

Abstract
Low copy number (LCN) typing, particularly for current short tandem repeat (STR) typing, refers to the analysis of any sample that contains less than 200 pg of template DNA. Generally, LCN typing simply can be defined as the analysis of any DNA sample where the results are below the stochastic threshold for reliable interpretation. There are a number of methodologies to increase sensitivity of detection to enable LCN typing. These approaches encompass modifications during the polymerase chain reaction (PCR) and/or post-PCR manipulations. Regardless of the manipulations, when processing a small number of starting templates during the PCR exaggerated stochastic sampling effects will occur. The result is that several phenomena can occur: a substantial imbalance of 2 alleles at a given heterozygous locus, allelic dropout, or increased stutter. With increased sensitivity of detection there is a concomitant increased risk of contamination. Recently, a commission reviewed LCN typing and found it to be “robust” and “fit for purpose.” Because LCN analysis by its nature is not reproducible, it cannot be considered as robust as that associated with conventional DNA typing. The findings of the commission seem inconsistent with the nature of LCN typing. While LCN typing is appropriate for identification of missing persons and human remains and for developing investigative leads, caution should be taken with its use in other endeavors until developments are made that overcome the vagaries of LCN typing. A more in-depth evaluation by the greater scientific community is warranted. The issues to consider include: training and education, evidence handling and collection procedures, the application or purpose for which the LCN result will be used, the reliability of current LCN methods, replicate analyses, interpretation and uncertainty, report writing, validation requirements, and alternate methodologies for better performance.
DNA typing has become a mainstay in the characterization of forensic biological evidence which allows analyses of a wide range of biological materials for direct and indirect (or kinship) identity testing. The exquisite sensitivity of the DNA typing assays permits even very minute quantities of DNA to be genotyped. This is the result of the use of the polymerase chain reaction (PCR), an exponential enzymatic cycling process that increases the number of target DNA sites from sub-analytical to analytical levels. Short tandem repeat (STR) loci are currently the primary genetic markers used for typing because of their reasonably high polymorphic nature and the ability to produce multiplex assays of up to 15 loci which substantially increases the power of discrimination and reduces sample consumption. In addition, the small amplicon size of STRs (typically ranging from 100 to 400 bps in length) makes them amenable to the analysis of degraded DNA samples. These forensic DNA typing assays have been invaluable for helping to resolve the source(s) of biological evidence. Their success is due to the robustness and reliability associated with the suite of technologies and methodologies that have been validated for forensic use (1-6).
Extensive validation studies have been conducted on the commercial DNA typing kits, and the conditions under which these kits produce reliable results are well described (1-6). Optimal template amounts are well defined and typically range from 200 pg to 2-3 ng of input DNA (1 ng is considered the optimum amount for most commercial kits). About a decade ago, several investigators attempted to increase the sensitivity of the assays by developing the technique known as low copy number (LCN) typing (7-12); however, the robustness of the assays was compromised with use of reduced template quantities (9,10,13,14). Originally, LCN typing of STRs with the typical commercial kit formats referred to the analysis of any sample that contained less than 100 pg of template DNA (9,13,15). More recently, the maximum template value for a LCN analysis has been raised to less than 200 pg (16), which is more consistent with the stochastic threshold DNA amounts described by Moretti et al (17,18) for conventional STR typing. These quantitative threshold values are based on an amount of template DNA where peak height imbalance becomes exaggerated and are relative to specific assays, kits, and methodologies. The value will change with technology and genetic markers typed and the 200-pg threshold therefore will not necessarily apply to all systems. More likely, a heterozygote peak height imbalance ratio may be a better criterion for a stochastic threshold. The stochastic threshold values need to be determined within the laboratory through proper validation studies for each system (17,18). Regardless, LCN typing simply can be defined as the analysis of any DNA sample where the results are below the stochastic threshold for reliable interpretation (13). “Touch DNA” (19) is now becoming the euphemistic in vogue term for LCN typing. Some touch DNA samples, however, do not qualify as LCN samples in that they contain sufficient DNA for routine conventional analyses. Conversely, many crime scene samples do meet the criteria of LCN samples. Such samples should be clearly distinguished and analyzed and interpreted accordingly.
In 1998, the city of Omagh in Northern Ireland experienced a terrorist bombing in a busy market area in which 29 people died and 200 were wounded. Sean Hoey, a 38-year-old electrician was arrested and placed on trial for the murders. His arrest was partially based on DNA evidence. He was placed on trial for the murders and ultimately was found not guilty. The Judge (Justice Weir) in the case was critical of the handling of the DNA evidence which included a LCN analysis. Particularly, Justice Weir found that the recording, packaging, storage, and transmission of some of the items of evidence were “thoughtless” and “slapdash.” He remarked that the crime scene investigators, police, and forensic laboratory did not take appropriate protective precautions for LCN typing (20).
Courtrooms are far from the best place to evaluate the reliability or validity of a particular application of science (21,22), and one should be cautious about taking at face value the specific criticisms of the “soundness” of LCN typing raised in that arena. Indeed, the necessary precautions were not in place for the collection and handling of evidence that might be subjected to LCN typing. The Omagh bombing (and related cases) pre-dates the implementation of LCN typing and thus the more stringent recommendations required to further reduce possible contamination were not in place. However, these more stringent collection protocols by themselves do not address the reliability of LCN typing. The criticisms levied in Queen v. Hoey resulted in a commissioned review of the LCN typing technology (16). The reviewers found that LCN typing as practiced specifically in the United Kingdom was “robust” and “fit for purpose” but offered a number of recommendations to improve the methodology. The findings of the commission seem inconsistent with the nature of LCN typing and LCN typing warrants a more in-depth evaluation by the greater scientific community. Some of the pertinent issues raised in the case and the review are:
1) There is a greater potential for error (compared with conventional STR typing protocols).
