

Abstract
The Children's Oncology Group (COG) recently celebrated the milestone of 50 years of pediatric clinical trials and collaborative research in oncology. Our group had its origins in the four legacy pediatric clinical trials groups: the Children's Cancer Group, the Pediatric Oncology Group, the National Wilms' Tumor Study Group and the Intergroup Rhabdomyosarcoma Study Group which merged in 2000 to form the COG. Over the last 50 years, the survival rates for childhood cancer have risen from 10% to almost 80%. Outcome in Acute Lymphoblastic Leukemia (ALL) has gone from a six month median survival to an 85% overall cure rate. We have modified therapies in most major diseases to induce remission with the least amount of long term sequelae. Here we look back on our advances but also look forward to the next 50 years which will produce even more successful treatments that will be tailored to the specific patient translating the tools of molecular genetics. Experience has clearly proven that everything we know about the diagnosis and management of childhood cancer is a result of research and the dramatic historical decrease in mortality from childhood cancer is directly related to cooperative group clinical research.
The Children's Oncology Group (COG) is an international research organization, supported principally by the National Cancer Institute (NCI). This group has evolved from the very first of the cooperative cancer clinical trials groups in 1955. Primarily through the conduct of clinical trials, COG is devoted to the development of new treatments and cures for the cancers of infants, children, adolescents, and young adults. In addition, the Group conducts research on the biology of these cancers, their causes, and the long-term outcomes of patients treated on group studies. COG membership includes over 5,000 pediatric cancer specialists in approximately 230 pediatric medical centers in the United States, Canada, Switzerland, the Netherlands, Australia, and New Zealand. At COG institutions, multidisciplinary teams consisting of physicians, basic scientists, nurses, psychologists, pharmacists and other specialists use their specialty skills in the diagnosis, management and investigation of childhood cancer. The mission of the COG is to cure and prevent childhood and adolescent cancer through scientific discovery and compassionate care. To accomplish its mission, the COG has several objectives that include the design and conduct of clinical trials to define the optimal treatment of pediatric cancers, the conduct of correlative laboratory investigations which are expected to translate into new, more effective and less toxic therapies, the conduct of epidemiologic investigations to identify possible associations and causes of childhood cancer for eventual therapeutic exploitation, and the conduct of research to improve the quality of life and survivorship. This last goal is a direct derivative of our ability to cure nearly 80 percent of childhood cancer patients.
Most member institutions of COG are pediatric research and/or teaching hospitals, integral to distinguished medical schools or cancer centers. At each institution, eligible patients who choose to enroll in the active trials of the Group are treated in accordance with COG protocols. The comparatively low incidence of cancer in children and resulting sample size constraints mandate multi-center and multi-disciplinary clinical trials. No single institution has sufficient patient volume to perform definitive, randomized, controlled trials. In addition, with the increasing complexity of the clinical and biologic sub- classification of even major types of childhood cancer, coupled with the current attention to risk-adjusted therapy approaches, the issue of sample size becomes even more complex and has lead in recent years to more international collaboration. Through the pooling of scientific ideas, patient data, and other resources, COG has made rapid progress with such scientific inquiry. In addition, children and adolescents gain access to state-of-the-art cancer therapy by being enrolled on these COG studies, which incorporate the most effective, proven treatment approaches to date. Thus, COG is able to offer children the best hope for cure while continuing to advance medical science through the conduct of clinical trials.
In the United States, 90-95 percent of all children under age 15 with a newly diagnosed malignancy are seen at a COG institution. If a clinical trial is available, 50-60 percent of children eligible are enrolled. For young children (less than 5 years of age) enrollment rates are much higher, 90 percent. In 2007, the COG has over 70,000 children with cancer who were being managed with research protocols or were in active follow up. COG provides state-of-the-art treatment as well as innovative experimental treatments. In the last 4 years 14,000 children have been enrolled on therapeutic clinical trials and 16,000 on non-therapeutic clinical trials. In 2007, 4300 children will be enrolled on a therapeutic trial and an additional 6000 on a non-therapeutic trial. More than any other factor, clinical research of this type has been responsible for dramatic improvements in the survival rates of children with cancer. Cure rates have improved from less than 10%—when the cooperative groups were founded—to nearly 80 percent at present. What is now known about curing children with cancer has been gleaned from research conducted through the pediatric cooperative groups.
Through the COG, this established and successful tradition of improving childhood cancer treatment through clinical trials continues. In late 1998, the existing pediatric cancer clinical trials groups based in North America (the Children's Cancer Group, the Pediatric Oncology Group, the Intergroup Rhabdomyosarcoma Study Group, and the National Wilms' Tumor Study Group) considered combining their efforts in order to accelerate progress and use resources wisely in their shared fight against cancer. The vision and dedication required to merge these entities into a single cooperative group—the Children's Oncology Group—marked the beginning of a new era in the effort to conquer childhood cancer. The spirit of collaboration signified by this event remains visible through the continued involvement of clinical investigators, laboratory scientists, patients, parents and other advocates.
Since the merger in 2000, over 1000 Manuscripts reporting the results of COG research were published in peer-reviewed scientific journals and numerous presentations have been made at national and international scientific meetings each year. Through these mechanisms, the results of COG research are made available to physicians around the world who treat children with cancer. COG is considered the premier childhood cancer research organization in the world. It has treated more children with cancer than any other organization in history, and has been responsible for many advances during the past 50 years. As part of this stewardship, the Group has active and planned affiliations with cooperative groups in Europe, Israel, and Latin and South America. (1)
The history of the cooperative group approach to childhood cancer began with one of the founders of the very first cooperative group, Dr. Joseph Burchenal. In 1948, he reported the effect of nitrogen mustard compounds on mouse leukemia. At about the same time, Dr. Sidney Farber had achieved temporary remissions in children with leukemia with folic acid antagonists. (2) Subsequently, investigators identified additional chemotherapeutic and hormonal agents that induced temporary remission, including ACTH, cortisone, and anti-purine analogues. (3,4) The median survival for children treated with anti-folic agents and/or folic acid antagonists between 1948 and 1952 was 8 months. By the mid-1950s, the addition of purine antagonists increased the median survival to one year, with a ten percent survival rate at 22 months. (5)
Seven years after the original Burchenal publication, the Cancer Chemotherapy National Service Center of the National Institute of Health sponsored programs to organize institutions to cooperate in evaluating new potential anti-leukemic agents for children. The Acute Leukemia Chemotherapy Cooperative Study Group A (ALCCSGA) was formed in 1955. The original members included Dr. Frank Bethell of the University of Michigan; Dr. Joseph H. Burchenal of Memorial Hospital Sloan Kettering Institute and Cornell University Medical College; Dr. Byron Hall of Stanford University Medical School; Dr. Charles D. May of State University of Iowa; Dr. E. Clarence Rice of the Children's Hospital of the District of Columbia; Dr. Carl Smith of The New York Hospital; Dr. Phillip Strugeon of the Children's Hospital Society of Los Angeles and Dr. James Wolff of Babies Hospital in New York.
