

Abstract
Introduction
Serotonin 1A receptor (5-HT1AR) function appears decreased in primary major depressive disorder (MDD) based upon physiological responses to 5-HT1AR agonists in vivo and 5-HT1AR receptor binding in brain tissue post- or antemortem. We previously assessed 5-HT1AR binding potential (BP) in depression using PET and [carbonyl-11C]WAY-100635, and demonstrated reduced 5-HT1AR BP in the mesiotemporal cortex (MTC) and raphe in depressives with primary, recurrent, familial mood disorders (n=12) versus controls (n=8)[1]. These findings were replicated by some, but not other studies performed in depressed samples more generally selected using criteria for MDD. In the current study we attempted to replicate our previous findings in an independent sample of subjects selected according to the criteria for primary, recurrent depression applied in our prior study.
Methods
Using PET and [carbonyl-11C]WAY-100635 the 5-HT1AR BP was assessed in 16 depressed subjects and 8 healthy controls.
Results
Mean 5-HT1AR BP was reduced 26% in MTC (p<0.005) and 43% in raphe (p<0.001) in depressives versus controls.
Conclusions
These data replicated our original findings, which showed BP reduced 27% in MTC (p<0.025) and 42% in raphe (p<0.02) in depression. The magnitudes of these reductions in 5-HT1AR binding were similar to those found post mortem in 5-HT1AR mRNA concentrations in hippocampus in MDD [2] and in 5-HT1AR binding capacity in the raphe in depressed suicide victims [3]. Disagreement exists within the literature, however, regarding the presence and direction of 5-HT1AR binding abnormalities in depression, which may be explained in some cases by differences in anatomical location (e.g., [4]) and in others by pathophysiological heterogeneity within MDD (e.g., some depressives hypersecrete cortisol, which would be expected to down-regulate 5-HT1AR expression[2]). Antidepressant drug treatment does not alter these abnormalities in 5-HT1AR binding [5, 6], but may compensate for blunted 5-HT1AR function by increasing post-synaptic 5-HT1AR transmission [7].
Keywords: PET, major depressive disorder, bipolar disorder
The central serotonergic system has received great interest in depression research, based initially on observations that selective serotonin reuptake inhibitors (SSRI) exert antidepressant effects, and that most other antidepressant drugs also increase serotonin (5-HT) transmission. This effect was thought to compensate for deficient serotonergic function in major depressive disorder (MDD), since remitted MDD subjects who are unmedicated or are being treated with SSRI agents experience depressive relapse under tryptophan depletion (which putatively decreases central serotonergic function (reviewed in [8]). The most promising evidence for a serotonin system deficiency that is compensated by antidepressant pharmacotherapy involves post-synaptic serotonin type 1A receptors (5-HT1AR). Chronic administration of antidepressant drugs with diverse primary pharmacological actions enhances post-synaptic 5-HT1AR function through a variety of receptor pharmacological mechanisms [7, 9]. Moreover, post mortem, neuroimaging and pharmacological challenge studies of depression have reported abnormalities in the 5-HT1AR density and mRNA expression [2-4, 10]. These abnormalities appear to be associated with reduced 5-HT1AR function, as depressed subjects show blunted thermic and endocrine responses to 5-HT1AR agonist challenge (reviewed in [1]).
The development of 5-HT1AR selective PET radioligands has enabled in vivo measures of pre- and post-synaptic 5-HT1AR binding in depression. In a previous study we demonstrated that mean 5-HT1AR binding potential (BP) was reduced in the mesiotemporal cortex (MTC) and raphe in unmedicated-depressives relative to controls using PET and [carbonyl-11C]WAY-100635[1]. Similar reductions were evident in the parietal and medial occipital/ posterior cingulate cortices. These data were consistent with those of Sargent et al [5], who found decreased 5-HT1AR BP, measured using PET and [11C]WAY 100635, in unmedicated depressives relative to controls in the raphe, MTC, insula, anterior cingulate cortex, temporal polar cortex and orbital cortex. Similarly, Meltzer et al.[11] found significantly decreased [11C]WAY-100635 binding in the raphe in elderly depressed patients (n=17) relative to age-matched controls (n=17). Several studies reported that these reductions in 5-HT1AR binding also were evident in currently remitted patients with a history of MDD [5, 6, 12], suggesting that this abnormality persists across illness episodes in recurrent depression.
Not all PET studies of 5-HT1AR in depression are in agreement, however. Parsey et al[13] found no difference in BP between depressed MDD subjects and controls. Moreover, they reported that in antidepressant drug naive MDD subjects, the BP values exceeded those of controls. These data contrasted with our own finding that antidepressant naïve cases had similar (or lower) BP than previously treated patients[14], as well as with those of longitudinal studies showing that antidepressant drug treatment exerted no significant effect on the abnormal reductions in BP in MDD [5, 6]. Nevertheless, disagreement regarding the presence and direction of abnormalities also exists within the post mortem literature (see Discussion), raising the possibility that clinical and pathophysiological heterogeneity within the conditions encompassed by the major depressive syndrome contributes to discrepancies in the results across studies.
Lopez et al[2] hypothesized that the reduced hippocampal 5-HT1AR mRNA found in depressed subjects post mortem specifically arose in response to the cortisol hypersecretion associated with MDD, since hippocampal 5-HT1AR mRNA expression is under tonic inhibition by adrenal corticosteroids. In rats and tree shrews hippocampal 5-HT1AR density and mRNA levels decrease in response to corticosterone administration and chronic stress, and increase following adrenalectomy [15-22]. Adrenalectomy also prevents stress-induced down-regulation of 5-HT1AR expression[2].
The sensitivity for detecting 5-HT1AR abnormalities in depressed samples may thus be enhanced by selecting subjects with a higher likelihood of having glucocorticoid hypersecretion. Imaging data have generally added to a variety of other types of psychobiological information to indicate that, in the absence of other entrance criteria, the DSM-series[23] criteria for MDD by themselves provide low sensitivity and specificity for detecting biological abnormalities. In contrast, depressives with bipolar disorder (BD) or with the MDD (unipolar depression) subtypes of melancholia or familial pure depressive disease (FPDD; i.e., primary MDD subjects who have first degree relatives with MDD, but not BD, alcoholism or antisocial personality disorder; where primary indicates that the depression did not arise in the setting of pre-existing medical or psychiatric disorders) proved more likely to show evidence of limbic-hypothalamic-pituitary-adrenal (LHPA) axis hyperactivity than depressed cases who did not meet criteria for these subtypes [24-28]. Moreover, Lewis & McChesney[29] showed that the mean platelet [3H]imipramine uptake was reduced in BD and FPDD but not in other MDD subtypes, suggesting the former subtypes may more likely manifest serotonergic dysfunction. Thus, to enhance the likelihood of detecting abnormalities in 5-HT1AR BP in depression our initial PET study [1] selected an “enriched sample” of depressed subjects who had a primary mood disorder and also fell within either the FPDD or BD spectrum categories.
This previous study assessed 5-HT1AR binding in 12 depressives with primary, recurrent depression, of whom 8 had MDD, and 4 had BD [1]. Of the MDD subjects, 4 met criteria for FPDD, while 4 had a first degree relative with BD, and thus were considered within the bipolar spectrum of affective illness (the offspring or monozygotic twin of an index case with BD can develop MDD, BD or schizoaffective disorder, suggesting that these conditions reflect the inheritance of a genotype which can be expressed as a spectrum of phenotypes of differing severities).[30] While differences in 5-HT1AR binding with respect to controls were evident when the 8 bipolar spectrum cases (BD subjects plus MDD subjects with bipolar relatives) were considered alone, the BP values in the small sample of 4 FPDD subjects did not differ significantly from controls when considered alone. Thus, in the current study we attempted to replicate our previous findings using an independent sample of depressed subjects with primary, recurrent mood disorders which included sufficient numbers of MDD cases within the FPDD or melancholic subtypes to permit extension of these findings to an enriched sample of unipolar depressives.
Methods
Depressed subjects (n=16; 10 female; mean age 32±10 years, range= 19 to 51 years) met DSM-IV criteria[23] for primary, recurrent MDD or BD, and had experienced depression onset prior to age 40 years. Fourteen subjects met DSM-IV criteria for MDD and two had BD, most recent episode depressed. Based upon family history, 3 of the MDD cases had a first degree relative with BD. Of the 11 MDD cases who did not have first degree relatives with BD, 7 were included for meeting FPDD criteria (of whom 4 also met melancholic subtype criteria) and 4 for meeting DSM-IV melancholic subtype, but not FPDD, criteria. Exclusion criteria included medical or neurological illnesses, MRI evidence of neuromorphological abnormalities, suicidal intent, substance abuse within 1 year, lifetime history of substance dependence (other than nicotine), and exposure to psychotropic medications within 3 weeks (8 weeks for fluoxetine).
Healthy controls (n=8; 4 female; mean age 32±12 years, range=18 to 50 years) met the same exclusion criteria and did not meet criteria for past or current psychiatric disorders.
Depression severity was rated using the Hamilton Rating Scale for Depression (HRSD [31]), and anxiety severity using the Spielberger State-Trait Anxiety Inventory (State; STAI) [32]. The plasma cortisol concentrations during the stress related to scanning (late morning to early afternoon for all subjects) was measured as described previously [1, 33].
None of the subjects in the current study participated in our previous study[1]. The depressed subjects were recruited from the out-patient psychiatric services of the University of Pittsburgh Medical Center (UPMC). All subjects provided written informed consent as approved by the UPMC Institutional Review Board.
