [PubMed] [Google Scholar]
- Davson H, Segal MB. Physiology of the CSF and blood–brain barriers. CRC Press; New York: 1996. [Google Scholar]
- Draper HH, Squires EJ, Mahmoodi H, Wu J, Agarwal M, Hadley M. A comparative evaluation of thiobarbituric acid methods for the determination of malondialdehyde in biological materials, Free Radic. Biol Med. 1993;15:353–363. doi: 10.1016/0891-5849(93)90035-s. [DOI] [PubMed] [Google Scholar]
- Ernst T, Chang L, Leonido-Yee M, Speck O. Evidence for long-term neurotoxicity associated with methamphetamine abuse: a 1H MRS study. Neurology. 2000;54:1344–1349. doi: 10.1212/wnl.54.6.1344. [DOI] [PubMed] [Google Scholar]
- Fiala M, Liu QN, Sayre J, Pop V, Brahmandam V, Graves MC, Vinters HV. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer's disease brain and damage the blood–brain barrier, Eur. J Clin Investig. 2002;32:360–371. doi: 10.1046/j.1365-2362.2002.00994.x. [DOI] [PubMed] [Google Scholar]
- Fitzmaurice P, Tong J, Yazdanpanah M, Liu PP, Kalasinsky KS, Kish SJ. Levels of 4-hydroxynonenal and malondialdehyde are increased in brain of human chronic users of methamphetamine. J Pharmacol Exp Ther. 2006;319:703–709. doi: 10.1124/jpet.106.109173. [DOI] [PubMed] [Google Scholar]
- Frey K, Kilbourn M, Robinson T. Reduced striatal vesicular monoamine transporters after neurotoxic but not after behaviorally-sensitizing doses of methamphetamine. Eur J Pharmacol. 1997;334:273–279. doi: 10.1016/s0014-2999(97)01152-7. [DOI] [PubMed] [Google Scholar]
- Grinberg L, Fibach E, Amer J, Atlas D. N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radic Biol Med. 2005;38:136–145. doi: 10.1016/j.freeradbiomed.2004.09.025. [DOI] [PubMed] [Google Scholar]
- Haorah J, Knipe B, Leibhart J, Ghorpade A, Persidsky Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J Leukoc Biol. 2005;78:1223–1232. doi: 10.1189/jlb.0605340. [DOI] [PubMed] [Google Scholar]
- Hong H, Joseph JA. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader, Free Radic. Biol Med. 1999;27:612–616. doi: 10.1016/s0891-5849(99)00107-0. [DOI] [PubMed] [Google Scholar]
- Ilzecka J. The structure and function of blood-brain barrier in ischaemic brain stroke process. Ann Univ Mariae Curie Sklodowska. 1996;51:123–127. [PubMed] [Google Scholar]
- Janero DR. Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury, Free Radic. Biol Med. 1990;9:515–540. doi: 10.1016/0891-5849(90)90131-2. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan S, Aslinkeel R, Sykes DE, Reynolds JL, Bindukumar B, Adal A, Qi M, Toh J, Xu G, Prasad PN, Schwartz SA. Methamphetamine alters blood brain barrier permeability via the modulation of tight junction expression: Implication for HIV-1 neuropathogenesis in the context of drug abuse. Brain Res. 2008;1203:133–148. doi: 10.1016/j.brainres.2008.01.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Immune and inflammatory responses in the nervous system. Oxford, UK: University Press; Oxford, UK: University Press; 2002. Inflammatory responses to traumatic brain injury: an overview for the new millennium; pp. 106–126. [Google Scholar]
- Moszczynska A, Turenne S, Kish S. Rat striatal levels of the antioxidant glutathione are decreased following binge administration of methamphetamine. Neurosci Lett. 1998;255:49–52. doi: 10.1016/s0304-3940(98)00711-3. [DOI] [PubMed] [Google Scholar]
- Park MJ, Lee SK, Lim MA, Chung HS, Cho SI, Jang CG, Lee SM. Effect of alpha-tocopherol and deferoxamine on methamphetamine-induced neurotoxicity. Brain Res. 2006;1109:176–182. doi: 10.1016/j.brainres.2006.06.030. [DOI] [PubMed] [Google Scholar]
- Penugonda S, Mare S, Goldstein G, Banks WA, Ercal N. Effects of N-acetylcysteine amide (NACA), a novel thiol antioxidant against glutamate-induced cytotoxicity in neuronal cell line PC-12. Brain Res. 2005;1056:132–138. doi: 10.1016/j.brainres.2005.07.032. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Stins M, Way D, Witte MH, Weinand M, Kim KS, Bock P, Gendelman HE, Fiala M. A model for monocyte migration through the blood–brain barrier during HIV-1 encephalitis, J. Immunol. 1997;158:3499–3510. [PubMed] [Google Scholar]