2) Errors of interpretation can be caused by allele drop-in, allele drop-out, peak height imbalance, and large stutter peaks.
3) There is a need for a robust and reliable quantitation assay in order to determine the amount of DNA available for analysis.
4) LCN profiles are not generally reproducible. Because of the potential error, the probative value of the results may not be estimated reliably.
5) The interpretation of mixture profiles from LCN typing is problematic. Interpretation guidelines based upon reliable validation studies do not exist.
6) Because of the sensitivity of the assay and the types of samples analyzed (ie, touch samples), the LCN profile may not be relevant to a case.
7) The evidence cannot be used for exculpatory purposes.
8) Proper evidence collection and handling protocols have not been well established or at least communicated.
9) Reagents and consumables may contain low level amounts of extraneous DNA that can complicate the interpretation of LCN typing results.
Methods of LCN typing
There are a number of ways to carry out LCN typing to increase the sensitivity of the assay. These include increasing the PCR cycle number, post-PCR sample clean-up prior to genotype analysis, or for that matter any manipulation that increases the signal that is below a stochastic threshold (13). Examples of LCN methods include:
1) Increasing PCR cycle number (9,10,12,13,15);
2) Nested PCR (23);
3) Reducing the volume of the PCR (13,24,25);
4) Whole genome amplification prior to the PCR (26);
5) Enhanced fluorescent dye signal;
6) Use of higher purity formamide in sample preparation for capillary electrophoresis (13);
7) Post-PCR clean-up to remove ions that compete with DNA during electrokinetic injection (13,27,28);
8) Increasing injection time (13).
LCN typing should not be confused with the observation of a weak profile and attempts to improve those results by re-extraction of another sample to obtain additional DNA or by concentrating a DNA extract. Because of inherent limitations, several investigators have urged caution in the practice and interpretation of LCN typing (9,10,13,15). Budowle et al (13) expanded the call for caution and suggested that LCN typing should be used only for identifying missing persons (including victims of mass disasters) and for investigative leads. We do not favor advocating use of current LCN methodologies in criminal proceedings, but recognize that all scenarios cannot be predicted that may warrant use of LCN typing and that technology is always changing and many concerns raised herein may soon be addressed. While LCN typing is appropriate for identification of missing persons and human remains and for developing investigative leads, caution should be taken with its use in other endeavors until developments are made that overcome the vagaries of LCN typing.
There is no doubt that LCN typing is and has proven to be an invaluable tool and the issues relating to its use warrant further investigation. These need to be discussed in the open scientific forum so that LCN typing can be reviewed critically to ensure that it can be used properly and effectively. For example, “fit for purpose” is used generically (16), but LCN typing robustness should be considered in context with the application. LCN typing may be more fit for some purposes than others. These should be sufficiently delineated and caveats defined. It is timely to discuss the state-of-the-art of LCN typing and provide suggestions or recommendations for improving the methodology, developing appropriate interpretation guidelines, augmenting information contained within reports, and adequately communicating limitations.
LCN applications
The discussion on the “soundness” of the science of LCN typing should properly begin with its application. Budowle et al (13) were the first to suggest that the use of LCN typing be limited to providing investigative leads and the identification of human remains. Moreover, they were not advocates of using it in criminal proceedings as is done currently with robust conventional STR typing data. Since LCN typing does not yield reproducible results, ie, the same result would not be expected if the sample were analyzed twice, it cannot be considered robust by conventional standards. When used to develop investigative leads, the loss of an allele(s) or the observation of an extraneous allele(s) in a profile is not necessarily detrimental in generating candidates as long as the limitation is understood and built into the evaluation process and other meta data are used to resolve the case. One area where LCN typing could be of value is that of identification of missing persons. In essence, identifying the remains of missing persons is practiced no differently than evidence used in an investigative lead. The genetic information obtained often is used to lead to other meta data to make an identification. Moreover, in many analyses of human remains multiple samples (ie, more than 2) can be analyzed. This is not to suggest that in a closed population disaster, such as an airplane crash, LCN typing cannot be used as the sole source of identification. Indeed LCN typing should be pursued for victim identification in those cases where the biological evidence is so compromised that conventional DNA typing would be fruitless. In the context of a closed population, some error in typing results can be tolerated and yet proper identifications could still be made. A notable difference between most human remains and other forensic biological evidence is that the surfaces of bones, teeth, and hairs can be cleansed so that in many cases it is reasonable to infer that these samples are a single source in nature. In addition, cleansing reduces the impact that low level exogenous DNA might have on the interpretation of LCN results (see sporadic contamination and drop-in below). Thus, LCN typing should be “fit for purpose” for this application.
Laboratory practices
There are aspects of the laboratory analytical portion of LCN typing that are robust. These include the practices to minimize laboratory-induced contamination. Recommendations for contamination prevention in laboratory controlled environments include pressurized facilities, appropriate laboratory gear, the analysis of a single sample at a time, DNA-free consumables, and decontamination practices (eg, exposure to UV light and/or ethylene oxide) (29). Unfortunately, similar constraints have not made their way into the protocols for evidence collection and handling (16,20). Proper contamination prevention practices need to be implemented not only in the laboratory but also at the crime scene. Extensive training in proper collection procedures will be required for first responders and crime scene investigators.