Data Acquisition
A dynamic emission scan (29 frames of increasing length over 60 min) was acquired following i.v. bolus administration of 10 to 20 mCi of high specific activity [carbonyl-11C]WAY-100635], synthesized using a modification of the McCarron et al [34] method. Images were acquired using a Siemens/CTI HR+ (63 contiguous slices over 15.2 cm) in 3D mode [FWHM resolution = 5±0.5 mm transverse and 4.5±0.5 mm axially]. A Neuro-insert™ (CTI PET Systems, Knoxville, TN) was locked into the scanner gantry to reduce random coincidences. Images were reconstructed using a Hanning window with cut-off = Nyquist frequency, resulting in an estimated (including the effect of scatter) true image FWHM resolution of 7.1 mm transverse and 6.7 mm axially.
MRI scans were obtained using a 1.5 T GE Signa Scanner and a 3-D spoiled gradient recalled (SPGR) sequence optimized for delineating gray matter/white matter/CSF boundaries.
Data Analysis
PET and MR images were aligned using AIR [35]. Because power was relatively low in this small sample size, hypothesis-testing was limited to the MTC and raphe. The MTC region-of-interest (ROI) was defined by manual tracing around the hippocampus, amygdala, and adjacent parahippocampal and periamygdaloid cortex bilaterally using ImageTool™ (CTI PET Systems, Knoxville, TN) in the 6 to 7 planes where both amygdala and hippocampus were evident, as described in Drevets et al.[1](Figure 1). The raphe ROI was centered over the raphe nuclei located within the midbrain and rostral-most slices of the pons, collectively evident in [11C]WAY-100635 images because of their very high 5HT1AR density relative to that of surrounding tissues [36]. The raphe is inadequately visualized in MR images for boundary tracing, so this ROI was defined directly on PET images (Figure 1). The midbrain raphe and upper pons were selected to emphasize measures from the dorsal and median raphe nuclei, which innervate the forebrain. These relevant horizontal sections were identified in the co-registered MRI slices, and circular ROI (5 mm radius) were centered over the brainstem area of high tissue radioactivity in each slice as described in Drevets et al.[1]. The diameter of this ROI was larger than that of the actual raphe to accommodate the blurring of the raphe 5-HT1AR-specific signal in PET images and reduce the effects of possible position differences in the raphe signal due to movement between frames.
Figure 1.
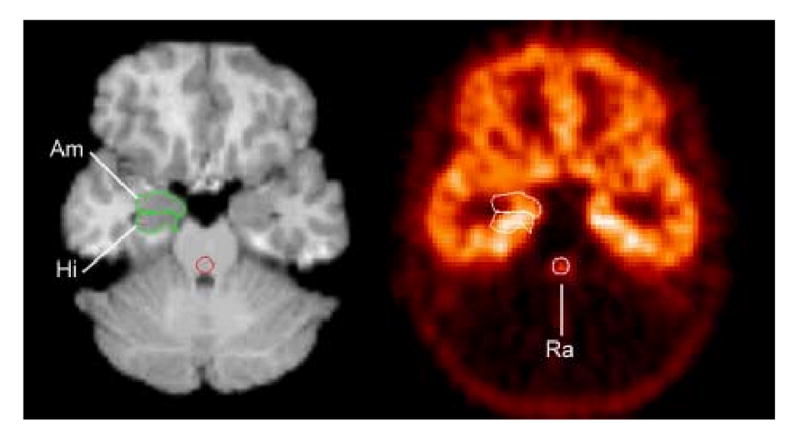
Co-registered PET and MRI sections through the mesiotemporal cortex and midbrain. This region included areas of high 5HT1AR density (hippocampus) along with areas of moderate density (amygdala, periamygdaloid cortex). By including amygdala (Am) and hippocampus (Hi) together, errors in distinguishing these two structures are avoided. The raphe (Ra) ROI was defined on the PET, but in the co-registered MRI it is evident that this circular ROI overlies the midbrain area where the dorsal and median raphe nuclei are known to be situated. Reproduced from [1].
The reference region for measuring free and nonspecifically bound radioligand concentrations was the cerebellar cortex. The cerebellum is most suitable as the reference tissue because it is nearly devoid of 5-HT1AR protein and mRNA, and is large enough that ROI can be situated far from tissues with abundant 5-HT1AR expression [36]. The cerebellar ROI was defined in 3 MRI planes where the cortex sampled was located at least twice the FWHM ventral and medial to ventral occipital cortex and posterior and medial to basal temporal cortex to avoid “spilling in” of radioactive counts from areas with abundant 5HT1A receptors [36, 37]. The posterior border of the cerebellar ROI also was situated >one cm from the brain edge to avoid spilling in of radioactive counts from the venous sinuses on the posterior surface of the cerebellum (which contain a higher concentration of radioactive polar metabolites than the cerebellar cortex, which only has a 4 to 5% blood volume)[38-40]. ROI placement was limited to axial planes at or dorsal to plane 56 (of 63) to measure from planes where the signal-to-noise ratio was relatively uniform in 3D images[41].
Decay-corrected, time-radioactivity curves were obtained from the dynamic PET image for each ROI using a calibrated phantom standard to convert tomographic counts to μCi/ml for each time point. Regional 5-HT1AR BP values were fit with a 3-parameter (simplified) reference tissue model [38, 42]. The BP values obtained using this model are less variable than those obtained using compartmental modeling approaches in which the plasma [11C]WAY-100635 concentration is the input function, because the rapid clearance of [11C]WAY-100635 from plasma limits the accuracy of quantitating metabolites at late imaging times [38].
The mean BP values were compared between the depressed and control groups using unpaired t-tests. Because the aim was to replicate previous results, one-tailed tests were used, with p-values corrected (Bonferroni) for comparisons in two ROI. In ROI where the original results[1] were replicated by the independent data set assessed herein, the power of post hoc clinical correlations was enhanced by combining the depressed samples from the current study with that from the original study (n=28) to assess relationships between regional BP values and illness severity ratings and plasma cortisol levels.
Results
The demographic characteristics and mean 5-HT1AR BP values of the study samples appear in table 1. Mean depression severity was in the moderately depressed range (mean HRSD17=18±7.0 and HRSD25 = 23±9.3) and mean STAI score was 23±10. When the MDD subjects with only MDD relatives was considered alone, their depression ratings were comparable (mean HRSD17=21±4.3; mean HRSD25 = 27±4.9; mean STAI=26±9.0). The mean stressed plasma cortisol concentrations trended toward being higher (0.05<p<0.1) in the depressives (13.1±7.77 μg/dl) than the controls (10.4±4.08 μg/dl). The mean time of day for assaying these levels did not differ significantly between groups.
Table 1.
Demographic and clinical characteristics and regional binding potentials for [11C]WAY-100635 of the study samples.
| Parameter | Control | Depressed | MDD (with MDD relatives) | BD Spectrum |
|---|---|---|---|---|
| Sample size (n) | 8 | 16 | 11 | 5 |
| Mean Age (SD) | 32.4 (12.1) | 32.4 (10.0) | 30.8 (7.8) | 35.8 (14.1) |
| % female | 50 | 63 | 55 | 80 |
| Mean (SD) BP: MTC | 9.17 (2.51) | 6.82 (1.20) a | 6.63 (1.29) b | 7.26 (0.94) c |
| Mean (SD) BP: raphe | 4.56 (1.26) | 2.60 (0.65) a | 2.66 (0.68) a | 2.47 (0.64) a |
p < 0.005, relative to controls
p = 0.01, relative to controls
0.05 < p < 0.10, relative to controls
Abbreviations: BD – bipolar disorder; BP – binding potential; MDD- major depressive disorder; MTC – mesiotemporal cortex
The mean 5-HT1AR BP was reduced 25.6% in the MTC (t=2.51; p<0.005) and 43.0% in the raphe (t=4.07; p<0.001) in depressives versus controls (table 1). The magnitudes of these differences were similar when the MDD cases who had only MDD first-degree relatives were considered alone, in whom the 5-HT1AR BP was reduced 27.7% in the MTC (t=2.62; p=0.010) and 41.7% in the raphe (t=3.88; p<0.001) in depressives versus controls. The difference in the raphe also remained significant when considering only the small sample of BD spectrum cases alone (p<0.005).
The 5-HT1AR BP values in the MTC correlated with those in the midbrain (r =0.553; p<0.01). In the combined sample of depressed patients from our current and previous studies (n=28) no relationship was evident between HRSD scores (17 item) and 5-HT1AR BP values in the raphe (r = -0.27; p>0.1), but the corresponding correlation involving BP values in the MTC trended toward significance (r= -0.33; 0.1<p<.05). The anxiety ratings were not correlated with BP values in the MTC or the raphe (p>0.2). The plasma cortisol levels were not correlated with the BP values in either ROI (p>0.2).
Discussion
In this independent subject sample, the mean 5-HT1AR BP was reduced 26% in the MTC (p<0.005) and 43% in the raphe (p<0.001) in depressives versus controls, replicating the findings in our original sample, which showed BP reduced 27% in MTC (p<0.025) and 42% in the raphe (p<0.02) in depressives versus controls [1]. Importantly, these differences remained significant when the MDD cases with only MDD first-degree relatives were considered alone (table 1).
These data were compatible with the results of 5-HT1AR agonist challenge studies, which showed that unmedicated MDD subjects have blunted hypothermic and adrenocorticotropin (ACTH) and cortisol release relative to controls in response to ipsapirone or buspirone [43-45]. Since 5-HT1AR stimulation induced hypothermia and ACTH/cortisol release are thought to distinguish pre- and postsynaptic 5-HT1AR stimulation, respectively, in humans and rats, 47 these findings were compatible with the PET data implicating both pre- and postsynaptic 5-HT1AR in MDD.