The first clinical trial of the ALCCSGA was a comparison of 6-MP versus 6-MP plus azaserine. It ran between 1955 and 1956 and its results were published in Blood in 1960, with Dr. Ruth Heyn as the lead author.(6)There were 168 patients entered without previous therapy but 30 were subsequently removed for the trial due to progression and the need for corticosteroid treatment. There was no statistical difference in the number of remission obtained or the duration of the complete remissions between the two arms. In addition the group was conducting phase I studies of actinomycin D, mitomycin C, 5-fluoro-2′-deoxyuridine, endoxan, 6-mercaptopurine riboside, miracil D and azauracil. By 1959, there were 12 member institutions and the meetings convened during the national meetings of the Society for Pediatric Research and the American Pediatric Society. This group evolved over the years into the Children's Cancer Study Group and later the Children's Cancer Group, one of the four legacy groups that agreed in 1998 to become the Children's Oncology Group.
Other Cooperative groups were also formed during the same era, with a focus on adult leukemia and other cancers—including the Acute Leukemia Chemotherapy Cooperative Group B (ALCCGB), The Eastern Cooperative Oncology Group and the Southwest Cancer Chemotherapy Study Group (SWOG). ALCCGB and SWOG both included pediatric components that preceded those of the legacy Pediatric Oncology Group.
The first progress report of the ALCCSGA, prepared by Dr. M. Lois Murphy in 1959, reported a group of twelve institutions. Five combination protocols in 542 patients were reviewed (with an additional 326 patients considered to be ineligible). Dr. Murphy reported on the occurrence of all types of cancer in children and noted that the group was preparing a protocol for metastatic solid tumors in children. By 1964, 1485 newly-diagnosed children with leukemia had been placed on a series of protocols and the group now had 3 geographical subgroups. Each subgroup chair had the responsibility of organizing Phase I and II sub-studies. Different ALL maintenance programs were assigned to the individual geographic subgroups. The average annual accrual for the group was now 200-250 patients. Children with metastatic solid tumors were now receiving triple therapy with chlorambucil, methotrexate and Actinomycin D (7); 72 patients were accrued onto this study that was the precursor to the Intergroup Rhabdomyosarcoma and Ewing's Study Groups as well as the National Wilm's Tumor Study Group. The progress report also cited the need to address other common solid tumors in children, including neuroblastoma and brain tumors, and recommended a multi-disciplinary approach. Ancillary studies were placed under the auspices of Dr. Mila Pierce and included evaluations of children with Down Syndrome and leukemia, siblings of children with leukemia, and infants with leukemia.
In the progress report for the ALCCSGA for the period from December 1955 to April 1964 there is a report that in leukemia patients induced with prednisone and maintained with 6 MP or 9-ethyl-6-MP that remission lasted an average of 230-300 days as compared to a 4-6 week prednisone induction with further cessation of therapy leading to a remission duration of 60 days. The planned follow up study was to look at the addition of methotrexate, vincristine and cyclophosphamide given concurrently versus the standard of one drug plus prednisone until relapse then reinduction with the next drug plus prednisone in a sequential manner.(8, 9) Dr. Sanford Leikin was in charge of a study that evaluated the optimal dose and schedule of prednisone in newly-diagnosed patients from 1964-1965(10). Five dose schedules were used in previously untreated patients. Three dose levels divided every 8 hours and two higher dose levels given at either 2 or 4 day intervals.
By September of 1967, Dr. Hartmann was the Chair of the ALCCSGA and its first constitution had been developed. The name had been shortened to the Children's Cancer Study Group. The first randomized trial for non-metastatic Wilms' tumor was completed—comparing a single 5-day course of Actinomycin-D plus surgery and radiation to repeated 5-day courses over 15 months, with 113 patients on study.(11) Eighty-six percent of those treated with the repeated course Actinomycin –D versus 48 percent with a single course of Actinomycin-D continued in complete remission. This trial led to the formation of the National Wilms' Tumor Study Group (NWTS).
The first meeting of the Wilms' Tumor Integrated Study Group was held in May 1968 at Babies Hospital in New York. The first site visit was in December of 1969, with Drs. D'Angio, Breslow, Beckwith and Evans in attendance. Over the next thirty years, five National Wilms' Tumor studies were developed and completed, with a profound effect on the therapy for this tumor and other malignancies of the renal system. This group was a model of interdisciplinary cooperation between pediatric oncologists, pediatric surgeons, and radiation therapists. The NWTS 1 detailed that post-operative abdominal radiation therapy was not necessary for children younger than 2 years of age with Stage I Wilms' tumor who were treated with dactinomycin. The combination of vincristine and dactinomycin was more effective for the treatment of those with Stage II or III Wilms' tumor than either drug alone. (12) NWTS 2 demonstrated that a reduced duration of chemotherapy—six months with vincristine and dactinomycin for children with Stage I Wilms' tumor who did not receive abdominal irradiation—did not affect prognosis. The addition of doxorubicin to the combination of vincristine and dactinomycin improved the event-free survival of irradiated children with stages II-IV Wilms' tumor. (13).
NWTS 3 further defined therapy for the Stage III patient with its proof of the benefit of the addition of a third drug (doxorubicin), and that an abdominal radiation dose of 1000 cGy was as effective as 2000 cGy. Cyclophosphamide did not benefit Stage IV patients as a fourth chemotherapeutic agent. (14) NWTS 4 looked at the economics and duration of treatment. This study found that pulses of actinomycin were as effective as the conventional schedule and markedly simplified treatment regimens. In addition, 6 months of therapy was found to be as effective as 15 months for patients with Stage III and IV disease with favorable histology. (15, 16)
With the superior survival statistics for this disease, the Wilms' Tumor Study Group initiated the study of late effects.(17) The consequences of abdominal irradiation, nephrectomy, and exposure to chemotherapy in this population of cancer survivors were reviewed, paving the way for the more extensive follow-up programs integral to the care of every current pediatric oncology patient. NWTS 5 built on the success of treatment for this disease and looked at the biology of the tumor. With prescribed uniform therapy for all stages, the tumors were evaluated for prognostic factors. Loss of heterozygosity on chromosomes 1p and 16q were found to be adverse factors in favorable histology Wilms' tumor. This knowledge will direct current therapy in this disease. (18)The next sequence of therapeutic trials has instituted a classification phase to determine the type and length of therapy for patients based on stage and biologic characteristics.