Issues associated with low template amounts
There are a number of issues which are raised by the analysis of sub-optimal amounts of DNA template in a PCR. These issues become more problematic as the amount of template decreases. In addition, mixture interpretation has yet to be well-addressed and this will be alluded to in a number of sections. The topics are:
1) Stochastic effects
a. Detection threshold
b. Profile interpretation
c. Allele drop-out and heterozygote peak imbalance
d. Stutter
2) Contamination
3) Replicate analyses
4) Appropriate controls
5) Application limitations
Stochastic effects
Due to the kinetics of the PCR process, a small number of starting templates will experience stochastic sampling effects. When only a few target templates are available, primer binding may not occur equally for each allele at a locus during the first few cycles and thus result in a notable imbalance between allelic products or, in some cases, total loss of one or both alleles. In other words, LCN DNA templates in a PCR will experience stochastic amplification that may result in either a substantial imbalance of 2 alleles at a given heterozygous locus, allelic dropout, or increased stutter (9,10,12,13,27,28) (Figures 1-3).
Figure 1.
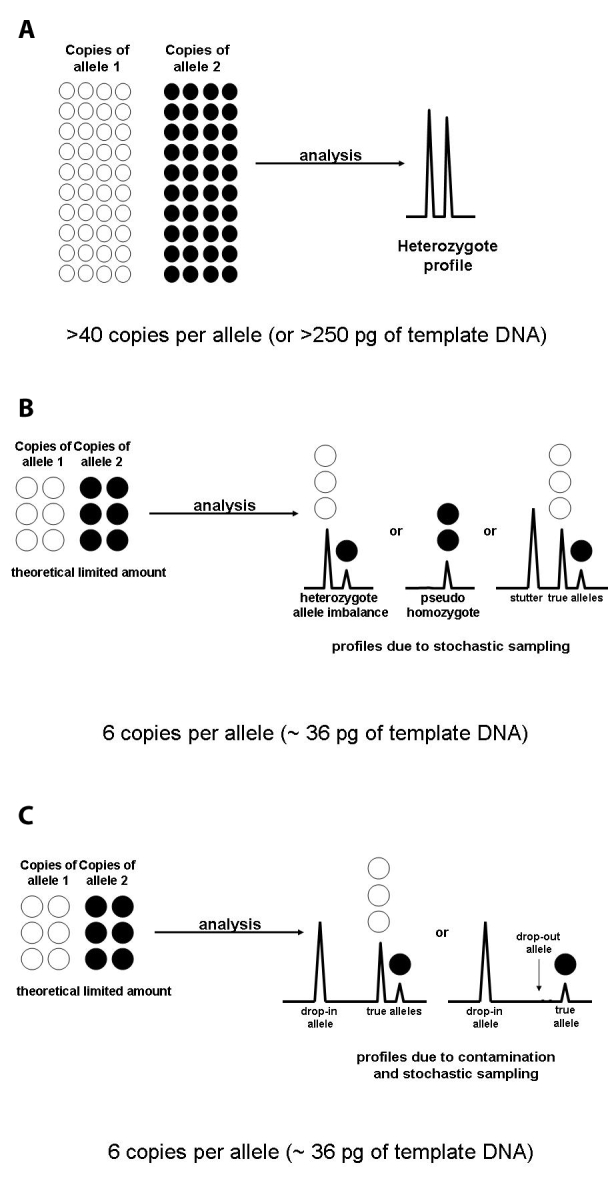
(A) With sufficient DNA, eg, 250 pg, faithful reproduction of the alleles can be generated. (B) Possible stochastic effects when a low copy number (LCN) sample containing approximately 36 pg of DNA is analyzed. The result can be heterozygote peak imbalance, allele drop-out (or a pseudo-homozygote profile), increased stutter, or combinations thereof. (C) The possible stochastic effects when a LCN sample containing approximately 36 pg of DNA is analyzed. Due to increased sensitivity of detection the risk of allele-drop in (and allele drop-out) is exacerbated.
Figure 3.
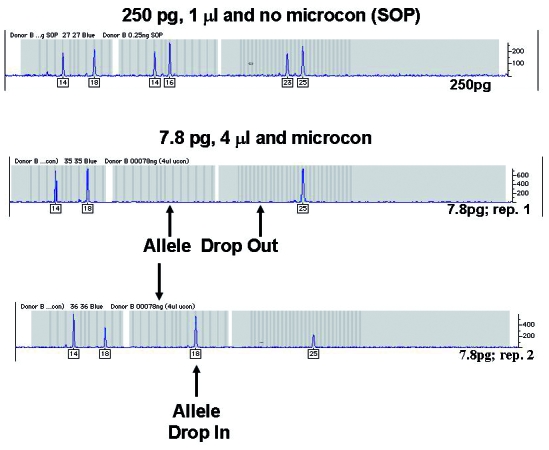
There are a number of ways to increase sensitivity of detection for low copy number (LCN) typing. The one displayed here is post-polymerase chain reaction (post-PCR) purification using microcon dialysis to remove ions that compete with DNA during electrokinetic injection. A known 250 pg DNA sample was analyzed under standard operating profile conditions. The same sample was diluted to 7.8 pg and 2 replicates were independently amplified using the AmpFlSTR® Profiler® PCR Amplification Kit (Applied Biosystems, Foster City, CA, USA). To increase sensitivity of detection, the 7.8 pg samples were subjected to post-PCR purification and a 4-fold increase of sample was used for analysis. The example shows the results for a subset of the full profile (D3S1358, vWA, and FGA loci). Typical stochastic effects are observed for the LCN samples.
Detection thresholds
Typically, minimum amounts of DNA template are recommended for a PCR, so that stochastic effects can be reduced to manageable levels. However, since variation in the quantitation of template DNA and pipetting volume inaccuracies can impact the amount of template DNA placed in a PCR, a stochastic interpretation threshold is used instead for STR typing (called the MIT by Budowle et al, ref. 30). A minimum peak height (or area), which is established by in-house laboratory validation studies, serves as a stochastic control. Those peaks below this threshold are not interpreted or are interpreted with extreme caution for limited purposes.