Moreover, the magnitudes of the differences we found in 5-HT1AR BP between depressives and controls appear similar to those reported by several other neuroimaging studies. For example, relative to healthy controls the post-synaptic 5-HT1AR BP values were decreased by 20 to 28% in women with post partum depression[46], and by 18 to 26% in depressed subjects with BD [47], and the 5-HT1AR BP in the raphe was reduced 37% in elderly depressives [11]. However, other studies reported reductions that were smaller in magnitude [5] or differences that were not significant with respect to controls [13], suggesting that heterogeneity exists with respect to the presence of abnormal 5-HT1AR binding in the conditions encompassed by DSM-IV criteria for MDD.
Specificity of 5-HT1AR binding abnormalities in mood and anxiety disorders
The specificity of 5-HT1AR binding abnormalities to mood disorders has not been entirely established. Studies in experimental animals have associated reductions in post-synaptic 5-HT1AR in multiple brain regions (including the MTC) following repeated stress in rats and tree shrews (see below) and in monkeys which show depressive-like behaviors [48]. In addition, humans with chronic fatigue syndrome have been shown to have reductions in 5-HT1AR binding that resemble those in MDD [49]. Nevertheless, some psychiatric disorders which are accompanied by physiological or psychological stress, such as post-traumatic stress disorder[50], schizophrenia [51], or restricting-type anorexia nervosa[52] have shown no differences in 5-HT1AR binding relative to healthy controls. In addition, women recovered from bulimia-type anorexia nervosa had significantly increased [11C]WAY-100635 binding potential in the MTC, raphe, cingulate, temporoparietal and prefrontal cortices relative to controls.[52] Furthermore, the data reviewed below on post mortem studies of suicide victims suggest that nonspecific depressive symptoms or stress exposure does not account for reductions in 5-HT1AR binding abnormalities in mood disorders.
We assessed the specificity of 5-HT1AR binding abnormalities by additional studies of depressive subtypes that were distinct from those studied either in our original and current studies, and anxiety disorders that commonly occur co-morbidly with depression. In each of these studies regional 5-HT1AR distribution volumes (DV) were measured along with BP values using arterial plasma input functions corrected for plasma protein binding of the parent radiotracer to ensure that the differences found in depression were not attributable to differences in free and nonspecific radioligand binding in the reference tissue:
Post Partum Depression
The 5-HT1AR binding was assessed in women who developed post partum depression (PPD) to elucidate mechanisms underlying the greater risk of MDD in women and the increased risk for MDD and BD onset in the post partum period. Ovarian steroids exert robust effects on 5-HT1AR and 5-HT transporter (5-HTT) expression in monkeys[53]. Moses et al.[46] compared 5-HT1AR BP between 9 unmedicated women with PPD (5 MDD, 4 BD) and 9 post partum control women using PET, [11C]WAY-100635, 90-min emission scans, and arterial input functions. Age, time since delivery, and reproductive hormones did not differ between groups. Postsynaptic 5-HT1AR receptor binding in PPD was reduced 20 to 28% relative to controls, with the most significant reductions (p<0.001) located in the MTC and anterior cingulate cortices.
Bipolar Disorder
To address the limitations in quantitating distribution volumes of [carbonyl-11C]WAY100635, we used PET and the 5-HT1AR selective radioligand, [18F]FCWAY, to assess the specificity of 5-HT1AR binding abnormalities in BD [47]. The mean 5-HT1AR BP was decreased in unmedicated, depressed BD subjects (n=14) vs HC (n=22) in anterior cingulate cortex (ACC; p=0.005), posterior cingulate cortex (PCC; p=0.002), left parietal cortex (p<0.001), and left (p=0.008) and right anterior insula (p=0.017). In contrast, the difference between groups in the raphe did not reach significance. The differences in post-synaptic 5-HT1AR BP remained significant when a range of modeling approaches and reference tissues was applied (no significant differences existed between groups in references tissues), and were similar in magnitude to those found herein in the MTC (18% to 26%).
Panic Disorder
The 5-HT1AR also has been implicated in the some pathological anxiety states, as thermic and ACTH/cortisol responses to 5-HT1AR agonist administration are blunted in panic disorder as well as in MDD, and 5-HT1AR “knock-out” mice manifest exaggerated anxiety responses (reviewed in[14, 54]). Using PET and [18F]FCWAY we demonstrated that the mean 5-HT1AR BP was decreased in unmedicated panic disorder subjects (n=16) relative to healthy controls (n=16) in ACC (p<0.001), PCC (p<0.001), and midbrain raphe (p<0.001)[14]. These abnormalities extended both to panic disorder subjects with comorbid depression (n=8) and to those without comorbid depression (n=8). These abnormalities also were significant in subjects who were naïve to psychotropic drug treatment (n=9).
Temporal Lobe Epilepsy (TLE)
The risks for depression and suicide are increased in TLE compared to control groups who are healthy or who suffer from other types of epilepsy. The 5-HT1AR binding in depressed TLE (n=16) versus TLE cases with no history of major depressive episodes (n=21) were compared using PET and [18F]FCWAY. The mean 5-HT1AR DV (corrected for protein binding) and BP were decreased in the depressed TLE cases versus the nondepressed cases in the anterior (p=.006) and posterior (p=.03) cingulate cortices, anterior insula (p=.009), raphe (p=.008), right hippocampus (p<.001), right amygdala (p=.03), and right parahippocampal cortex (p=.03)[55, 56].
Post-Traumatic Stress Disorder (PTSD)
The mean 5-HT1AR BP and DV did not differ significantly between unmedicated subjects with PTSD (n=12) vs. matched, never traumatized controls in any region studied. No difference in 5-HT1AR DV or BP was observed between PTSD patients with current or past diagnosis of depression versus those without depression. PTSD constitutes a particularly useful comparator condition for considering the neurobiological basis of 5-HT1AR abnormalities in MDD, because most studies find no evidence for glucocorticoid hypersecretion in PTSD.[57]
Treatment Effects in MDD, BD
Alterations in 5-HT1AR function (especially enhancement of post-synaptic 5-HT1AR function) appear critical to the efficacy of ADT in animal models that predict antidepressant efficacy (e.g., [58]). This effect does not involve changes in 5-HT1AR density, however [7, 9, 59]. Nevertheless, because treatment may reduce cortisol hypersecretion, we assessed effects of chronic SSRI treatment on 5-HT1AR binding. The 5-HT1AR BP did not significantly change between the pre- and post-treatment scans in the raphe, MTC, lateral orbital cortex, medial occipital/ posterior cingulate cortex, or parietal cortex.[6] Among the 22 subjects for whom the clinical response-to-treatment was established, the treatment non-responders (n=7) had higher baseline BP values in the left (p=0.01) and right orbital cortex (p=0.02) than the responders (n=15), implying that higher baseline 5-HT1AR binding was associated with poorer response to treatment, consistent with the previous report of Parsey et al.[60]. These data suggest the hypothesis that those patients who have reduced 5-HT1AR binding are more likely to benefit from treatments that enhance 5-HT1AR function/ transmission.
The observation that chronic antidepressant drug treatment did not significantly change cerebral 5-HT1AR binding is consistent with preclinical evidence that the alterations in serotonergic function associated with ADT are not accompanied by changes in 5-HT1AR density [7, 9, 59, 61, 62]. The preclinical effects of antidepressant drugs on 5-HT1AR function nevertheless suggest that these agents would compensate for a reduction in postsynaptic 5-HT1AR function in primary mood disorders, even if these agents did not correct abnormal 5-HT1AR binding [7, 9, 58]. This hypothesis is compatible with the clinical observation that major depressive episode (MDE) typically recur following the cessation of antidepressant drug treatment.
In contrast, some mood stabilizers (i.e., lithium, divalproex) increases gene expression of BAG-1, which would reduce the translocation of cortisol-GR into cell nuclei, and attenuate the effect of glucocorticoid receptor stimulation.[63] In both cross-sectional and longitudinal studies we demonstrated that mood stabilizer treatment with lithium or divalproex is associated with increased (toward normative levels) 5-HT1AR binding in BD samples [64].
This increased 5-HT1AR binding during mood stabilizer treatment may alternatively reflect neurotrophic/ neuroprotective effects on the neuronal processes which express 5-HT1AR protein. Lithium and divalproex also robustly increase the levels of the cytoprotective protein bcl-2 in various areas of the rodent brain and in cells of human neuronal origin.[65] Bcl-2 exerts both neuroprotective and neurotrophic effects in the mammalian CNS.[65]
Finally, the elevation in 5-HT1AR binding could reflect enhancement of 5-HT1AR promoter activity, since high affinity AP-1 sites exist in the 5-HT1AR promoter and both lithium and divalproex robustly increase AP-1 DNA binding activity [66, 67]. Nevertheless, lithium does not directly increase 5-HT1AR density in the cortex of experimental animals, implying the effect in BD may be specific to the pathophysiological state[68].
Interpretation of reduced 5-HT1A receptor binding in mood disorders
Abnormally decreased 5-HT1AR BP in FPDD/BD most likely reflects either down-regulation of receptor density or a reduction in the number of brain cells expressing 5-HT1AR. In contrast, reduced [11C]WAY-100635 and [18F]FCWAY binding in depression are not expected to reflect a compensatory responses to abnormal 5-HT release. The 5-HT1AR density and mRNA expression appear insensitive both to reducing 5-HT transmission by lesioning the raphe or administering the 5-HT depleting agent, PCPA, and to increasing 5-HT concentrations using SSRI or monoamine oxidase inhibitors (MAOI) [61, 62, 69-73]. Moreover, [11C]WAY-100635 and [18F]FCWAY binding appear insensitive to endogenous 5-HT concentrations, as in baboons administration of agents which increase 5-HT release, including fenfluramine, citalopram, and amphetamine, does not alter [11C]WAY-100635 binding.[74] The insensitivity to endogenous 5HT is partly related to the ∼50-fold greater 5-HT1A receptor affinity of WAY-100635 relative to endogenous 5HT (based upon KD).