A most interesting and intriguing accomplishment by this group has been the definition of renal malignancies. In 1978, Dr. Beckwith published the early results of the NWTS pathological reviews of patients. The Group defined favorable and unfavorable histology and identified the clear cell sarcoma of the kidney and rhabdoid tumor of the kidney. (19)
In June of 1968, Dr. Denman Hammond was elected Chair of the Children's Cancer Study Group, which had grown to 25 institutional members. He established an operations center to include data and statistical departments. Standing disease and discipline committees were established. CCSG was governed by an executive committee, in conjunction with the Group Chair. In 1969, 291 newly-diagnosed patients with leukemia were entered onto trials, in addition to 591 previously-treated patients and 130 patients with solid tumors. The first group epidemiology study was initiated to evaluate the survival of 1,770 children with leukemia treated by the Children's Cancer Study Group A between 1957 and 1964. (20) The patients were primarily Caucasian with a slight male predominance. The peak incidence was 3.1 years. They found that survival time was dependent on the morphology of the leukemia with acute lymphoid leukemia at a median of 278 days, acute granulocylic leukemia 98 days, and acute monocytic leukemia 101 days. In all morphologic types, the patients with WBC less than 4,000 fared better. The first pharmacology study assessed the kinetics of L-asparaginase via intravenous versus intramuscular administration. Drs Mark E. Nesbit and Ronald Chard hypothesized a relation of anaphylactic reaction to asparaginase to loss of effect based on serum drug levels. Both methods of administration achieved similar effects if evaluated over 48 hours. (21) In the early part of the 1970's, the computer entered the group sphere and was used for data management on CCA-903 and for an immune stimulation study in ALL/AUL.
Dr. Ruth Heyn was the chair of the Rhabdomyosarcoma Study Committee which studied the effectiveness of Actinomycin-D and vincristine in addition to surgery and radiation therapy. (22) In May of 1970 at a Pediatric Tumor Intergroup Liaison Meeting, a Rhabdomyosarcoma task force was formed with Dr. Ruth Heyn, Dr. Hal Mauer (ALGB) and Dr. Teresa Vietti (Southwest Cancer Chemotherapy Study Group, SWCCG) at the suggestion of Dr. Lawrence Foye, Chief of the Clinical Investigations Branch of the NCI. This group put together the mechanisms that allowed the first Intergroup Rhabdomyosarcoma Study Group (IRSG) protocol to be activated in 1972. The first Chair of the IRSG was Dr. Hal Mauer, and their first meeting occurred in Salt Lake City in February, 1973. At that meeting Dr. Mauer obtained the drug doxorubicin from the NIH for future studies, while vincristine, cyclophosphamide and dactinomycin were felt to be commercially available. Other physicians and investigators crucial to the success of the IRSG include Drs. Beverly Rainey, Frederick Ruymann, Wataru Sutow, Edward Gehan, William Crist, Edward Soule, and William Newton.
Over the next thirty years four national studies were conducted by the IRSG. IRSG I treated 686 patients with Rhabdomyosarcoma or undifferentiated sarcoma, with results published in 1988. (23) Patients with low-stage disease (Clinical Group 1, who presented with local disease and had complete resections) did not benefit from radiation added to vincristine, dactinomycin and cyclophosphamide (VAC). Those with regional disease who received radiation did not benefit from the addition of cyclophosphamide to vincristine and dactinomycin. Those with more extensive disease did not benefit from the addition of doxorubicin to aggressive VAC plus irradiation. Overall, distant metastatic recurrence was more common than local recurrence. Tumors of the orbit and GI tract had the best prognosis. (23) The goal of the IRSG II was to improve overall survival in all groups. Nine hundred and ninety-nine patients were treated, with an overall 5-year survival rate of 71%, compared to 63% for IRSG I (24) Pathology and clinical group were found to be variables for prognosis upon analysis of IRS I and II. (25) The clinical group was most important in both studies. With respect to pathology, alveolar tumors had the poorest survival and botyroid/embryonal tumors the best. In IRSG III, intensification of treatment again correlated with improved outcome when a risk-based study design was utilized. The best results were seen in patients with gross residual disease after biopsy??. This study also proved that decreased therapy in selected subsets was possible without an affecting overall survival, in particular, Group 1 patients with favorable histology. (26)
IRSG IV (27,28) expanded upon previous studies, and determined that for patients with local or regional disease, multiple drug combinations were effective with surgery and radiation therapy if indicated. Younger patients with Group 1 Para testicular embryonal primaries, and all patients with Group 1 or 2 orbit or eyelid tumors, could be cured with vincristine and dactinomycin, with the addition of radiation for patients with Group 2 disease. Patients with embryonal tumors who received intensive three-drug chemotherapy and appropriate other treatment modalities had a 3-year event-free survival of 83%. As part of the design, patients with Group III disease could be randomized to hyperfractionated radiotherapy compared to conventional radiotherapy, but no benefit was noted.
During the 1970's and early 1980's, the Intergroup Ewing's Sarcoma Group (IESS) systematically approached patients with this disease and with two large studies defined major therapeutic treatment advances. IESS -1 utilized multi agent chemotherapy (cyclophosphamide, vincristine and dactinomycin) with doxorubicin and localized radiotherapy to improve long term survival in patients with localized disease.(29,30,31). Prophylactic pulmonary irradiation or the addition of doxorubicin to the VAC regimen also reduced the subsequent incidence of pulmonary metastases. (30) IESS-2 looked at high dose intermittent four drug combinations from IESS-1 to moderate dose continuous therapy with local control radiation and recommended surgical resection. The high dose arm was superior with metastatic disease as the primary reason for failure in both groups. Relapse free survival was 73% compared to 56%. Cardiovascular toxicity was significantly greater on the high dose regimen.(32)
While the Children's Cancer Study Group continued to evolve, the Pediatric Oncology Group also became an entity in 1980. In 1956 the SWCCSG was organized with a pediatric component under Dr. Grant Taylor at MD Anderson with Dr. Ken Griffith as their group statistician. Dr. Taylor was succeeded by Dr. Emil Frei III as chair in 1969; Dr. Ed Gehan was the new group statistician. In 1972, Dr. Barth Hoogstraten became the new chair, and in 1973 the group became known as the Southwestern Oncology Group (SWOG). In 1980, the pediatric division of SWOG separated and became the Pediatric Oncology Group (POG), with Dr. Teresa Vietti as Chair. Drs. Jon Shuster and Jeff Krischer were the statisticians. In 1993, the POG operations office moved from St. Louis to Chicago when Dr. Sharon Murphy assumed the Chairmanship.