With too little input DNA, the height of allelic peaks by definition would fall below the established stochastic threshold for conventional STR typing and, at times, the signal may be too low to observe. With LCN typing, the allele peak heights are substantially increased (by for example additional PCR cycles or post-PCR clean-up) and then interpreted. Since LCN typing inherently refers to the interpretation of results that would normally be below the stochastic interpretation threshold, there is no minimum peak height criterion for interpretation that is similar to that of standard STR typing (with samples containing 250 pg to 1 ng of DNA). Indeed, the manipulation of LCN samples such that allelic peaks meet or exceed the in-house stochastic threshold established for conventional STR typing has no meaning with regard to generating a reliable result. This stochastic threshold was established and validated with typing analyses using conditions very different than those for LCN typing. Currently, there is no purported way to establish a threshold for LCN typing and this will continue to be a weakness of the application.
Profile interpretation
Two factors that impact the robustness of LCN typing are stochastic effects and sensitivity of detection. They collectively result in allele drop-out, exaggerated heterozygote peak height imbalance, exaggerated peak height difference between loci within a profile exaggerated stutter (stochastic effects), and allele drop-in (sensitivity to contamination). Protocols for the interpretation of LCN generated profiles have been proposed which consider all of these phenomena (9). However, these suggested interpretation guidelines are based on studies of single source profiles developed using relatively pristine samples. The poor quality of evidence samples and mixtures exacerbate the interpretation of LCN data. The presence of 3 or more alleles at a given locus is indicative of a mixed sample. Also, the presence of 2 alleles at a locus, with substantially different peak heights, may be indicative of a mixed sample. These conventional criteria for mixture interpretation (30-32) and confirmation of a mixture are not reliable with LCN typing, because of the peak height imbalance of heterozygote alleles, the increased production of stutter products, and allele drop-in. To date well-developed LCN interpretation guidelines for mixtures have not been described. Since many touch samples are mixtures (15,16), a lack of validation studies and interpretation guidelines is a serious deficiency.
Allele drop-out
Allele drop-out is the simplest LCN-related phenomenon to address. If a single source profile presents itself, one can legitimately assume that a single peak at a locus may not be a true homozygote. For example if an allele 15 was observed in the LCN evidence, then based on that evidence any individual that was a 15 homozygote or a 15,X heterozygote (where X can be any allele) could not be excluded as a potential contributor of the sample. For placing weight on the evidence, the 2p rule could be used for any single allele at a locus (33), and this calculation would be conservative. The observation of 2 alleles at a locus might be assumed to represent a heterozygous profile and thus would suggest that allele drop-out has not occurred. The likelihood ratio for a single source sample can be calculated using1/2fa for homozygotes and 1/2fafb for heterozgotes, where fa is the frequency of allele a and fb is the frequency of allele b (9). Gill et al (9) also recommended modifying calculations by considering the probability of allele drop-out (p(D)). The p(D) is based on experimental observation. However, it is difficult to justify p(D) based solely on experimental studies using pristine samples and applying it to any specific case. Drop-out is related to sample quantity and quality. These parameters often are undefined in LCN samples and are sample-specific. Allele drop-out cannot be predicated only on current laboratory-controlled validation studies. More research needs to be carried out before providing values for p(D).
Stutter
When generating LCN STR profiles, the percent stutter is variable and is not informative since a stutter peak may actually exceed the height/area of the associated allelic peak (9,15). Although some investigators (9) have attempted to factor the probability of stutter into statistical calculations, the probability of stutter and percent of stutter to the true allele currently are not predictable. It is possible that a peak due to stutter may be seen twice in replicate analyses and deemed a “true” allele. The likelihood of stutter being observed twice in replicate analyses has not been addressed and there has yet to be a proposal on how to handle stutter with mixed samples.
Contamination
Allele drop-in is the appearance of an allele that is not from the evidence and is attributed to contamination due to laboratory processes (9). This is a very narrow definition of the source of contamination. Indeed, low level contaminating DNA could originate from reagents and other laboratory consumables, from laboratory personnel, and from sample-to-sample cross contamination. A compilation of data comprising spurious bands from negative controls might not be a good estimate of the probability of drop-in as has been suggested (9). Many LCN samples are touch samples. Therefore, low level DNA could exist in the evidence from background contamination at the crime scene. Also contamination could occur during evidence collection and handling. Thus, the appearance of allele drop-in may be inherent in the samples or induced during crime scene evidence collection. Predicting the probability of drop-in based solely on pristine experimental data may not be useful in approximating the circumstances where drop-in may have occurred. Additionally, allele drop-in has been characterized as a random event. This contention may not be supportable. It is known that some manufacturers of tubes, for example, have produced products that are contaminated with human DNA (our unpublished observation). These tubes would more likely have DNA that is not random and likely would be in multiple tubes very different in frequency than what occurs in the population. Another difficulty, particularly with mixtures, is determining what allele constitutes a drop-in. In fact, these vagaries tend to create bias in deciding whether there is support for contamination. For example, Gill et al (9) recommended for assessing contamination “If a locus shows ab alleles in the crime stain and the suspect is an ab genotype…no contamination has occurred.” Assessments of the evidence profile to determine what is a true evidence allele and what is allele drop-in need to be made without knowledge of the suspect profile. A quality unbiased approach requires the interpretation of the evidence profile in the absence of knowledge of the suspect profile. Interpretation of the evidence profile contemporaneously with the reference profile is indicative of bias and is anathema to the objective nature of forensic science. Because of the limitations of LCN typing, we need to be vigilant not to over-step quality interpretation practices to ensure that interpretation bias is minimized as best as is possible (22).