In the current study the specificity of the 5-HT1AR BP measures was limited by the dependency of the modeling approach on cerebellar [11C]WAY-100635 concentrations. The abnormal 5-HT1AR BP values in depression thus may have been accounted for either by reduced 5-HT1AR specific binding in the target ROI or by elevated cerebellar [11C]WAY-100635 concentrations. However, there is no reason to expect that abnormal 5-HT1AR expression exists in the cerebellum in mood disorders, and the PET studies reviewed above which involved arterial input functions did not find differences in the radioligand distribution volume in the cerebellum.
The abnormal 5-HT1AR binding in primary mood disorders is also unlikely to be accounted for by differences in nonspecific binding because of the very high selectivity of [carbonyl-11C]WAY-100635 for 5-HT1AR. WAY-100635 shows greater than 100-fold selectivity for 5-HT1AR relative to a variety of other receptors, reuptake sites, and ion channels [75, 76]. These data are consistent with the high specific-to-nonspecific binding ratio, the rank order of uptake across regions, and the very low binding in cerebellum in [carbonyl-11C]WAY-100635 images.[38, 77] In humans [carbonyl-11C]WAY-100635 is almost exclusively metabolized to [11C]cyclohexane-carboxylic acid and more polar radioactive metabolites[40, 77]. The only radioactive metabolite of [carbonyl-11C]WAY-100635 in humans with high affinity for 5-HT1A receptors is [carbonyl-11C]desmethyl-WAY-100635, which Osman et al.[40] found detectable at only a “possible trace level” in plasma 60 min after injection. It is also unlikely that differences in radiolabeled metabolite concentrations contributed to the abnormal BP values in the depressives, since we showed previously that depressives metabolized [carbonyl-11C]WAY-100635 at a similar rate as healthy controls.[1] Nevertheless, a disadvantage to [carbonyl-11C]WAY-100635 is that its rapid clearance from plasma limits the accuracy of quantitating metabolites at late imaging times [38], so that BP values obtained using compartmental modeling approaches in which the plasma [11C]WAY-100635 concentration is the input function are highly variable.
To address this limitation of [carbonyl-11C]WAY-100635 with respect to quantitative modeling, Lang, Eckelman and colleagues[78] developed an analog of CWAY labeled with [18F] to take advantage of the longer radioisotope half-life (110 min). The compound used in these studies is the trans 4-fluorocyclohexanecarboxylic acid analog of CWAY and is referred to as FCWAY. The in vitro affinity of this ligand for the 5-HT1A receptor is ∼ 1 nM, comparable to WAY 100635. FCWAY specific binding could be blocked by co-injection of CWAY [78]. The primary metabolites are [18F]fluorocyclohexanecarboxylic acid and [18F]fluoride. As with the previous studies with [11C]WAY-100635, there was evidence that the carboxylic acid crosses the blood-brain-barrier to some extent, although this contributed to no more than 7% of the signal in the cerebellum and less in other regions with specific binding. Tracer kinetic modeling analysis showed that the data were well described by a model with two tissue compartments and the total DV was a useful measure of receptor binding. Across brain regions, DV estimates for [18F]FCWAY correlated (r= 0.99) with those measured from [11C]CWAY, excepting brain tissues located near the skull (the [18F]fluoride secondary metabolite of FCWAY binds to bone, confounding measures near skull).
Correlation with in vitro studies of depression conducted post mortem or antemortem
The sensitivity and specificity for measuring 5-HT1AR binding using neuroimaging are limited by the low spatial resolution of PET. For example, the 5-HT1AR specific binding measured over the raphe appears lower than expected based upon post mortem autoradiographic data[36], since the small size of the raphe relative to the spatial resolution of PET results in extensive partial volume effects[37]. Moreover, while extant PET radioligands measure binding to an 5-HT1AR antagonist, many post mortem studies assessed binding to the 5-HT1AR agonist, [3H]8-OH-DPAT. In healthy humans, 5-HT1AR Bmax measured using [3H]WAY-100635 correlate tightly with those measured using [3H]8-OH-DPAT, with Bmax measured using [3H]WAY-100635 being 60 to 70% higher in most regions than when measured using [3H]8-OH-DPAT[79]. Thus, the results of post mortem and antemortem studies of brain tissue obtained from individuals with primary mood disorders provides important confirmatory and, in some cases, complementary information regarding the nature of 5-HT1AR abnormalities in depression.
In Vitro Studies of 5-HT1AR Density in Primary Mood Disorders
In the hippocampus and neocortex, post mortem studies of primary MDD have reported abnormal reductions of post-synaptic 5-HT1AR density and mRNA expression, although these studies have the limitation of having even smaller sample sizes than those of neuroimaging studies. The 5-HT1AR mRNA levels have been shown to be significantly reduced in the hippocampus [2, 80] and the dorsolateral PFC [80] in MDD subjects post mortem. Bowen et al. [10] found reduced [3H]8-OH-DPAT binding in the temporal polar and lateral orbital cortices in 7 elderly subjects (6 MDD, 1 BD) who died of natural causes. Bowen et al [10] found the reduction in 5-HT1AR in two regions was associated with a reduction in “grey matter wet weight”, which was decreased 29% in the pars opercularis (ventrolateral PFC) and 38% in temporal polar C. In the entire pars opercularis and temporal polar C the magnitude of the significant reduction in 5-HT1AR Bmax (45% and 59%, respectively; p<0.05) was greater than the reduction in grey matter weight (29% and 38%, respectively) in depressives versus controls, and the 5-HT1AR density was also reduced in these regions (24% in both regions) when measured per unit of grey matter tissue. Finally, Francis et al.[81] reported that 5-HT1AR density measured antemortem by [3H]8-OH-DPAT binding was decreased 29% (p<0.01) in superficial layers of the dorsal anterolateral PFC tissue (BA 9) removed during neurosurgical intervention for intractable depression in 4 subjects with primary MDD or BD relative to neurosurgical controls.
In the raphe, the post mortem literature suggests a more complex pattern of 5-HT1AR binding abnormalities exists in depression. In suicide victims who were both depressed prior to suicide and had no history of alcoholism, Arango et al.[3] observed a significant, 38% reduction of the 5-HT1AR binding capacity (reflecting a reduction of both 5-HT1AR density and of the total area of the DRN labeled by [3H]OH-DPAT) relative to controls. This finding was consistent with the 37 to 43% reductions in 5-HT1AR binding we (table 1; also [1]) and others[11] found in the raphe in primary depressives using PET. However, in the dorsal and ventrolateral subnuclei of the dorsal raphe nucleus the 5-HT1AR binding was abnormally increased in depressed in another study of depressed suicide victims relative to controls[4].
In contrast to the agreement between the results of most post mortem studies, PET imaging studies and 5-HT1AR agonist challenge studies regarding the status of post-synaptic 5-HT1AR binding in primary MDD, the results of post mortem studies of 5-HT1AR binding in unselected suicide victims vary widely (reviewed in[1]). The highly variable results across suicide studies suggest that the nonspecific presence of depressive symptoms or suicidal behavior provides neither sensitivity nor specificity for identifying subjects with abnormal 5-HT1AR binding. Suicidal behavior and depressive symptoms are nonspecific clinical signs/symptoms occurring in a variety of medical and psychiatric conditions. Only 1/3 to 1/2 of suicides appear to have suffered from a MDE prior to death and many depressed suicides have MDE arising secondary to other psychiatric of medical conditions[30, 82]. The “primary” versus “secondary” affective disease distinction thus may relate to the sensitivity for identifying 5-HT1AR abnormalities in depression, since nearly all studies of primary mood disorders found reduced post-synaptic 5-HT1AR mRNA expression, binding or sensitivity.
Neurobiological Significance of Decreased 5-HT1AR Binding in Depression
In humans the 5-HT1AR is expressed presynaptically on 5-HT cell bodies in the raphe (as a somatodendritic autoreceptor) and postsynaptically in other brain regions [83]. In humans and monkeys, the density of 5-HT1AR is very high in the raphe and parts of the hippocampal formation, high in hypothalamus, insula, temporal, cingulate and ventral prefrontal cortices, moderately high in occipital and parietal cortices, and very low in cerebellum, striatum, thalamus and white matter.[84, 85] In the raphe stimulation of presynaptic 5-HT1A autoreceptors reduces 5-HT release by inhibiting 5-HT neuronal firing, and reduces 5-HT synthesis by inhibiting tryptophan hydroxylase.[73, 86] Alterations in 5-HT1AR density in the raphe could thus influence the amount or timing of serotonin release in widespread brain areas.
In the cortex, hippocampus and amygdala postsynaptic 5-HT1AR are located on pyramidal cells and interneurons, and stimulation of these receptors generally inhibits glutamate-mediated depolarization of parent neuron[59, 86]. Altered 5-HT1AR function exerts a major influence on neuronal activity in the amygdala and hippocampus [87-89]. Reduced 5-HT1AR function in the amygdala is hypothesized to play a role in disinhibiting amygdala responses to emotional stumuli [89], potentially contributing to the pathological emotional behavior associated with mood disorders [87, 90]. In addition, stimulation of 5-HT1AR in the PFC plays a major role in modulating 5-HT release during stress,[91] so reduced 5-HT1AR binding in this cortex may alter the amount of 5-HT release during stress.