Numerous scientific advances were accomplished by the POG. Their investigators, in conjunction with members of the Children's Cancer Group, collaborated with an international panel to establish a staging system for Neuroblastoma that reflected the biology of the tumor as well as the clinical presentation. (33) This collaboration continues to evolve and is now the basis of the classification protocol for the COG that is a prerequisite for treatment determination for all patients with this disease. Utilizing intergroup resources, the importance of MYCN was described as a prognostic indicator for survival for patients with high-risk Neuroblastoma. Treatment protocols for acute lymphoblastic leukemia emphasized the use of methotrexate (34) and the identification of biologic prognostic factors for this disease. POG investigators described the poor prognostic indicator of the Philadelphia Chromosome in ALL. (35) They documented that the presence of hyperdiploidy was a good prognostic indicator for Acute Lymphoblastic Leukemia. (36) Further cytogenetic investigations of ALL also defined that trisomies of chromosomes 4 and 10 (37) were independent indicators of improved prognosis with anti-metabolite therapy. Overall, the POG developed risk stratification for ALL with biologic markers that were identified in serial studies throughout the 1980's and 1990's. Down Syndrome (DS) patients with AML were identified as a group responsive to therapy, unlike other non-DS patients with this disease. (38)
Post-surgical chemotherapy for patients with osteogenic sarcoma was proven valuable by a small POG study of only 36 patients. (39) but this remains the standard for this disease group. POG published extensively on the topic of infant CNS tumors and the profound effects of radiation therapy in this age group. This legacy group worked to modify treatment for infant brain tumors in order to decrease sequelae in survivors by utilizing aggressive post-operative chemotherapy in order to delay radiotherapy. (40)
POG and the Children's Cancer Group (CCG) performed many joint studies. They participated in NIH-sponsored panels to advance the classification of acute lymphoblastic leukemia (41) and developed intergroup studies in Ewing's sarcoma after the IESS group ended, and hepatoblastoma. The Ewing's study defined the importance of ifosfamide and etoposide in the treatment of that disease. (42) The legacy CCG established the therapeutic benefit of cisplatin and continuous infusion doxorubicin in Stage III and IV hepatoblastoma while POG had similar results for cisplatin, vincristine and fluorouracil. (43,44) An intergroup study for hepatoblastoma was completed that showed no significant difference in event free survival or survival for either regimen. (45)
The POG Statistics and Data Center was one of the first to use remote data entry which allowed for enrollments to clinical trials continuously. They established the POG log, in which all patients with a pediatric malignancy at their member institutions were registered. This is the precursor to the Children's Cancer Research Network, a major goal of the merged COG. This mechanism will establish a population based pediatric cancer registry for North America.
The Children's Cancer Study Group—also known as the Children's Cancer Group (CCG)—also achieved many milestones in the last 50 years. Through its collaboration with the POG and participation with the IRS and NWTS Groups, it has helped increase the survival rate of children with cancer, now approaching 78% at 5 years.
In the early 1970s, the CCG initiated CCG 101 to evaluate the sanctuary therapy for ALL that had been systematically developed during the prior decade.(33,34) This multi-agent approach for ALL was first pioneered with MOPP therapy in Hodgkin disease. It provided the future foundation for the use of multi-agents induction, consolidation, and maintenance phases of current leukemia therapy. CCG 101 was developed to define the length of therapy (46,47) and to focus on disease in the CNS and testes. Subsequent analysis of these leukemia patients revealed the impact of initial white blood cell count and age on prognosis. Prophylactic CNS therapy was determined to be important, and CNS radiation to be effective for prophylaxis. With the advent of late effects studies revealing cognitive and other deficits related to CNS radiation, the CCG slowly eliminated CNS radiation prophylaxis in favor of intrathecal chemotherapy for all but the most aggressive forms of ALL.
The CCG continued its ALL focus and incorporated the Berlin-Frankfurt-Munich (BFM) delayed intensification phase for all risk groups.(48)In the 1980s and 1990s, serial protocols proved the benefit of delayed intensification with decadron, L-asparaginase, doxorubicin, vincristine, cyclophosphamide, cytarabine and 6-thioguanine for low, average, and high risk ALL patients. CCG 1922(49) introduced decadron into induction for the average risk ALL patient, which proved to be another significant improvement in event-free survival. While the POG focused on prognostic indicators and later minimal residual disease (MRD), the CCG assessed early response based on early marrow improvement measured during induction therapy. CCG measured bone marrow response on day seven which dictated further therapy for high risk patients and proved the benefit of further augmented therapy in slow early responders.(50) This response based model is the basis of our current open ALL trials that are identifying risk group based on age, wbc, minimal residual disease (MRD), cytogenetics and morphological response.
In figure 1, we see the overall effect of the serial ALL studies of the CCG that addressed sanctuary therapy, the integration of delayed intensification and the use of response based therapy.
Fig 1.
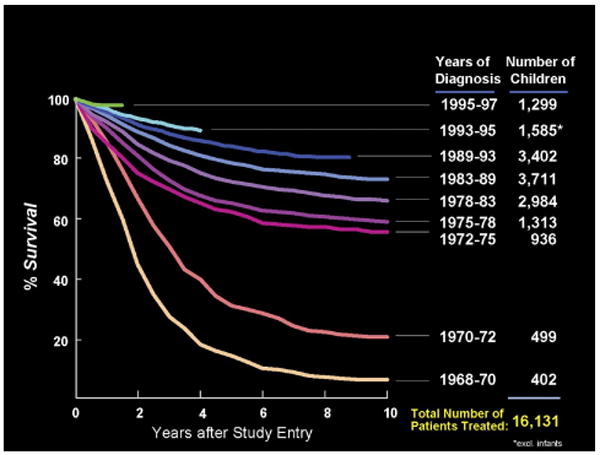
Survival of CCG Patients with ALL 1968 -1997
The CCG was the first American group show the benefit of chemotherapy in patients with Medulloblastoma. (51) This was followed by studies utilizing adjuvant chemotherapy to reduce the overall dose of craniospinal radiation without loss of efficacy for children with non-disseminated Medulloblastoma (52).
International collaboration has been vital to the CCG approach to Burkitt lymphoma, which most recently has produced a marked improvement in disease-free survival with aggressive therapy. (53,54) In collaboration with the French LMB (write out) group, an international study was developed that looked at a reduction in therapy for intermediate risk Burkitt patients, and there was no difference in the group given half dose cyclophosphamide. In addition, the high risk patients with central nervous system disease, leukemia or poor response the reduction phase of cyclophosphamide, prednisone and vincristine required the standard therapy and did significantly worse if randomized the reduced dose therapy.
Effective treatment of high-risk Neuroblastoma improved with autologous stem cell transplant, and significant relapse-free survival was noted in patients receiving aggressive chemotherapy or transplants with the addition of cis-retinoic acid during remission. (55) Lower risk children with Neuroblastoma were studied extensively, resulting in recommendations to eliminate chemotherapy and radiation after surgery in low stage disease. (56) Children with acute myelogenous leukemia (AML) were found to have significantly increased survival rates when treatment included allogeneic bone marrow transplant (biologic randomization) versus maintenance chemotherapy.(57) Subsequent studies revealed an improved outcome for those without transplant with aggressive chemotherapy and intensified timing. (58) Infant acute lymphoblastic leukemia was studied with an intergroup approach (CCG and POG). Infants benefited from an aggressive four-drug induction phase and a high-dose intravenous methotrexate consolidation phase in lieu of cranial irradiation. Infants with germ line mutations and those younger than 6 months of age remain difficult to treat. (59)
Throughout the therapeutic studies of all of the legacy groups, treatment protocols were augmented by biologic research agendas that allowed for the development of exquisite classification systems for Neuroblastoma, ALL, AML, Wilms' tumor and Rhabdomyosarcoma. With ongoing research targeted at CNS tumors, Ewing sarcoma and osteogenic sarcoma, similar improvements are anticipated.