While the appearance of spurious alleles in negative controls can be attributed to contamination, the same degree of ascription may not be so for allele drop-in for evidence samples and known samples with low quantities of template DNA. Some drop-in alleles differ by one or 2 repeats from true known alleles in pristine sample validation studies and may be the result of slippage during the PCR. Two hypotheses can be proffered regarding the presence of a drop-in allele: 1) it is the result of contamination; or 2) it is due to slippage (following a random genetic drift model). Currently, these 2 possible causes for drop-in cannot be distinguished. However, if the latter hypothesis was found to account for a significant proportion of drop-in alleles, then LCN allele drop-in could be modeled and assessed quantitatively. The possibility that some allele drop-ins may be due to slippage, such as increased stutter, suggests that the probability of contamination values suggested by Gill et al (9) may be inappropriate for addressing allele drop-in.
Replicate analyses
The approach most widely used for the designation of an allele in a LCN sample requires the division of the sample into 2 or more aliquots and reporting only the alleles that are common in at least 2 replicates (9,14,15). There is some appeal to this redundancy approach in that if drop-in occurs randomly and infrequently, then observing an allele multiple times increases the confidence that the allele is truly derived from the evidentiary sample (assuming that contamination did not occur during evidence collection). The supposition of randomness may not be justified, but if drop-in is infrequent lack of randomness may be inconsequential. Most practitioners of LCN typing advocate 2-3 replicate analyses and that an allele must be observed twice to be sufficient for recording it as an allele. Taberlet et al (34) advocated more (up to 7) replicates to increase the confidence of allele calls. Thus, redundancy of alleles in replicates is the basis for reliability in LCN typing. Obviously, the more replicates that show the same allele(s) will increase the confidence that the observation is less likely due to laboratory contamination. Suppose 5 replicates were carried out and 2 showed the same allele, would that be considered sufficient to report the allele? Clearly, if all 5 replicates showed the same allele the confidence regarding an identified allele would be greater than if it were observed in only 2 replicates. The number of replicate analyses, the number of times an allele is observed, and the degree of confidence (quantitatively or qualitatively) need to be better defined. However, there is a practical realization that more than 2-3 replicates may not be possible with a limited sample. Therefore, most interpretation guidelines and degree of confidence assessments must be predicated on 2-3 replicate analyses. Presumably, the more replicates that show the same alleles at a locus are more reliable than less redundancy. So, consider 3 replicates showing the same 2 alleles (eg 13,15) in all 3 analyses; these results for designating the 2 alleles 13 and 15 should be more reliable than 3 replicates where the alleles are seen twice but parsed over the 3 replicates (eg, replicate 1 – 13; replicate 2 – 13,15; and replicate 3 – 15). Both of these would be reported as a 13,15 and yet these 2 scenarios are treated with the same degree of confidence with current procedures. A third scenario could be – replicate 1 – 13,14; replicate 2 – 13,15; and replicate 3 – 15,16. This scenario would also be reported as a 13,15 and be treated with the same degree of confidence as the other 2 scenarios. More work is needed to provide guidelines for reporting results and their significance.
Common sense would intimate that splitting a sample into multiple aliquots exacerbates the limitations of LCN typing (13) and all efforts should be made to concentrate as much template as possible into one reaction. However, redundancy has been the only approach advocated. Studies on dilution and redundancy have been based on relatively pristine samples which do not approximate evidence based samples that have undetermined quantities of (possibly degraded) DNA and may contain inhibitors of the PCR that also can impact allele drop-out. There has been little discussion on the number of replicates, the degree of confidence related to the uncertainty of the nature of the samples, and how these should be reported given the quality of the sample. Additionally, the confidence of a result may differ for the number of replicates used for assessing drop-in than for evaluating allele drop-out. Given the uncertainty and decision process that alleles seen for example only once in say 2 replicate analyses are not reported, should these single observation alleles be placed in the final report (and not just the case file) to assist anyone involved in the legal process? If so, what about single allele observations in 3 replicate analyses? The data to be included in a report should be defined since LCN typing is not a robust assay. We strongly recommend that the issue of replicates be addressed. Should replicates be done? If so, the number of replicates that are sufficient for LCN typing needs to be addressed. The degree of confidence that can be placed on the replicate analyses and the allelic profile results obtained over these replicate analyses should also be discussed.
Because of the issues surrounding interpretation of LCN profiles and the lack of confidence that exists in defining true profile alleles, LCN typing cannot be used for exculpatory purposes.
Controls
Another issue of LCN typing is the number and type of control samples that should be used. For LCN typing, the negative control does not serve well as an indicator of sporadic low level contamination within associated processed samples of the same batch (9). There has been no discussion on the number of negative controls one should run to have confidence that allele drop-in is sporadic (a balance between confidence and cost will have to be considered). Additionally, little discussion has been raised about what constitutes a proper positive control(s). It would seem reasonable that positive controls approximate the same quantity of DNA as that in the evidence, which is a difficult practice to achieve since the amount of DNA in an evidence sample is unknown and difficult to approximate for mixed samples. Perhaps a range of template quantities should be attempted (such as ranging from 20 pg to 200 pg). If a positive control sample approximates in quantity that of a LCN sample, then allele drop-out (and possibly drop-in) will occur. Does the loss of an allele or appearance of an aberrant allele suggest that the positive control failed? Not necessarily, since an analysis is expected to fail, where failure means the complete profile is not observed. However, if a positive control sample does not reliably produce a known result, then the control does not serve its function well. These thorny issues on what constitutes appropriate control samples need to be addressed.
Disclosing LCN limitations
Some of the differences that LCN has compared with conventional STR typing can impact its utility (13,15). Since LCN samples are low in quantity and because of the extreme sensitivity of detection, background level DNA and DNA from casual contact may and will be detected. Thus, profiles that are observed may not be relevant to the case.