- Plateel M, Dehouck MP, Torpier G, Cecchelli R, Teissier E. Hypoxia increases the susceptibility to oxidant stress and the permeability of the blood–brain barrier endothelial cell monolayer. J Neurochem. 1995;65:2138–2145. doi: 10.1046/j.1471-4159.1995.65052138.x. [DOI] [PubMed] [Google Scholar]
- Price TO, Uras F, Banks WA, Ercal N. A novel antioxidant, N-acetylcysteine amide prevents gp120 and tat-induced oxidative stress in brain endothelial cells. Exp Neurol. 2006;201:193–202. doi: 10.1016/j.expneurol.2006.03.030. [DOI] [PubMed] [Google Scholar]
- Ridnour LA, Winters RA, Ercal N, Spitz DR. Measurement of glutathione, glutathione disulfide, and other thiols in mammalian cell and tissue homogenates using high-performance liquid chromatography separation of N-(1-pyrenyl)maleimide derivatives. Methods Enzymol. 1999;299:258–267. doi: 10.1016/s0076-6879(99)99025-0. [DOI] [PubMed] [Google Scholar]
- Sattler R, Tymianski M. Molecular mechanisms of calcium dependent excitotoxicity. J Mol Med. 2000;78:3–13. doi: 10.1007/s001090000077. [DOI] [PubMed] [Google Scholar]
- Scott JC, Woods SP, Matt GE, Meyer RA, Heaton RK, Atkinson JH, Grant I. Neurocognitive effects of methamphetamine: A critical review and meta-analysis. Neuropsychol Rev. 2000;17:275–297. doi: 10.1007/s11065-007-9031-0. [DOI] [PubMed] [Google Scholar]
- Seiden L, Ricaurte G. Neurotoxicity of methamphetamine and related drugs psychopharmacology: a third generation of progress. New York: Raven; 1987. pp. 359–366. [Google Scholar]
- Staszewski RD, Yamamoto BK. Methamphetamine-induced spectrin proteolysis in the rat striatum. J Neurochem. 2006;96:1267–1276. doi: 10.1111/j.1471-4159.2005.03618.x. [DOI] [PubMed] [Google Scholar]
- Tayarani I, Chaudiere J, Lefauconnier JM, Bourre JM. Enzymatic protection against peroxidative damage in isolated brain capillaries. J Neurochem. 1987;48:1399–1402. doi: 10.1111/j.1471-4159.1987.tb05677.x. [DOI] [PubMed] [Google Scholar]
- Villemagne V, Yuan J, Wong DF, Dannals RF, Hatzidimitriou G, Mathews WB, Ravert HT, Musachio J, McCann UD, Ricaurte GA. Brain dopamine neurotoxicity in baboons treated with doses of methamphetamine comparable to those recreationally abused by humans: evidence from [11C] WIN-35,428 positron emission tomography studies and direct in vitro determinations. J Neurosci. 1998;18:419–427. doi: 10.1523/JNEUROSCI.18-01-00419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren MW, Kobeissy FH, Liu MC, Hayes RL, Gold MS, Wang KK. Concurrent calpain and caspase-3 mediated proteolysis of alpha II-spectrin and tau in rat brain after methamphetamine exposure: a similar profile to traumatic brain injury. Life Sci. 2005;78:301–309. doi: 10.1016/j.lfs.2005.04.058. [DOI] [PubMed] [Google Scholar]
- Wise R, Hoffman DC. Localization of drug reward mechanisms by intracranial injections. Synapse. 1992;10:247–263. doi: 10.1002/syn.890100307. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Amphetamine-like stimulants: a report from the World Health Organization meeting on amphetamines, MDMA, and other psychostimulants; Geneva. Substance Abuse Department, World Health Organization; 1997. [Google Scholar]
- Wu CW, Ping YH, Yen JC, Chang CY, Wang SF, Yeh CL, Chi CW, Lee HC. Enhanced oxidative stress and aberrant mitochondrial biogenesis in human neuroblastoma SH-SY5Y cells during methamphetamine induced apoptosis. Toxicol Appl Pharmacol. 2007;220:243–251. doi: 10.1016/j.taap.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Wu W, Abraham LS, Ogony J, Matthews R, Goldstein G, Ercal N. Effects of N-acetylcysteine amide, a novel thiol antioxidant on radiation induced cytotoxicity in chinese hamster ovary cells. Life Sci. 2008;82:1122–1130. doi: 10.1016/j.lfs.2008.03.016. [DOI] [PubMed] [Google Scholar]
- Yamamoto BK, Zhu W. The Effects of methamphetamine on the production of free radicals and oxidative stress. J Pharmacol Exp Ther. 1998;287:107–114. [PubMed] [Google Scholar]