Late effects of therapy have more recently been the focus of research; many members of the legacy groups have participated in the Childhood Cancer Survivor Study. CCG outcomes and sequelae studies of patients treated for ALL revealed an increase in the incidence of brain tumors in patients treated on early protocols that included prophylactic cranial irradiation and intrathecal methotrexate. (60) Analyses of intellectual outcome are integral to current treatment protocols, particularly for patients with ALL and CNS tumors. (61)
In addition to the success of our clinical trials program, we have also facilitated the progress of the NCI's Pediatric Central Intuitional Review Board such that greater than 60 percent of our eligible institutions are fully affiliated after only two years. COG has also continued its focus of international collaboration with the European-American Osteosarcoma study that is now open and includes the COG as well as several European clinical trials groups. The negotiations and interactions that preceded the opening of this study, which includes commercial and an investigational agent, led to the development of CTEP guidelines with input for OHRP for the conduct of clinical trials
Since the merger of the legacy groups, many new achievements have occurred. In ALL the prognostic significance of MRD and its relation to other risk factors is the cornerstone of a new risk classification system for ALL. All patients with ALL now undergo multiple cytogenetic, MRD, and soon to be molecular analysis to determine their risk group and subsequent treatment after an initial four week induction regimen that build on the classic factor of age and white blood cell count. (62, 63). The merged group has also allowed for a renewed interest in retinoblastoma and the ability to provide a Group approach to multidisciplinary treatment for this disease. The Hodgkin Disease studies are focused on the use of CT and PET response to early cycles of therapy to determine length and aggressiveness of continued therapy.
Even with our now encouragingly successful outcome in ALL (figure 2); there has been continued improvement in the last 5 years with survival approaching 90%. In figure 3, we see that
Fig 2.
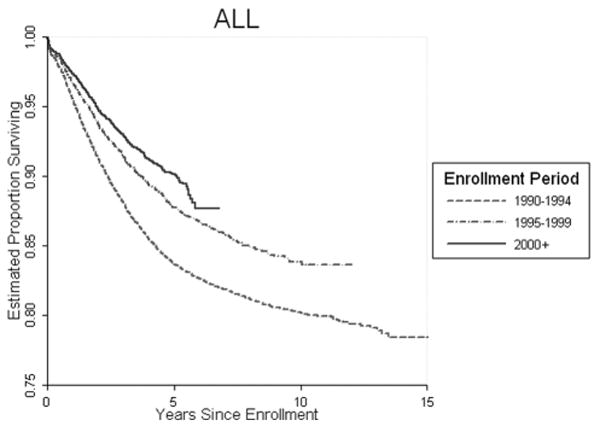
Overall COG Treatment Trials ALL
Fig 3.
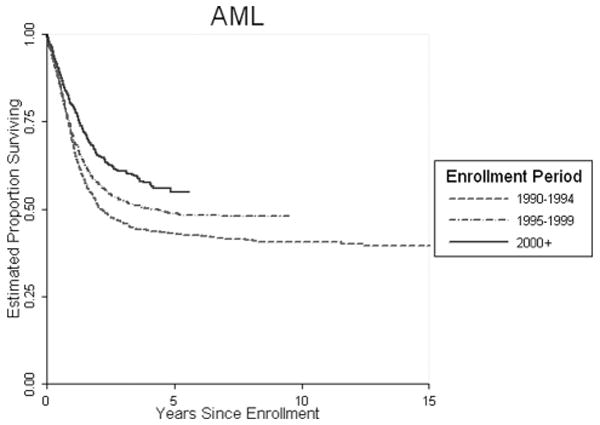
Overall COG Treatment Trials AML
With the more aggressive strategies, the survival of AML with and without bone marrow transplantation is over 50 percent. The use of targeted therapy approaches, including monoclonal antibodies in this disease, is the focus of our next generation of studies.
In figure 4, we see the benefit of aggressive surgery, chemotherapy and more targeted radiation therapy for patients with Medulloblastoma treated by the COG in the last 5 years, with survival approaching 70 percent.
Fig 4.
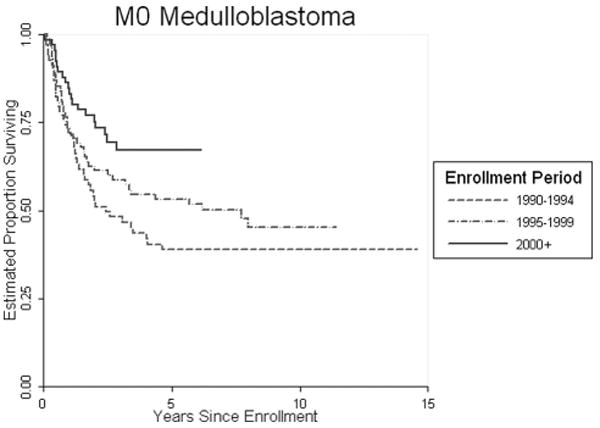
Overall COG Treatment Trials Medulloblastoma
But despite achievements in other aggressive disease we continue to have challenges. Stage IV Neuroblastoma continues to prove resistant to therapy and in figure 5 we see that the overall survival in this disease while improved remain below 50 percent.
Fig 5.
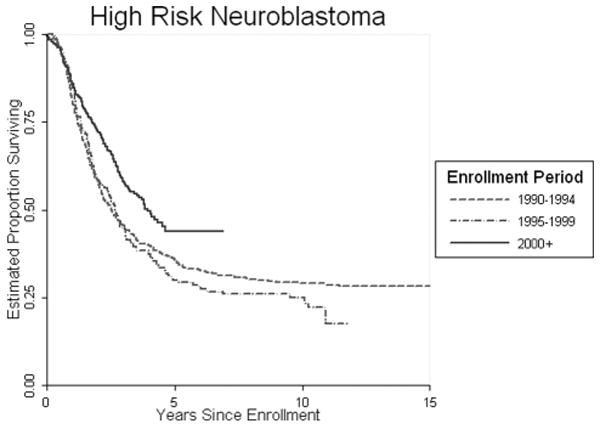
Overall COG Treatment Trials High Risk Neuroblastoma
The last fifty years have been replete with scientific discoveries and accomplishments in the treatment of childhood cancer. The members of COG anticipate the next fifty, in hopes of applying the many discoveries in translational research, developing a pediatric cancer registry and long-term survivor registry, improving outcomes for patients with diseases such as high-risk Neuroblastoma, CNS tumors, relapsed acute lymphocytic leukemia, infant leukemia, and AML in those lacking stem cell donors, and improving the quality of life for survivors of childhood cancer.(64) As new versions of old standards become available we anticipate the continued modification of therapies, as in the new look at asparaginase.(65) COG, as the successful cooperative group treating most children in North America, Australia and New Zealand under the age of 15, is now focused on strategies to extend this success to adolescents and young adults with cancer.(66) Another fascinating fact revealed in a review of the historical reports of the legacy groups is the commitment of senior pediatric hematologist-oncologists to training the next generation. It remains a focus today, succession planning for the future physicians and scientists that will lead the next generation of therapeutic and non-therapeutic trials.