Even though LCN typing is better suited as an investigative tool, analyses have ended up in court and likely are to continue to do so. If so, the limitations and vagaries of LCN typing should be documented and made available (in the report or in an accompanying document) so all involved in the investigative and legal process are aware of the limitations that may impact the significance in a specific case result. Publicizing the potential of the application of LCN typing without describing its limitations is not a responsible role for the forensic scientist to take (13). Topics to consider for this documentation are:
1) LCN typing is not a reproducible technique. A statement about this limitation and all LCN replicate results should be disclosed in the report.
2) LCN results cannot be used to exclude an individual. LCN typing should not be applied to post-conviction analyses and examination of old cases without substantial consideration. LCN contamination from handling may have occurred and this possibility needs to be considered.
3) A concentrated sample may perform better in an analysis than replicates that use allele redundancy for interpretation.
4) The number and type of controls used should be defined and related confidence be provided quantitatively or qualitatively.
5) There are stochastic effects and the potential of contamination which impact LCN typing. The interpretation guidelines are not well-established, but those that exist are better suited for single-source samples. Mixture interpretation has not been validated.
6) Contamination or allele drop-in can come from several sources.
7) Due to the enhanced sensitivity, secondary transfer cannot be ruled out as a possible explanation for LCN typing results.
8) STR kits, some reagents, and other consumables may not have been subjected to sufficiently stringent quality control conditions to detect contamination from extraneous DNA similar to the rigor required for LCN typing.
9) Statistical interpretations, and supporting data for probabilities, need to be better defined and developed to convey the uncertainty associated with LCN typing.
10) Because the analysis yields results from very minute samples, the tissue source of the DNA cannot currently be inferred.
Conclusions
LCN typing by its nature cannot be considered robust. However, currently it does have a place in the forensic science toolbox, primarily for developing investigative leads and in the identification of human remains. The success rate has been reported to be low (16) and thus routinely carrying out LCN analysis will be a high resource expense. However, in some cases exigency and need may outweigh expense and success expectations. Therefore, it is incumbent upon scientists to define what constitutes a LCN analysis, disclose its limitations, disclose more information in reports, and carry out more validation studies. A summit meeting should be held with all due speed to define deficiencies, identify needs, and propose direction for LCN typing with the hope that a consensus could be achieved.
While the efforts to use LCN typing primarily have focused on reducing laboratory contamination and employing redundancy for confidence, a more sound approach would be to improve the recovery, extraction, and PCR. Approaches to consider include:
1) Improving crime scene collection methodology and educating crime scene investigation personnel.
2) Increasing efficiency of recovery and yield from a collection device and/or the extract to attempt to increase the amount of template DNA recovered so that a sample contains sufficient DNA to no longer qualify as a LCN sample and then can be analyzed conventionally (35-37). Some portion of current LCN samples may fall into this category.
3) Improving the PCR so that stochastic affects are less incurred with limited template DNA. Thus, the minimal amount of template DNA could be lowered for the PCR and obtain robust amplification.
4) Evaluating SNPs as a primary genetic marker suite for low copy and degraded DNA (38-40). The amplicons for SNPs can be shorter than are those for conventional and mini-STRs. Thus, amplification could be more robust for SNPs and stochastic affects may be less than for larger amplicons STRs.
5) Improve the quality of (and hence quantity of available template) the sample DNA by using DNA repair and/or whole genome amplification methods (28,41).
Figure 2.
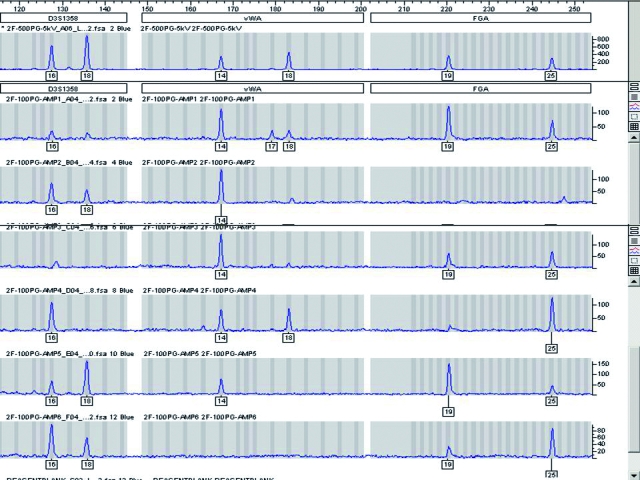
A known DNA sample divided into 6 reactions, each containing 100 pg of DNA and independently amplified using the AmpFlSTR® Profiler Plus® ID PCR Amplification Kit (Applied Biosystems, Foster City, CA, USA) and following the manufacturer’s recommended amplification conditions including 28 cycles for the polymerase chain reaction, but using a 40 relative fluorescence unit (RFU) detection threshold. The example shows the results for a subset of the full profile (D3S1358, vWA, and FGA loci). The top panel is a control sample run with sufficient DNA for conventional typing. The 6 panels below the control are the 100 pg replicates. This example demonstrates that replicates of pristine control samples containing 100 pg of DNA or less may not yield reproducible results. The problems associated with the amplification and interpretation of low copy number (LCN) samples are greatly compounded when evidentiary mixtures are analyzed. The lack of reproducibility will persist with methods employed to increase sensitivity of detection for LCN typing.