Postsynaptic 5-HT1AR also are expressed abundantly by astrocytes and some other glia [83, 92, 93]. Stimulation of astrocyte-based 5-HT1AR causes astrocytes to acquire a more mature morphology and to release the trophic factor, S-100B, which promotes growth and arborization of serotonergic axons.[94] S-100B is primarily released by astroglia in the developing brain, when it plays a role in the development of the serotonergic system[83, 95]. In the adult brain S-100B plays major roles in the plasticity of 5-HT neurons[96]. The importance of 5-HT1AR function on 5-HT system development is evident in 5-HT1AR knockout mice, which showed elevated anxiety behaviors specifically as a result of deficient 5-HT1AR function during development.[97]
S-100B also plays a role in maintaining the cytoskeleton in adult animals by promoting tubulin polymerization and inhibiting PKC-mediated breakdown of microtubules.[98] In addition, stimulation of neuron-based 5-HT1AR inhibits PKA-mediated disassociation of the proteins comprising the tubulin polymers of the cytoskeleton. Administration of 5-HT1AR antagonists, antibodies to S-100B, or agents that deplete 5-HT all produce similar losses of dendrites, spines and/or synapses in adult and developing animals, effects which are blocked by administration of 5-HT1AR agonists or SSRIs.[98] The role of postsynaptic 5-HT1AR function in maintenance of the cytoskeleton has led to the hypothesis that a reduction of 5-HT1AR function may comprise a risk factor for developing the neuropathological abnormalities identified in limbic and paralimbic cortical areas in mood disorders (reduced cortex volume, reduced synaptic proteins, increased neuronal density, reduced glial counts) [99, 100].
Regulation of 5-HT1A Receptor Expression by Glucocorticoid Hormones
Although 5-HT1AR density and mRNA expression is relatively insensitive to reductions in 5-HT transmission associated with raphe lesions or serotonin depletion[59, 69-71], or to elevations of 5-HT transmission resulting from SSRI or MAOI[59, 61, 62, 72], the 5-HT1AR expression is potently inhibited by adrenal corticosteroids. Postsynaptic 5-HT1AR gene expression is under tonic inhibition by adrenal steroids in the hippocampus and some other regions. Thus, in rodents the hippocampal 5-HT1AR mRNA expression is increased by adrenalectomy and decreased by corticosterone (CORT) administration or chronic stress.[2, 18, 19, 22, 101, 102]. The stress-induced down-regulation of 5-HT1AR expression is prevented by adrenalectomy. Mineralocorticoid receptor stimulation has the most potent effect on down-regulating 5-HT1AR, although glucocorticoid receptor stimulation also contributes to this effect [17, 18, 103].
Thus one factor which may contribute to the reduction in 5-HT1AR binding in depression is increased cortisol secretion associated with primary mood disorders[2]. The magnitude of the reduction in 5-HT1AR density and mRNA levels induced by stress-induced glucocorticoid secretion in rodents is similar to that of the differences between depressed and healthy humans. For example, in tree shrews chronic social subordination stress decreased the density of 5-HT1AR in the posterior cingulate, parietal cortex, prefrontal cortex, and hippocampus (by 11% to 34%),[22] similar to the magnitude of reduced 5-HT1AR BP found by Sargent et al.[5] and Drevets et al.[1] in these regions. Thus, elevated cortisol concentrations may conceivably induce a relatively widespread reduction of 5-HT1AR expression.
Relative to healthy controls, depressives show elevated levels of cortisol in plasma, hypertrophy of the adrenal and pituitary glands, and exaggerated cortisol response to ACTH stimulation (presumably reflecting adrenal hypertrophy)[26, 104, 105]. The relationship between 5-HT1AR binding in depression and current measures of cortisol secretion may prove complex, however. Because 5-HT1AR stimulation increases plasma ACTH and cortisol secretion, down-regulation of 5-HT1AR gene expression is thought to comprise a compensatory mechanism for inhibiting cortisol release[43, 106]. To the extent that this response maintains cortisol levels near the normative range, peripheral endocrine measures may no longer reflect the history of excessive cortisol secretion and reduced 5-HT1AR BP may instead reflect the recent propensity to hypersecrete cortisol in depression. This interaction may explain the lack of correlation between hippocampal 5-HT1AR BP and current cortisol concentrations in our current and previous studies [1]. Assessments of the pathophysiological diathesis to hypersecrete cortisol may, therefore, provide more sensitive correlates of reduced 5-HT1AR binding. It thus is noteworthy that our previous study[1] found reduced 5-HT1AR BP in a depressed sample who showed significantly elevated stressed plasma cortisol levels[1], and that in the current study we also found reduced 5-HT1AR BP in an independent depressed sample whose stressed plasma cortisol levels trended toward being elevated with respect to controls (0.05<p<0.10; the mean values in our original sample of 12.5±2.8 and 9.7±2.1 μg/dl, in depressives and controls, respectively, were similar to those obtained in the current sample of 13.1±7.8 and 10.4±4.1 in depressives and controls, respectively).
Two PET studies attempted to assess the effect of glucocorticoid hormone on 5-HT1AR using PET and [11C]WAY-100635. The first showed no change in 5-HT1AR BP at 12 hours after infusion of hydrocortisone 100 mg [107]. However, while mRNA expression decreases within hours, the reduction in 5-HT1AR protein is not evident until 2 to 4 days later [2]. The other study showed no reduction in 5-HT1AR BP in patients with medical disorders for which they received prednisolone >7.5 mg [108]. However, prednisolone predominantly stimulates glucocorticoid receptors and has relatively low affinity for mineralocorticoid receptors, and it is the latter steroid receptor subtype that exerts the most prominent effect on 5-HT1AR[17] mRNA expression. Thus the design of these studies may have reduced the sensitivity for detecting glucocorticoid-mediated effects on 5-HT1AR binding.
Genetic Correlations
Finally, the amount of variability of 5-HT1AR binding that is modulated by gene polymorphisms associated with the vulnerability to depression remains unclear. A functional polymorphism in the 5-HT1AR promoter region was associated with increased risk for MDD and suicide.[109] The allele associated with MDD leads to decreased activity by the repressor protein NUDR, which may in turn lead to increased 5-HT1AR expression in the raphe. In contrast, the NUDR repressor may exert opposite effects on hippocampal and cortical postsynaptic 5-HT1AR, as decreased NUDR function reduced the expression of postsynaptic 5-HT1AR in vitro[109]. One PET study of 5-HT1AR BP in depressed subjects reported that the G-allele of this 5-HT1AR promoter polymorphism was associated with higher 5-HT1AR binding[13], although this relationship was not replicated by another group [110]. The latter group also assessed the relationship of the common variable number tandem repeat polymorphism [short (S) and long (L) alleles] of the 5-HT transporter gene to 5-HT1AR BP, and found that binding was lower in all brain regions in subjects with short (SS or SL) genotypes than in those with long (LL) genotypes. The short genotype has been associated with an increased vulnerability for developing depression within the context of stress[111], suggesting the hypothesis that this increased risk for depression is mediated by a heritable diathesis to more profoundly down-regulate 5-HT1AR expression in response to stress.
Summary
It remains unclear whether the reduction in 5-HT1AR function and expression in mood disorders constitutes a neurodevelopmental or an acquired abnormality. This issue is of interest because interruption of 5-HT1AR function during neurodevelopment persistently alters the function of emotion modulating systems [97]. Nevertheless, the reduction in 5-HT1AR binding and mRNA expression in depression may arise secondarily to cortisol hypersecretion, as the 5-HT1AR mRNA expression and density are under tonic inhibition by glucocorticoid receptor (GR) stimulation[2]. Moreover, the mood and anxiety disordered subgroups with reduced 5-HT1AR binding may be limited to those with a diathesis to hypersecrete cortisol (e.g., [1, 2].
Although antidepressant drug treatment does not normalize the abnormalities in 5-HT1AR in depression [5, 6], evidence from preclinical studies suggests that chronic administration of these agents results in enhancement of 5-HT1AR function[7, 9]. Potentially consistent with this evidence, treatment responders have lower 5-HT1AR binding than nonresponders[6, 60], suggesting the hypothesis that those patients who have reduced 5-HT1AR binding are more likely to benefit from treatments that enhance 5-HT1AR function/ transmission.
Figure 2.

Regional 5HT1AR binding for the combined samples from our current and previous[1] studies of recurrent depression.
Table 2. Specificity of 5-HT1AR Imaging abnormalities in mood disorders.