Our future will build on the past. We will continue with clinical trials but are now committed to look at the biologic parameters of the cancers and therapies we deal with. COG is evaluating gene expression signatures in our leukemia and solid tumor banked specimens. We have evidence that gene expression patterns can refine diagnosis and predict outcome and will prove useful for future risk group classification. Pharmacogenomic investigations will allow us to predict toxicities in homogeneously treated groups of patients, leading to individualized therapy models. COG has been an active participant in the unique NCI intramural and extramural collaboration with the Cancer Genome Atlas and Cancer Genome Anatomy Project through the TARGET (Therapeutically Applicable Research to Generate Effective Treatments) initiative. We are able to participate in such initiative because of the Biopathology Center that banks specimens from our patients on our biology and therapeutic studies. This is the world's largest repository of clinically annotated biospecimens of both tumor and germ line DNA which will make it possible for COG to design, coordinate, and undertake genomic and transcriptomic analysis in collaboration with other scientists nationally and internationally. This will ultimately lead to the identification of specific genes and/or gene regions for sequencing. Information from the sequencing of informative genes can then direct us to potential therapeutic targets.
The COG is dedicated to discovery and committed to care for children and adolescents. Our accomplishments have been made possible by the willingness and dedication of our patients and families to participate in treatment and research protocols.
Acknowledgments
Supported in part by: CA 98543, CA98413 and National Childhood Cancer Foundation
Footnotes
Financial Disclosure Obligations: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Children's Oncology Group. Internal documents [Google Scholar]
- 2.Burchenal JH, Lester RA, Riley JB, et al. Studies on the chemotherapy of leukemia. Cancer. 1948;1:399–412. doi: 10.1002/1097-0142(194809)1:3<399::aid-cncr2820010305>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 3.Farber S, Diamond LK, Mercer RD, et al. Temporary remissions in acute leukemia in children produced by folic acid antagonists, 4-Aminopteroyl-glutamic acid (Aminopterin) N Engl J Med. 1948;238:787. doi: 10.1056/NEJM194806032382301. [DOI] [PubMed] [Google Scholar]
- 4.Pearson OH, Eliel LP. Use of adrenocorticotropic hormone (ACTH) and cortisone in lymphomas and leukemias. JAMA. 1950;144:1349. doi: 10.1001/jama.1950.02920160023005. [DOI] [PubMed] [Google Scholar]
- 5.Burchenal JH, Murphy ML, Ellison RR, et al. Clinical evaluation of a new antimetabolite, 6-mercapto purine in the treatment of leukemia and allied diseases. Blood. 1953;8:965–99. [PubMed] [Google Scholar]
- 6.Heyn RM, Brubaker CA, Burchenal JH, et al. The comparison of 6-mercaptopurine with the combination of 6-mercaptopurine and azaserine in the treatment of acute leukemia in children: results of a cooperative study. Blood. 1960;15:350–9. [PubMed] [Google Scholar]
- 7.Sitarz A, Heyn R, Murphy ML, et al. Triple drug therapy with Actinomycin-D (NSC 3053), Chlorambucil (NSC 3088) and Methotrexate (NSC 740) in Metastatic Solid Tumors in Children. Ca Chem Rep. 1965;45:45. [PubMed] [Google Scholar]
- 8.Wolff JA, Brubaker CA, Murphy ML, et al. Maintenance of Prednisone induced remissions of Acute Childhood Leukemia by Thiopurines [Google Scholar]
- 9.Krivit W, Brubaker C, Hartmann JR, et al. Cyclic vs. Sequential Use of Chemotherapeutic Agents in Acute Leukemia of Childhood. Proc Of Am Assoc of Ca Res; 57th Annual Meeting; Denver. 1966. Abstract. [Google Scholar]
- 10.Leikin SL, Brubaker C, Hartmann JR, et al. Varying Prednisone dosage in remission induction of previously untreated childhood leukemia. Cancer. 1968 Mar;21(3):346–51. doi: 10.1002/1097-0142(196803)21:3<346::aid-cncr2820210303>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 11.Wolff JA, Krivit W, Newton WA, et al. Single versus multiple dose dactinomycin therapy of Wilms' Tumor. N Engl J Med. 1968;279:290–4. doi: 10.1056/NEJM196808082790605. [DOI] [PubMed] [Google Scholar]
- 12.D'Angio GJ, Evans AE, Breslow N, et al. The treatment of Wilms' tumor: Results of the National Wilms' Tumor Study. Cancer. 1976;38:633–46. doi: 10.1002/1097-0142(197608)38:2<633::aid-cncr2820380203>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 13.D'Angio GJ, Evans A, Breslow N, et al. The treatment of Wilms' tumor: Results of the second National Wilms' Tumor Study. Cancer. 1981;47:2302. doi: 10.1002/1097-0142(19810501)47:9<2302::aid-cncr2820470933>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 14.D'Angio GJ, Breslow N, Beckwith JB, et al. The treatment of Wilms' tumor. Results of the Third National Wilms' Tumor Study. Cancer. 1989;64:349–60. doi: 10.1002/1097-0142(19890715)64:2<349::aid-cncr2820640202>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 15.Green D, Breslow N, Beckwith J, et al. The effect of duration of treatment on outcome and cost of the treatment for Wilms' Tumor. A report from the National Wilms' Tumor Study Group. J Clin Oncol. 1998;16:3744–51. doi: 10.1200/JCO.1998.16.12.3744. [DOI] [PubMed] [Google Scholar]
- 16.Green D, Breslow N, Beckwith J, et al. A comparison between single dose and divided dose administration of dactinomycin and doxorubicin. A report from the National Wilms' Tumor Study Group. J Clin Oncol. 1998;16:237–44. doi: 10.1200/JCO.1998.16.1.237. [DOI] [PubMed] [Google Scholar]
- 17.Evans AE, Nrokool P, Evan I, et al. Late effects of treatment for Wilms' Tumor. A report from the National Wilms' Tumor Study Group. Cancer. 1991;67:331–6. doi: 10.1002/1097-0142(19910115)67:2<331::aid-cncr2820670202>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 18.Grundy PE, Breslow NE, Li S, et al. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms' tumor: A report from the National Wilms' Tumor Study Group. J Clin Oncol. 2005;23:7312–21. doi: 10.1200/JCO.2005.01.2799. [DOI] [PubMed] [Google Scholar]