References
- 1.Collins PJ, Hennessy LK, Leibelt CS, Roby RK, Reeder DJ, Foxall PA. Developmental validation of a single-tube amplification of the 13 CODIS STR loci, D2S1338, D19S433, and amelogenin: the AmpFlSTR Identifiler PCR Amplification Kit. J Forensic Sci. 2004;49:1265–77. doi: 10.1520/JFS2002195. [DOI] [PubMed] [Google Scholar]
- 2.Cotton EA, Allsop RF, Guest JL, Frazier RR, Koumi P, Callow IP, et al. Validation of the AMPFlSTR SGM plus system for use in forensic casework. Forensic Sci Int. 2000;112:151–61. doi: 10.1016/S0379-0738(00)00182-1. [DOI] [PubMed] [Google Scholar]
- 3.Krenke BE, Tereba A, Anderson SJ, Buel E, Culhane S, Finis CJ, et al. Validation of a 16-locus fluorescent multiplex system. J Forensic Sci. 2002;47:773–85. [PubMed] [Google Scholar]
- 4.Holt CL, Buoncristiani M, Wallin JM, Nguyen T, Lazaruk KD, Walsh PS. TWGDAM validation of AmpFlSTR PCR amplification kits for forensic DNA casework. J Forensic Sci. 2002;47:66–96. [PubMed] [Google Scholar]
- 5.Micka KA, Amiott EA, Hockenberry TL, Sprecher CJ, Lins AM, Rabbach DR, et al. TWGDAM validation of a nine-locus and a four-locus fluorescent STR multiplex system. J Forensic Sci. 1999;44:1243–57. [PubMed] [Google Scholar]
- 6.Mulero JJ, Chang CW, Lagace RE, Wang DY, Bas JL, McMahon TP, et al. Development and validation of the AmpFlSTR MiniFiler PCR Amplification Kit: a MiniSTR multiplex for the analysis of degraded and/or PCR inhibited DNA. J Forensic Sci. 2008;53:838–52. doi: 10.1111/j.1556-4029.2008.00760.x. [DOI] [PubMed] [Google Scholar]
- 7.Barbaro A, Falcone G, Barbaro A. DNA typing from hair shaft. Progress in Forensic Genetics. 2000;8:523–5. [Google Scholar]
- 8.Findlay I, Taylor A, Quirke P, Frazier R, Urquhart A. DNA fingerprinting from single cells. Nature. 1997;389:555–6. doi: 10.1038/39225. [DOI] [PubMed] [Google Scholar]
- 9.Gill P, Whitaker J, Flaxman C, Brown N, Buckleton J. An investigation of the rigor of interpretation rules for STRs derived from less than 100 pg of DNA. Forensic Sci Int. 2000;112:17–40. doi: 10.1016/S0379-0738(00)00158-4. [DOI] [PubMed] [Google Scholar]
- 10.Kloosterman AD, Kersbergen P. Efficacy and limits of genotyping low copy number (LCN) DNA samples by multiplex PCR of STR loci. J Soc Biol. 2003;197:351–9. doi: 10.1051/jbio/2003197040351. [DOI] [PubMed] [Google Scholar]
- 11.Wiegand P, Kleiber M. DNA typing of epithelial cells after strangulation. Int J Legal Med. 1997;110:181–3. doi: 10.1007/s004140050063. [DOI] [PubMed] [Google Scholar]
- 12.Whitaker JP, Cotton EA, Gill P. A comparison of the characteristics of profiles produced with the AMPFlSTR SGM Plus multiplex system for both standard and low copy number (LCN) STR DNA analysis. Forensic Sci Int. 2001;123:215–23. doi: 10.1016/S0379-0738(01)00557-6. [DOI] [PubMed] [Google Scholar]
- 13.Budowle B, Hobson DL, Smerick JB, Smith JA. Low copy number – consideration and caution. Available from: http://www.promega.com/geneticidproc/ussymp12proc/contents/budowle.pdf Accessed: April 22, 2009.
- 14.Gill P, Kirkham A, Curran J. LoComatioN: a software tool for the analysis of low copy number DNA profiles. Forensic Sci Int. 2007;166:128–38. doi: 10.1016/j.forsciint.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 15.Gill P. Application of low copy number DNA profiling. Croat Med J. 2001;42:229–32. [PubMed] [Google Scholar]
- 16.Caddy B, Taylor DR, Lincare AM. A review of the science of low template DNA analysis. Available from: http://police.homeoffice.gov.uk/publications/operational-policing/Review_of_Low_Template_DNA_1.pdf?view=Binary Accessed: April 22, 2009.
- 17.Moretti TR, Baumstark AL, Defenbaugh DA, Keys KM, Smerick JB, Budowle B. Validation of short tandem repeats (STRs) for forensic usage: performance testing of fluorescent multiplex STR systems and analysis of authentic and simulated forensic samples. J Forensic Sci. 2001;46:647–60. [PubMed] [Google Scholar]
- 18.Moretti TR, Baumstark AL, Defenbaugh DA, Keys KM, Brown AL, Budowle B. Validation of STR typing by capillary electrophoresis. J Forensic Sci. 2001;46:661–76. [PubMed] [Google Scholar]
- 19.van Oorschot RA, Jones MK. DNA fingerprints from fingerprints. Nature. 1997;387:767. doi: 10.1038/42838. [DOI] [PubMed] [Google Scholar]
- 20.The Queen v Sean Hoey. Neutral Citation Number [2007] NICC 49. Available from: http://www.xproexperts.co.uk/newsletters/feb08/R%20v%20Hoey.pdf Accessed: April 22, 2009.