Post-synaptic 5-HT1A R decreased in
5HT1AR not different or increased in
|
See text for references
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Drevets WC, et al. PET imaging of serotonin 1A receptor binding in depression. Biol Psychiatry. 1999;46(10):1375–87. doi: 10.1016/s0006-3223(99)00189-4. [DOI] [PubMed] [Google Scholar]
- 2.Lopez JF, et al. Regulation of serotonin 1A, glucocorticoid, and mineralocorticoid receptor in rat and human hippocampus: implications for neurobiology of depression. Biol Psychiatry. 1998;43:547–573. doi: 10.1016/s0006-3223(97)00484-8. [DOI] [PubMed] [Google Scholar]
- 3.Arango V, et al. Serotonin 1A receptors, serotonin transporter binding and serotonin transporter mRNA expression in the brainstem of depressed suicide victims. Neuropsychopharmacology. 2001;25(6):892–903. doi: 10.1016/S0893-133X(01)00310-4. [DOI] [PubMed] [Google Scholar]
- 4.Stockmeier CA, et al. Increase in serotonin-1A autoreceptors in the midbrain of suicide victims with major depression-postmortem evidence for decreased serotonin activity. J Neurosci. 1998;18(18):7394–401. doi: 10.1523/JNEUROSCI.18-18-07394.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sargent PA, et al. Brain serotonin1A receptor binding measured by positron emission tomography with [11C]WAY-100635: effects of depression and antidepressant treatment. Arch Gen Psychiatry. 2000;57(2):174–80. doi: 10.1001/archpsyc.57.2.174. [DOI] [PubMed] [Google Scholar]
- 6.Moses-Kolko EL, P J, Thase ME, Meltzer CC, Kupfer DJ, Mathis CA, Bogers WD, Berman SR, Houck PR, Schneider TN, Drevets WC. Measurement of 5-HT(1A) receptor binding in depressed adults before and after antidepressant drug treatment using positron emission tomography and [(11)C]WAY-100635. Synapse. 2007;61(7):523–30. doi: 10.1002/syn.20398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaput Y, de Montigny C, Blier P. Presynaptic and postsynaptic modifications of the serotonin system by long-term administration of antidepressant treatments. An in vivo electrophysiologic study in the rat. Neuropsychopharmacology. 1991;5(4):219–29. [PubMed] [Google Scholar]
- 8.Neumeister A, et al. Neural and behavioral responses to tryptophan depletion in unmedicated patients with remitted major depressive disorder and controls. Arch Gen Psychiatry. 2004;61(8):765–73. doi: 10.1001/archpsyc.61.8.765. [DOI] [PubMed] [Google Scholar]
- 9.Haddjeri N, Blier P, de Montigny C. Long-term antidepressant treatments result in a tonic activation of forebrain 5-HT1A receptors. J Neurosci. 1998;18(23):10150–6. doi: 10.1523/JNEUROSCI.18-23-10150.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bowen DM, et al. Circumscribed changes of the cerebral cortex in neuropsychiatric disorders of later life. Proc Natl Acad Sci U S A. 1989;86(23):9504–8. doi: 10.1073/pnas.86.23.9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meltzer CC, et al. Serotonin 1A receptor binding and treatment response in late-life depression. Neuropsychopharmacology. 2004;29(12):2258–65. doi: 10.1038/sj.npp.1300556. [DOI] [PubMed] [Google Scholar]
- 12.Bhagwagar Z, et al. Persistent reduction in brain serotonin1A receptor binding in recovered depressed men measured by positron emission tomography with [11C]WAY-100635. Mol Psychiatry. 2004;9(4):386–92. doi: 10.1038/sj.mp.4001401. [DOI] [PubMed] [Google Scholar]
- 13.Parsey RV, et al. Altered Serotonin 1A Binding in Major Depression: A [carbonyl-C-11]WAY100635 Positron Emission Tomography Study. Biol Psychiatry. 2006;59(2):106–13. doi: 10.1016/j.biopsych.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 14.Neumeister A, et al. Reduced serotonin type 1A receptor binding in panic disorder. J Neurosci. 2004;24(3):589–91. doi: 10.1523/JNEUROSCI.4921-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bagdy G, et al. Long-term cortisol treatment impairs behavioral and neuroendocrine responses to 5-HT1 agonists in the rat. Neuroendocrinology. 1989;50(3):241–7. doi: 10.1159/000125248. [DOI] [PubMed] [Google Scholar]
- 16.Chalmers DT, et al. Corticosteroids regulate brain hippocampal 5-HT1A receptor mRNA expression. J Neurosci. 1993;13(3):914–23. doi: 10.1523/JNEUROSCI.13-03-00914.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meijer OC, de Kloet ER. A role for the mineralocorticoid receptor in a rapid and transient suppression of hippocampal 5-HT1A receptor mRNA by corticosterone. J Neuroendocrinol. 1995;7(8):653–7. doi: 10.1111/j.1365-2826.1995.tb00804.x. [DOI] [PubMed] [Google Scholar]
- 18.Meijer OC, Van Oosten RV, De Kloet ER. Elevated basal trough levels of corticosterone suppress hippocampal 5- hydroxytryptamine(1A) receptor expression in adrenally intact rats: implication for the pathogenesis of depression. Neuroscience. 1997;80(2):419–26. doi: 10.1016/s0306-4522(97)00008-0. [DOI] [PubMed] [Google Scholar]
- 19.Mendelson SD, McEwen BS. Autoradiographic analyses of the effects of restraint-induced stress on 5-HT1A, 5-HT1C and 5-HT2 receptors in the dorsal hippocampus of male and female rats. Neuroendocrinology. 1991;54(5):454–61. doi: 10.1159/000125951. [DOI] [PubMed] [Google Scholar]
- 20.Mendelson SD, McEwen BS. Autoradiographic analyses of the effects of adrenalectomy and corticosterone on 5-HT1A and 5-HT1B receptors in the dorsal hippocampus and cortex of the rat. Neuroendocrinology. 1992;55(4):444–50. doi: 10.1159/000126160. [DOI] [PubMed] [Google Scholar]
- 21.Zhono P, C R. Transcription regulation of hippocampal 5-HT1A receptors by glucocorticoid hormones. Neuroscience. 1994;(29):1161. [Google Scholar]
- 22.Flugge G. Dynamics of central nervous 5-HT1A-receptors under psychosocial stress. J Neurosci. 1995;15(11):7132–40. doi: 10.1523/JNEUROSCI.15-11-07132.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.APA. Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) Washington, D.C.: APA Press; 1994. [Google Scholar]
- 24.Winokur G. The development and validity of familial subtypes in primary unipolar depression. Pharmacopsychiatria. 1982;15(4):142–6. doi: 10.1055/s-2007-1019527. [DOI] [PubMed] [Google Scholar]
- 25.Lewis DA, et al. Differentiation of depressive subtypes by insulin insensitivity in the recovered phase. Arch Gen Psychiatry. 1984;40:167–170. doi: 10.1001/archpsyc.1983.01790020061005. [DOI] [PubMed] [Google Scholar]
- 26.Arana GW, Baldessarini RJ, Ornsteen M. The dexamethasone suppression test for diagnosis and prognosis in psychiatry. Commentary and review. Arch Gen Psychiatry. 1985;42(12):1193–204. doi: 10.1001/archpsyc.1985.01790350067012. [DOI] [PubMed] [Google Scholar]
- 27.Rush AJ, Weissenburger JE. Melancholic symptom features and DSM-IV. Am J Psychiatry. 1994;151(4):489–98. doi: 10.1176/ajp.151.4.489. [DOI] [PubMed] [Google Scholar]
- 28.Contreras F, et al. Hormonal differences between psychotic and non-psychotic melancholic depression. Journal of Affective Disorders. 2007;100(13):65–73. doi: 10.1016/j.jad.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 29.Lewis DA, McChesney C. Tritiated imipramine binding distinguishes among subtypes of depression. Arch Gen Psychiatry. 1985;42(5):485–8. doi: 10.1001/archpsyc.1985.01790280067006. [DOI] [PubMed] [Google Scholar]
- 30.Drevets WC, Todd RD. Depression, mania and related disorders. In: Guze SB, editor. Adult Psychiatry. Mosby Press; St. Louis, MO: 1997. [Google Scholar]
- 31.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spielberger CD, G R, Lushene RE. Manual for the State_Trait Anxiety Inventory. Palso Alto, CA: Consulting Psychologists Press; 1970. [Google Scholar]
- 33.Drevets WC, P J, Bardgett ME, Reich T, Todd R, Raichle ME. Glucose metabolism in the amygdala in depression: relationship to diagnostic subtype and stressed plasma cortisol levels. Pharmacol Biochem Behav. 2002;71:431–447. doi: 10.1016/s0091-3057(01)00687-6. [DOI] [PubMed] [Google Scholar]
- 34.McCarron JA, et al. Remotely-controlled production of the 5-HT1A receptor radioligand, [carboxyl-11c] Way-100635, via 11C-carboxylation of an immobilized Grigard Reagent. J Lablled Compunds Radiopharmaceuticals. 1996;38:941–953. [Google Scholar]
- 35.Woods RP, Mazziotta JC, Cherry SR. MRI-PET registration with automated algorithm. J Comput Assist Tomogr. 1993;17(4):536–46. doi: 10.1097/00004728-199307000-00004. [DOI] [PubMed] [Google Scholar]
- 36.Hall H, et al. Autoradiographic localization of 5-HT1A receptors in the post-mortem human brain using [3H]WAY-100635 and [11C]way-100635. Brain Res. 1997;745(12):96–108. doi: 10.1016/s0006-8993(96)01131-6. [DOI] [PubMed] [Google Scholar]