- 19.Beckwith JB, Palmer N. Histopathology and prognosis of Wilms' tumor: Results of the National Wilms' Tumor Study. Cancer. 1978;41:1937. doi: 10.1002/1097-0142(197805)41:5<1937::aid-cncr2820410538>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 20.Pierce MI, Borges WH, Heyn RM, et al. Epidemiologic factors and survival experience in 1770 children with Acute Leukemia. Cancer. 1969;23:1296–1304. doi: 10.1002/1097-0142(196906)23:6<1296::aid-cncr2820230609>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 21.Nesbit M, Chard R, Evans A, et al. Evaluation of intramuscular versus intravenous administration of L-asparaginase in childhood leukemia. Am J of Pediatr Hematol Oncol. 1979 Spring;1(1):9–13. [PubMed] [Google Scholar]
- 22.Heyn R, Holland R. Treatment of rhabdomyosarcoma in children. Proc Am Assoc Cancer Res. 1970;2:36. [Google Scholar]
- 23.Mauer HM, Beltangady M, Gehan EA, et al. The Intergroup Rhabdomyosarcoma Study-I: A final report. Cancer. 1988;61:209–20. doi: 10.1002/1097-0142(19880115)61:2<209::aid-cncr2820610202>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 24.Maurer HM, Gehan EA, Beltangady M, et al. The Intergroup Rhabdomyosarcoma Study-II. Cancer. 1993;71:1904–22. doi: 10.1002/1097-0142(19930301)71:5<1904::aid-cncr2820710530>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 25.Crist WM, Garnsey L, Beltangady MS, et al. Prognosis in children with rhabdomyosarcoma: a report of the Intergroup Rhabdomyosarcoma Studies I and II. Intergroup Rhabdomyosarcoma Committee. J Clin Oncol. 1990;8:443–52. doi: 10.1200/JCO.1990.8.3.443. [DOI] [PubMed] [Google Scholar]
- 26.Crist W, Gehan EA, Ragab AH, et al. The Third Intergroup Rhabdomyosarcoma Study. J Clin Oncol. 1995;13:610–30. doi: 10.1200/JCO.1995.13.3.610. [DOI] [PubMed] [Google Scholar]
- 27.Crist WM, Anderson JR, Meza JL, et al. Intergroup Rhabdomyosarcoma Study-IV: results for patients with nonmetastatic disease. J Clin Oncol. 2001;19:3091–102. doi: 10.1200/JCO.2001.19.12.3091. [DOI] [PubMed] [Google Scholar]
- 28.Donaldson SS, Meza J, Breneman JC, et al. Results from the IRS-IV randomized trial of hyper fractionated radiotherapy in children with rhabdomyosarcoma- a report from the IRSG. Int J Radiation Oncol Biol Phys. 2001;51:718–28. doi: 10.1016/s0360-3016(01)01709-6. [DOI] [PubMed] [Google Scholar]
- 29.Nesbit ME, Jr, Gehan EA, Burgert EO, Jr, et al. Multimodal therapy for the management of primary, non-metastatic Ewing's sarcoma of bone: a long-term follow-up of the First Intergroup Study. J Clin Oncol. 1990 Oct;8(10):1664–74. doi: 10.1200/JCO.1990.8.10.1664. [DOI] [PubMed] [Google Scholar]
- 30.Razek A, Perez CA, Tefft M, et al. Intergroup Ewing's Sarcoma Study: local control related to radiation dose, volume and site of primary lesion in Ewing's sarcoma. Cancer. 1980 Aug 1;46(3):516–21. doi: 10.1002/1097-0142(19800801)46:3<516::aid-cncr2820460316>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 31.Perez CA, Tefft M, Nesbit ME, Jr, et al. Radiation therapy in the multimodal management of Ewing's sarcoma of bone: report of the Intergroup Ewing's Sarcoma Study. Natl Cancer Inst Monogr. 1981 Apr;(56):263–71. [PubMed] [Google Scholar]
- 32.Burgert EO, Jr, Nesbit ME, Garnsey LA, et al. Multimodal therapy for the management of nonpelvic, localized Ewing's Sarcoma of bone: Intergroup study IESS-II. J Clin Oncol. 1990 Sep;8(9):1514–24. doi: 10.1200/JCO.1990.8.9.1514. [DOI] [PubMed] [Google Scholar]
- 33.Brodeur GM, Seeger RC, Barrett A, et al. International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J Clin Oncol. 1988;6:1874–81. doi: 10.1200/JCO.1988.6.12.1874. [DOI] [PubMed] [Google Scholar]
- 34.Camitta B, Leventhal B, Lauer S, et al. Intermediate-dose intravenous methotrexate and mercaptopurine therapy for non-T, non-B acute lymphocytic leukemia of childhood: a Pediatric Oncology Group study. J Clin Oncol. 1989;7:1539–44. doi: 10.1200/JCO.1989.7.10.1539. [DOI] [PubMed] [Google Scholar]
- 35.Crist W, Carroll A, Shuster J, et al. Philadelphia chromosome positive childhood acute lymphoblastic leukemia: clinical and cytogenetic characteristics and treatment outcome. A Pediatric Oncology Group study. Blood. 1990;76:489–94. [PubMed] [Google Scholar]
- 36.Jackson JF, Boyett J, Pullen J, et al. Favorable prognosis associated with hyperdiploidy in children with acute lymphocytic leukemia correlates with extra chromosome 6. A Pediatric Oncology Group study. Cancer. 1990;66:1183–9. doi: 10.1002/1097-0142(19900915)66:6<1183::aid-cncr2820660618>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 37.Harris MB, Shuster JJ, Carroll A, et al. Trisomy of leukemic cell chromosomes 4 and 10 identifies children with B-progenitor cell acute lymphoblastic leukemia with a very low risk of treatment failure: a Pediatric Oncology Group study. Blood. 1992:3316–24. [PubMed] [Google Scholar]
- 38.Ravindranath Y, Abella E, Krischer JP, et al. Acute myeloid leukemia (AML) in Down's syndrome is highly responsive to chemotherapy: experience on Pediatric Oncology Group AML Study 8498. Blood. 1992;80:2210–4. [PubMed] [Google Scholar]
- 39.Link M, Goorin AM, Miser AW, et al. The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med. 1986;314:1600–2. doi: 10.1056/NEJM198606193142502. [DOI] [PubMed] [Google Scholar]
- 40.Duffner PK, Horowitz ME, Krischer JP, et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328:1725–31. doi: 10.1056/NEJM199306173282401. [DOI] [PubMed] [Google Scholar]
- 41.Smith M, Arthur D, Camitta B, et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J Clin Oncol. 1996;14:18–24. doi: 10.1200/JCO.1996.14.1.18. [DOI] [PubMed] [Google Scholar]
- 42.Grier HE, Krailo MD, Tarbell NJ, Link MP, et al. Addition of ifosfamide and etoposide to standard therapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003 Feb 20;348(8):694–701. doi: 10.1056/NEJMoa020890. [DOI] [PubMed] [Google Scholar]
- 43.Ortega JA, Krailo MD, Haas JE, et al. Effective treatment of unresectable or metastatic hepatoblastoma with cisplatin and continuous infusion doxorubicin: A report from the Children's Cancer Study Group. J Clin Oncol. 1991;9:2167–2176. doi: 10.1200/JCO.1991.9.12.2167. [DOI] [PubMed] [Google Scholar]
- 44.Douglass EC, Reynolds M, Finegold M, et al. Cisplatin, vincristine and fluorouracil therapy for hepatoblastoma: A pediatric Oncology Group Study. J Clin Oncol. 1993;11:96–99. doi: 10.1200/JCO.1993.11.1.96. [DOI] [PubMed] [Google Scholar]