- 21.Budowle B, Allard MW, Wilson MR, Chakraborty R. Forensics and mitochondrial DNA: applications, debates, and foundations. Annu Rev Genomics Hum Genet. 2003;4:119–41. doi: 10.1146/annurev.genom.4.070802.110352. [DOI] [PubMed] [Google Scholar]
- 22.Budowle B, Bottrell MC, Bunch SG, Fram R, Harrison D, Meagher S, et al. A perspective on errors, bias, and interpretation in the forensic sciences and direction for continuing advancement. J Forensic SciForthcoming2009 [DOI] [PubMed] [Google Scholar]
- 23.Strom CM, Rechitsky S. Use of nested PCR to identify charred human remains and minute amounts of blood. J Forensic Sci. 1998;43:696–700. [PubMed] [Google Scholar]
- 24.Gaines ML, Wojtkiewicz PW, Valentine JA, Brown CL. Reduced volume PCR amplification reactions using the AmpFlSTR Profiler Plus kit. J Forensic Sci. 2002;47:1224–37. [PubMed] [Google Scholar]
- 25.Leclair B, Sgueglia JB, Wojtowicz PC, Juston AC, Fregeau CJ, Fourney RM. STR DNA typing: increased sensitivity and efficient sample consumption using reduced PCR reaction volumes. J Forensic Sci. 2003;48:1001–13. [PubMed] [Google Scholar]
- 26.Hanson EK, Ballantyne J. Whole genome amplification strategy for forensic genetic analysis using single or few cell equivalents of genomic DNA. Anal Biochem. 2005;346:246–57. doi: 10.1016/j.ab.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 27.Forster L, Thomson J, Kutranov S. Direct comparison of post-28-cycle PCR purification and modified capillary electrophoresis methods with the 34-cycle “low copy number” (LCN) method for analysis of trace forensic DNA samples. Forensic Sci Int; Genet. 2008;2:318–28. doi: 10.1016/j.fsigen.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 28.Smith PJ, Ballantyne J. Simplified low-copy-number DNA analysis by post-PCR purification. J Forensic Sci. 2007;52:820–9. doi: 10.1111/j.1556-4029.2007.00470.x. [DOI] [PubMed] [Google Scholar]
- 29.Shaw K, Sesardic I, Bristol N, Ames C, Dagnall K, Ellis C, et al. Comparison of the effects of sterilisation techniques on subsequent DNA profiling. Int J Legal Med. 2008;122:29–33. doi: 10.1007/s00414-007-0159-5. [DOI] [PubMed] [Google Scholar]
- 30.Budowle B, Onorato AJ, Callaghan TF, Della Manna A, Gross AM, Guerrieri RA, et al. Mixture interpretation: defining the relevant features for guidelines for the assessment of mixed DNA profiles in forensic casework. J Forensic SciForthcoming2009 [DOI] [PubMed] [Google Scholar]
- 31.Gill P, Brown RM, Fairley M, Lee L, Smyth M, Simpson N, et al. National recommendations of the Technical UK DNA working group on mixture interpretation for the NDNAD and for court going purposes. Forensic Sci Int; Genet. 2008;2:76–82. doi: 10.1016/j.fsigen.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 32.Schneider PM, Fimmers R, Keil W, Molsberger G, Patzelt D, Pflug W, et al. The German Stain Commission: recommendations for the interpretation of mixed stains. Int J Legal Med. 2009;123:1–5. doi: 10.1007/s00414-008-0244-4. [DOI] [PubMed] [Google Scholar]
- 33.Budowle B, Giusti AM, Waye JS, Baechtel FS, Fourney RM, Adams DE, et al. Fixed-bin analysis for statistical evaluation of continuous distributions of allelic data from VNTR loci, for use in forensic comparisons. Am J Hum Genet. 1991;48:841–55. [PMC free article] [PubMed] [Google Scholar]
- 34.Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N, et al. Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res. 1996;24:3189–94. doi: 10.1093/nar/24.16.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Budimlija ZM, Lechpammer M, Popiolek D, Fogt F, Prinz M, Bieber FR. Forensic applications of laser capture microdissection: use in DNA-based parentage testing and platform validation. Croat Med J. 2005;46:549–55. [PubMed] [Google Scholar]
- 36.Elliott K, Hill DS, Lambert C, Burroughes TR, Gill P. Use of laser microdissection greatly improves the recovery of DNA from sperm on microscope slides. Forensic Sci Int. 2003;137:28–36. doi: 10.1016/S0379-0738(03)00267-6. [DOI] [PubMed] [Google Scholar]
- 37.Schiffner LA, Bajda EJ, Prinz M, Sebestyen J, Shaler R, Caragine TA. Optimization of a simple, automatable extraction method to recover sufficient DNA from low copy number DNA samples for generation of short tandem repeat profiles. Croat Med J. 2005;46:578–86. [PubMed] [Google Scholar]
- 38.Budowle B, van Daal A. Forensically relevant SNP classes. Biotechniques. 2008;44:603–10. doi: 10.2144/000112806. [DOI] [PubMed] [Google Scholar]
- 39.Kidd KK, Pakstis AJ, Speed WC, Grigorenko EL, Kajuna SL, Karoma NJ, et al. Developing a SNP panel for forensic identification of individuals. Forensic Sci Int. 2006;164:20–32. doi: 10.1016/j.forsciint.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 40.Sanchez JJ, Phillips C, Borsting C, Balogh K, Bogus M, Fondevila M, et al. A multiplex assay with 52 single nucleotide polymorphisms for human identification. Electrophoresis. 2006;27:1713–24. doi: 10.1002/elps.200500671. [DOI] [PubMed] [Google Scholar]
- 41.Nunez AN, Kavlick MF, Robertson JM, Budowle B. Application of circular ligase to provide template for rolling circle amplification of low amounts of fragmented DNA. Nineteenth International Symposium on Human Identification 2008, Promega Corporation, Madison, Wisconsin, 2008. Available from: http://www.promega.com/geneticidproc/ussymp19proc/oralpresentations/Nunez.pdf Accessed: May 4, 2009.