- 37.Links JM, et al. Influence of spatially heterogeneous background activity on “hot object” quantitation in brain emission computed tomography. J Comput Assist Tomogr. 1996;20(4):680–7. doi: 10.1097/00004728-199607000-00033. [DOI] [PubMed] [Google Scholar]
- 38.Gunn RN, et al. Tracer kinetic modeling of the 5-HT1A receptor ligand [carbonyl-11C]WAY- 100635 for PET. Neuroimage. 1998;8(4):426–40. doi: 10.1006/nimg.1998.0379. [DOI] [PubMed] [Google Scholar]
- 39.Osman S, Lundkvist C, Pike V. Radioactive metabolites of the 5-HT1A receptor radioligand. [O-methyl-11C]WAY-100635, in rat, monkey and humans plus evaluation of the brain uptake of the metabolite. [O-methyl-11C]WAY-100634 in monkey. J Label Compd Radiopharm. 1995;(37):283. [Google Scholar]
- 40.Osman S, et al. Characterisation of the appearance of radioactive metabolites in monkey and human plasma from the 5-HT1A receptor radioligand, [carbonyl-11C]WAY-100635--explanation of high signal contrast in PET and an aid to biomathematical modelling. Nucl Med Biol. 1998;25(3):215–23. doi: 10.1016/s0969-8051(97)00206-0. [DOI] [PubMed] [Google Scholar]
- 41.Townsend DW, Isoardi RA, Bendrium B. Volume imaging tomographs. In: Bendrium B, Townsend DW, editors. The Theory and Practice of 3D PET. Kluwer Academic Publishers; Boston: 1998. pp. 133–167. [Google Scholar]
- 42.Lammertsma AA, Hume SP. Simplified reference tissue model for PET receptor studies. Neuroimage. 1996;4(3 Pt 1):153–8. doi: 10.1006/nimg.1996.0066. [DOI] [PubMed] [Google Scholar]
- 43.Lesch K. The ipsapirone/5-HT1A receptor challenge in anxiety disorders and depression. In: Stahl S, et al., editors. Serotonin 1A Receptors in Depression and Anxiety. Raven Press; New York: 1992. pp. 135–162. [Google Scholar]
- 44.Lesch KP, Disselkamp-Tietze J, Schmidtke A. 5-HT1A receptor function in depression: effect of chronic amitriptyline treatment. J Neural Transm Gen Sect. 1990;80(2):157–61. doi: 10.1007/BF01257081. [DOI] [PubMed] [Google Scholar]
- 45.Rausch JL, Stahl SM, Hauger RL. Cortisol and growth hormone responses to the 5-HT1A agonist gepirone in depressed patients. Biol Psychiatry. 1990;28(1):73–8. doi: 10.1016/0006-3223(90)90434-4. [DOI] [PubMed] [Google Scholar]
- 46.Moses-Kolko E, et al. Serotonin 1A receptor reductions in postpartum depression: a PET study. Fertility and Sterility. 2007 doi: 10.1016/j.fertnstert.2007.03.059. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bain EE, et al. Decreased 5-HT1A receptor binding in bipolar depression. Biol Psychiatry. 2004;55:178S. doi: 10.1016/j.biopsych.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 48.Shively CA, et al. Behavioral depression and positron emission tomography-determined serotonin 1A receptor binding potential in cynomolgus monkeys. Arch Gen Psychiatry. 2006;63(4):396–403. doi: 10.1001/archpsyc.63.4.396. [DOI] [PubMed] [Google Scholar]
- 49.Cleare AJ, et al. Brain 5-HT1A receptor binding in chronic fatigue syndrome measured using positron emission tomography and [11C]WAY-100635. Biol Psychiatry. 2005;57(3):239–46. doi: 10.1016/j.biopsych.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 50.Bonne O, et al. No change in serotonin type 1A receptor binding in patients with posttraumatic stress disorder. Am J Psychiatry. 2005;162(2):383–5. doi: 10.1176/appi.ajp.162.2.383. [DOI] [PubMed] [Google Scholar]
- 51.Bantick RA, et al. A positron emission tomography study of the 5-HT1A receptor in schizophrenia and during clozapine treatment. J Psychopharmacol. 2004;18(3):346–54. doi: 10.1177/026988110401800304. [DOI] [PubMed] [Google Scholar]
- 52.Bailer UF, et al. Altered brain serotonin 5-HT1A receptor binding after recovery from anorexia nervosa measured by positron emission tomography and [carbonyl11C]WAY-100635. Arch Gen Psychiatry. 2005;62(9):1032–41. doi: 10.1001/archpsyc.62.9.1032. [DOI] [PubMed] [Google Scholar]
- 53.Lu NZ, Bethea CL. Ovarian steroid regulation of 5-HT1A receptor binding and G protein activation in female monkeys. Neuropsychopharmacology. 2002;27(1):12–24. doi: 10.1016/S0893-133X(01)00423-7. [DOI] [PubMed] [Google Scholar]
- 54.Drevets WC, et al. PET imaging of serotonin 1A receptor binding in depression. Biol Psychiatry. 1999;46(10):1375–87. doi: 10.1016/s0006-3223(99)00189-4. [DOI] [PubMed] [Google Scholar]
- 55.Theodore WH, et al. Reduced Hippocampal 5HT1A PET Receptor Binding and Depression in Temporal Lobe Epilepsy. Epilepsia. 2007 doi: 10.1111/j.1528-1167.2007.01089.x. [DOI] [PubMed] [Google Scholar]
- 56.Hasler G, et al. 5-HT1A receptor binding in temporal lobe epilepsy patients with and without major depression. Biological Psychiatry. doi: 10.1016/j.biopsych.2007.02.015. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Charney DS, Drevets WC. The Neurobiological Basis of Anxiety Disorders. In: Davis K, Charney DS, Coyle J, Nemeroff CB, editors. Psychopharmacology The Fifth Generation of Progress. Lippencott, Williams and Wilkins; New York: 2002. pp. 901–930. [Google Scholar]
- 58.Detke MJ, Wieland S, Lucki I. Blockade of the antidepressant-like effects of 8-OH-DPAT, buspirone and desipramine in the rat forced swim test by 5HT1A receptor antagonists. Psychopharmacology (Berl) 1995;119(1):47–54. doi: 10.1007/BF02246053. [DOI] [PubMed] [Google Scholar]
- 59.Frazer A, Hensler JG. 5-HT1A receptors and 5-HT1A-mediated responses: effect on treatments that modify serotonergic neurotransmission. In: Whitaker-Azmitia PM, Peroutka SJ, editors. The Neuropharmacology of Serotonin. The New York Academy of Sciences; New York: 1990. pp. 460–475. [DOI] [PubMed] [Google Scholar]
- 60.Parsey RV, et al. Higher 5-HT1A receptor binding potential during a major depressive episode predicts poor treatment response: preliminary data from a naturalistic study. Neuropsychopharmacology. 2006;31(8):1745–9. doi: 10.1038/sj.npp.1300992. [DOI] [PubMed] [Google Scholar]
- 61.Spurlock G, et al. Lack of effect of antidepressant drugs on the levels of mRNAs encoding serotonergic receptors, synthetic enzymes and 5HT transporter. Neuropharmacology. 1994;33(34):433–40. doi: 10.1016/0028-3908(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 62.Welner SA, et al. Autoradiographic quantification of serotonin1A receptors in rat brain following antidepressant drug treatment. Synapse. 1989;4(4):347–52. doi: 10.1002/syn.890040410. [DOI] [PubMed] [Google Scholar]
- 63.Zhou R, et al. The anti-apoptotic, glucocorticoid receptor cochaperone protein BAG-1 is a long-term target for the actions of mood stabilizers. J Neurosci. 2005;25(18):4493–502. doi: 10.1523/JNEUROSCI.4530-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carlson PJ, et al. Serotonin-1A receptor binding in bipolar depression before and after mood stabilizer treatment. Biological Psychiatry. 2007;61:57S. [Google Scholar]
- 65.Manji HK, Drevets WC, Charney DS. The cellular neurobiology of depression. Nature Medicine. 2001;7(5):541–7. doi: 10.1038/87865. [DOI] [PubMed] [Google Scholar]
- 66.Chen G, et al. Valproate robustly enhances AP-1 mediated gene expression. Brain Res Mol Brain Res. 1999;64(1):52–8. doi: 10.1016/s0169-328x(98)00303-9. [DOI] [PubMed] [Google Scholar]
- 67.Yuan PX, et al. Lithium stimulates gene expression through the AP-1 transcription factor pathway. Brain Res Mol Brain Res. 1998;58(12):225–30. doi: 10.1016/s0169-328x(98)00114-4. [DOI] [PubMed] [Google Scholar]
- 68.Mizuta T, Segawa T. Chronic effects of imipramine and lithium on postsynaptic 5-HT1A and 5- HT1B sites and on presynaptic 5-HT3 sites in rat brain. Jpn J Pharmacol. 1988;47(2):107–13. doi: 10.1254/jjp.47.107. [DOI] [PubMed] [Google Scholar]
- 69.Hensler JG, Kovachich GB, Frazer A. A quantitative autoradiographic study of serotonin1A receptor regulation. Effect of 5,7-dihydroxytryptamine and antidepressant treatments. Neuropsychopharmacology. 1991;4(2):131–44. [PubMed] [Google Scholar]
- 70.Pranzatelli MR. Dissociation of the plasticity of 5-HT1A sites and 5-HT transporter sites. Neurochem Res. 1994;19(3):311–5. doi: 10.1007/BF00971579. [DOI] [PubMed] [Google Scholar]
- 71.Verge D, et al. Quantitative autoradiography of multiple 5-HT1 receptor subtypes in the brain of control or 5,7-dihydroxytryptamine-treated rats. J Neurosci. 1986;6(12):3474–82. doi: 10.1523/JNEUROSCI.06-12-03474.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carli M, Afkhami-Dastjerdian S, Reader TA. [3H]8-OH-DPAT binding and serotonin content in rat cerebral cortex after acute fluoxetine, desipramine, or pargyline. J Psychiatry Neurosci. 1996;21(2):114–22. [PMC free article] [PubMed] [Google Scholar]
- 73.Frazer A, Hensler J. Serotonin. In: Siegel G, et al., editors. Basic Chemistry. Raven Press; New York: 1990. pp. 283–308. [Google Scholar]