- 45.Ortega JA, Douglass EC, Feusner JH, et al. Randomized Comparison of Cisplatin/Vincristine/Fluorouracil and Cisplatin/Continuous Infusion doxorubicin for Treatment of Pediatric Hepatoblastoma: A Report from the Children's Cancer Group and the Pediatric Oncology Group. J Clin Oncol. 2000;18(14):2665–2675. doi: 10.1200/JCO.2000.18.14.2665. [DOI] [PubMed] [Google Scholar]
- 46.Nesbit ME, Sather H, Robison LL, et al. Sanctuary therapy: a randomized trial of 724 children with previously untreated acute lymphoblastic leukemia: A Report from Children's Cancer Study Group. Cancer Res. 1982;42:674–80. [PubMed] [Google Scholar]
- 47.Nesbit ME, Jr, Sather HN, Robison LL, et al. Randomized study of 3 years versus 5 years of chemotherapy in childhood acute lymphoblastic leukemia. J Clin Oncol. 1983;1:308–16. doi: 10.1200/JCO.1983.1.5.308. [DOI] [PubMed] [Google Scholar]
- 48.Gaynon PS, Trigg ME, Heerema NA, et al. Children's Cancer Group Trials in Childhood acute lymphoblastic leukemia: 1983-1995. Leukemia. 2000 Dec;14(12):2223–33. doi: 10.1038/sj.leu.2401939. [DOI] [PubMed] [Google Scholar]
- 49.Bostrom BC, Sensel MG, Sather HN, et al. Dexamethasone versus prednisone and daily oral versus weekly intravenous mercaptopurine for patients with standard-risk acute lymphoblastic leukemia: a report from the Children's Cancer Group. Blood. 2003;101:3809–17. doi: 10.1182/blood-2002-08-2454. [DOI] [PubMed] [Google Scholar]
- 50.Gaynon PS, Bleyer WA, Steinherz PG, et al. Day 7 marrow response and outcome for children with acute lymphoblastic leukemia and unfavorable presenting features. Med Pediatr Oncol. 1990;18:273–9. doi: 10.1002/mpo.2950180403. [DOI] [PubMed] [Google Scholar]
- 51.Evans AE, Jenkin RD, Sposto R, et al. The treatment of medulloblastoma. Results of a prospective randomized trial of radiation therapy with and without CCNU, vincristine, and prednisone. J Neurosurg. 1990;72:572–82. doi: 10.3171/jns.1990.72.4.0572. [DOI] [PubMed] [Google Scholar]
- 52.Packer RJ, Goldwein J, Nicholson HS, et al. Treatment of children with Medulloblastoma with reduced-dose craniospinal radiation and adjuvant chemotherapy: A Children's Cancer Group Study. J Clin Oncol. 1999 Jul;17(7):2127–36. doi: 10.1200/JCO.1999.17.7.2127. [DOI] [PubMed] [Google Scholar]
- 53.Patte C, Auperin A, Gerrard M, Michon J, et al. Results of the randomized international FAB/LMB96 trial for intermediate risk B-cell non-Hodgkin lymphoma in children and adolescents: it is possible to reduce treatment for the early responding patients. Blood. 2007 Apr;109(7):2773–80. doi: 10.1182/blood-2006-07-036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cairo MS, Gerrard M, Sposto R, et al. Results of a randomized international study of high-risk central nervous system B non-Hodgkin lymphoma and B acute lymphoblastic leukemia in children and adolescents. Blood. 2007 Apr 1;109(7):2736–43. doi: 10.1182/blood-2006-07-036665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matthay KK, Villablanca JG, Seeger RC, et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children's Cancer Group. N Engl J Med. 1999;341:1165–73. doi: 10.1056/NEJM199910143411601. [DOI] [PubMed] [Google Scholar]
- 56.Matthay KK, Sather HN, Seeger RC, et al. Excellent outcome of stage II neuroblastoma is independent of residual disease and radiation therapy. J Clin Oncol. 1989;7:236–44. doi: 10.1200/JCO.1989.7.2.236. [DOI] [PubMed] [Google Scholar]
- 57.Nesbit M, Buckley L, Lampkin B, et al. Comparison of allogeneic bone marrow transplantation (BMT) with maintenance chemotherapy in previously untreated childhood acute nonlymphocytic leukemia. Proc Am Soc Clin Onc. 1987;6:163. (ANLL) [abstract] [Google Scholar]
- 58.Woods WG, Ruymann FB, Lampkin BC, et al. The role of timing of high-dose cytosine arabinoside intensification and of maintenance therapy in the treatment of children with acute nonlymphocytic leukemia. Cancer. 1990;66:1106–13. doi: 10.1002/1097-0142(19900915)66:6<1106::aid-cncr2820660605>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 59.Hilden JM, Dinndorf PA, Meerbaum SO, et al. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children's Oncology Group. Blood. 2006;108:441–51. doi: 10.1182/blood-2005-07-3011. Epub 2006 Mar 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Neglia JP, Meadows AT, Robison LL, et al. Second neoplasms after acute lymphoblastic leukemia in childhood. N Engl J Med. 1991;325:1330–6. doi: 10.1056/NEJM199111073251902. [DOI] [PubMed] [Google Scholar]
- 61.Bleyer WE, Fallavollita J, Robison L, et al. Influence of age, sex, and concurrent intrathecal methotrexate therapy on intellectual function after cranial irradiation during childhood: a report from the Children's Cancer Study Group. Pediatr Hematol Oncol. 1990;7:329–38. doi: 10.3109/08880019009033410. [DOI] [PubMed] [Google Scholar]
- 62.Borowitz M, Devidas M, Bowman WP, et al. Prognostic significance of minimal residual disease (MRD) in Childhood precursor ALL and its relation to other risk factors. A Children's Oncology Group (COG) Blood. 2006;108(11):#219. [Google Scholar]
- 63.Bhojwani D, Kang H, Moskowitz NP, et al. a Children's Oncology Group Study. Blood. 2006 Jul 15;108(2):711–7. doi: 10.1182/blood-2006-02-002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reaman GH, Haase GM. Quality of life research in childhood cancer. The time is now. Cancer. 1996;78:1330–2. doi: 10.1002/(SICI)1097-0142(19960915)78:6<1330::AID-CNCR23>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 65.Avramis VI, Sencer S, Periclou AP, et al. A randomized comparison of native Escherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-risk acute lymphoblastic leukemia: a Children's Cancer Group study. Blood. 2002;99:1986–94. doi: 10.1182/blood.v99.6.1986. [DOI] [PubMed] [Google Scholar]; Blood. 2002;100:1531. Erratum in. [Google Scholar]
- 66.Hammond GD, Nixon DW, Nachman JB, et al. American Cancer Society Workshop on Adolescents and Young Adults with Cancer. Workgroup #4: Clinical research implications. Cancer. 1993;71:2423. doi: 10.1002/1097-0142(19930401)71:7<2423::aid-cncr2820710740>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]