- 74.Parsey RV, et al. Kinetic derivation of serotonin 5-HT1A receptor binding potential with [C-11]Carbonyl-Way 100635 and competition studies with endogenous serotonin. J Nuclear Med. 1998;39(5 Suppl):167P. [Google Scholar]
- 75.Laporte AM, et al. Selective in vivo labelling of brain 5-HT1A receptors by [3H]WAY 100635 in the mouse. Eur J Pharmacol. 1994;271(23):505–14. doi: 10.1016/0014-2999(94)90812-5. [DOI] [PubMed] [Google Scholar]
- 76.Fletcher A, et al. Electrophysiological, biochemical, neurohormonal and behavioural studies with WAY-100635, a potent, selective and silent 5-HT1A receptor antagonist. Behav Brain Res. 1996;73(12):337–53. doi: 10.1016/0166-4328(96)00118-0. [DOI] [PubMed] [Google Scholar]
- 77.Pike VW, et al. Exquisite delineation of 5-HT1A receptors in human brain with PET and [carbonyl-11 C]WAY-100635. Eur J Pharmacol. 1996;301(13):R5–7. doi: 10.1016/0014-2999(96)00079-9. [DOI] [PubMed] [Google Scholar]
- 78.Lang L, et al. Development of fluorine-18-labeled 5-HT1A antagonists. J Med Chem. 1999;42(9):1576–86. doi: 10.1021/jm980456f. [DOI] [PubMed] [Google Scholar]
- 79.Burnet PW, Eastwood SL, Harrison PJ. [3H]WAY-100635 for 5-HT1A receptor autoradiography in human brain: a comparison with [3H]8-OH-DPAT and demonstration of increased binding in the frontal cortex in schizophrenia. Neurochem Int. 1997;30(6):565–74. doi: 10.1016/s0197-0186(96)00124-6. [DOI] [PubMed] [Google Scholar]
- 80.Lopez-Figueroa AL, et al. Serotonin 5-HT1A, 5-HT1B, and 5-HT2A receptor mRNA expression in subjects with major depression, bipolar disorder, and schizophrenia. Biol Psychiatry. 2004;55(3):225–33. doi: 10.1016/j.biopsych.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 81.Francis PT, et al. Antemortem measurements of neurotransmission: possible implications for pharmacotherapy of Alzheimer's disease and depression. J Neurol Neurosurg Psychiatry. 1993;56(1):80–4. doi: 10.1136/jnnp.56.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martin RL, et al. Mortality in a follow-up of 500 psychiatric outpatients. II. Cause-specif ic mortality. Arch Gen Psychiatry. 1985;42(1):58–66. doi: 10.1001/archpsyc.1985.01790240060006. [DOI] [PubMed] [Google Scholar]
- 83.Azmitia EC, Whitaker-Azmitia PM. Awakening the sleeping giant: anatomy and plasticity of the brain serotonergic system. J Clin Psychiatry. 1991;52 Suppl:4–16. [PubMed] [Google Scholar]
- 84.Palacios J, Pazos A, Hoyer D. Characterisation and mapping of 5-HT1A receptors in animals and in man. In: Dourish C, Ahlenius S, Hutson P, editors. Brain 5-HT1A Receptors - Behavioural and Neurochemical Pharmacology. Ellis Horwood; Chichester: 1987. Chapter 6. [Google Scholar]
- 85.Pazos A, Probst A, Palacios JM. Serotonin receptors in the human brain--III. Autoradiographic mapping of serotonin-1 receptors. Neuroscience. 1987;21(1):97–122. doi: 10.1016/0306-4522(87)90326-5. [DOI] [PubMed] [Google Scholar]
- 86.Sprouse JS, Aghajanian GK. Responses of hippocampal pyramidal cells to putative serotonin 5-HT1A and 5-HT1B agonists: a comparative study with dorsal raphe neurons. Neuropharmacology. 1988;27(7):707–15. doi: 10.1016/0028-3908(88)90079-2. [DOI] [PubMed] [Google Scholar]
- 87.Drevets WC, Raichle ME. Neuroanatomical circuits in depression: implications for treatment mechanisms. Psychopharmacol Bull. 1992;28(3):261–74. [PubMed] [Google Scholar]
- 88.Grasby PM, et al. Effects of the 5-HT1A partial agonists gepirone, ipsapirone and buspirone on local cerebral glucose utilization in the conscious rat. Psychopharmacology (Berl) 1992;106(1):97–101. doi: 10.1007/BF02253595. [DOI] [PubMed] [Google Scholar]
- 89.Fisher PM, et al. Capacity for 5-HT1A-mediated autoregulation predicts amygdala reactivity. Nat Neurosci. 2006;9(11):1362–3. doi: 10.1038/nn1780. [DOI] [PubMed] [Google Scholar]
- 90.Drevets WC. Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Curr Opin Neurobiol. 2001;11(2):240–9. doi: 10.1016/s0959-4388(00)00203-8. [DOI] [PubMed] [Google Scholar]
- 91.Haddjeri N, Lucas G, Blier P. Role of cholinergic and GABAergic systems in the feedback inhibition of dorsal raphe 5-HT neurons. Neuroreport. 2000;11(15):3397–401. doi: 10.1097/00001756-200010200-00026. [DOI] [PubMed] [Google Scholar]
- 92.Azmitia EC, et al. Cellular localization of the 5-HT1A receptor in primate brain neurons and glial cells. Neuropsychopharmacology. 1996;14(1):35–46. doi: 10.1016/S0893-133X(96)80057-1. [DOI] [PubMed] [Google Scholar]
- 93.Whitaker-Azmitia PM, Clarke C, Azmitia EC. Localization of 5-HT1A receptors to astroglial cells in adult rats: implications for neuronal-glial interactions and psychoactive drug mechanism of action. Synapse. 1993;14(3):201–5. doi: 10.1002/syn.890140303. [DOI] [PubMed] [Google Scholar]
- 94.Whitaker-Azmitia PM, Murphy R, Azmitia EC. Stimulation of astroglial 5-HT1A receptors releases the serotonergic growth factor, protein S-100, and alters astroglial morphology. Brain Res. 1990;528(1):155–8. doi: 10.1016/0006-8993(90)90210-3. [DOI] [PubMed] [Google Scholar]
- 95.Whitaker-Azmitia PM, Azmitia EC. Stimulation of astroglial serotonin receptors produces culture media which regulates growth of serotonergic neurons. Brain Res. 1989;497(1):80–5. doi: 10.1016/0006-8993(89)90972-4. [DOI] [PubMed] [Google Scholar]
- 96.Azmitia EC, Whitaker-Azmitia PM. Awakening the sleeping giant: anatomy and plasticity of the brain serotonergic system. J Clin Psychiatry. 1991;52:4–16. Suppl. [PubMed] [Google Scholar]
- 97.Gross C, et al. Serotonin1A receptor acts during development to establish normal anxiety-like behaviour in the adult. Nature. 2002;416(6879):396–400. doi: 10.1038/416396a. [DOI] [PubMed] [Google Scholar]
- 98.Azmitia EC. Serotonin neurons, neuroplasticity, and homeostasis of neural tissue. Neuropsychopharmacology. 1999;21(2 Suppl):33S–45S. doi: 10.1016/S0893-133X(99)00022-6. [DOI] [PubMed] [Google Scholar]
- 99.McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. 1999;22:105–22. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- 100.Drevets WC. Neuroimaging studies of mood disorders. Biol Psychiatry. 2000;48(8):813–29. doi: 10.1016/s0006-3223(00)01020-9. [DOI] [PubMed] [Google Scholar]
- 101.Meijer OC, de Kloet ER. Corticosterone suppresses the expression of 5-HT1A receptor mRNA in rat dentate gyrus. Eur J Pharmacol. 1994;266(3):255–61. doi: 10.1016/0922-4106(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 102.Watanabe Y, et al. Stress and antidepressant effects on hippocampal and cortical 5-HT1A and 5-HT2 receptors and transport sites for serotonin. Brain Res. 1993;615(1):87–94. doi: 10.1016/0006-8993(93)91117-b. [DOI] [PubMed] [Google Scholar]
- 103.Hesen W, Joels M. Modulation of 5HT1A responsiveness in CA1 pyramidal neurons by in vivo activation of corticosteroid receptors. J Neuroendocrinol. 1996;8(6):433–8. doi: 10.1046/j.1365-2826.1996.04724.x. [DOI] [PubMed] [Google Scholar]
- 104.Carroll BJ, et al. A specific laboratory test for the diagnosis of melancholia. Standardization, validation, and clinical utility. Arch Gen Psychiatry. 1981;38(1):15–22. doi: 10.1001/archpsyc.1981.01780260017001. [DOI] [PubMed] [Google Scholar]
- 105.Holsboer F. Neuroendocrinology of mood disorders. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: The Fourth Generation of Progress. Raven Press; New York: 1995. pp. 957–969. [Google Scholar]
- 106.McEwen BS. Stressful experience, brain, and emotions: developmental, genetic and hormonal influences. In: Gazzaniga MS, editor. The Cognitive Neurosciences. MIT Press; Cambridge, Ma: 1995. Chapter 74. [Google Scholar]
- 107.Bhagwagar Z, et al. Lack of effect of a single dose of hydrocortisone on serotonin(1A) receptors in recovered depressed patients measured by positron emission tomography with [11C]WAY-100635. Biol Psychiatry. 2003;54(9):890–5. doi: 10.1016/s0006-3223(03)00466-9. [DOI] [PubMed] [Google Scholar]
- 108.Montgomery AJ, et al. PET measurement of the influence of corticosteroids on serotonin-1A receptor number. Biol Psychiatry. 2001;50(9):668–76. doi: 10.1016/s0006-3223(01)01205-7. [DOI] [PubMed] [Google Scholar]
- 109.Lemonde S, et al. Impaired repression at a 5-hydroxytryptamine 1A receptor gene polymorphism associated with major depression and suicide. J Neurosci. 2003;23(25):8788–99. doi: 10.1523/JNEUROSCI.23-25-08788.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.David SP, et al. A functional genetic variation of the serotonin (5-HT) transporter affects 5-HT1A receptor binding in humans. J Neurosci. 2005;25(10):2586–90. doi: 10.1523/JNEUROSCI.3769-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Caspi A, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301(5631):386–9. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]