

Abstract
Aim
To test the reliability, robustness, and reproducibility of short tandem repeat (STR) profiling of low template DNA (LT-DNA) when employing a defined set of testing and interpretation parameters.
Methods
DNA from known donors was measured with a quantitative real time polymerase chain reaction (PCR) assay that consistently detects less than 1 pg/µL of DNA within a factor of 0.3. Extracts were amplified in triplicate with AmpFℓSTR® Identifiler® reagents under enhanced PCR conditions. Replicates were examined independently and alleles confirmed using a consensus approach. Considering observed stochastic effects inherent to LT-DNA samples, interpretation protocols were developed and their accuracy verified through examination of over 800 samples.
Results
Amplification of 100 pg or less of DNA generated reproducible results with anticipated stochastic effects. Down to 25 pg of DNA, 92% or more of the expected alleles were consistently detected while lower amounts yielded concordant partial profiles. Although spurious alleles were sometimes observed within sample replicates, they did not repeat. To account for allelic dropout, interpretation guidelines were made especially stringent for determining homozygous alleles. Due to increased heterozygote imbalance, stutter filters were set conservatively and minor components of mixtures could not be resolved. Applying the resultant interpretation protocols, 100% accurate allelic assignments for over 107 non-probative casework samples, and subsequently 319 forensic casework samples, were generated.
Conclusion
Using the protocols and interpretation guidelines described here, LT-DNA testing is reliable and robust. Implementation of this method, or one that is suitably verified, in conjunction with an appropriate quality control program ensures that LT-DNA testing is suitable for forensic purposes.
With the increased sensitivity of multiplex polymerase chain reaction (PCR)-based short tandem repeat (STR) DNA typing kits, low template DNA (LT-DNA) samples, such as those from sloughed skin cells deposited on touched objects, have become a viable source of DNA profiles for forensic casework. This application of LT-DNA testing has been illustrated in many publications (1-6). The poor success rate inherent to LT-DNA typing (4) may be improved with some modifications to routine protocols, such as adding PCR cycles (6-11). For example, Findlay et al (12), by altering the PCR conditions, was able to obtain a useful, accurate genetic profile from an isolated single cell sampled from a buccal swab.
With the opposite goal, aiming to avoid detecting small quantities of DNA, the manufacturer of the AmpFℓSTR® Identifiler® (ID) kit (Applied Biosystems, Foster City, CA, USA) selected their recommended cycle number, 28, following testing of 27 to 31 cycles (13). Thus, our efforts to heighten sensitivity, termed High Sensitivity Testing, included raising the cycle number, with 31 being optimal, reducing the reaction volume, and doubling the annealing time. The injection times and voltage were also altered for capillary electrophoresis (14). In addition, others have shown that purification of the PCR product (15,16), nested PCR (17), and whole genome amplification (18) enhance sensitivity.
Intensifying the DNA signal through such measures carries an increased risk of detecting contamination. Stochastic amplification also results in allelic dropout, increased heterozygote imbalance, and elevated stutter peaks (7-11,14-16,19). An allele from exogenous DNA or stutter may be perceived as a true allele; conversely, the absence of an allele due to dropout, may lead to a locus incorrectly interpreted as homozygous when it is actually heterozygous. Moreover, mixtures may be incorrectly resolved due to peak imbalance. In order to mitigate these risks, interpretation guidelines must be adapted and caution applied (20).
Efforts to minimize the occurrence of exogenous DNA in samples include specific quality control procedures designed to prevent laboratory based contamination (21,22), treatment of consumables to remove DNA (23,24), and the selection of reagents shown to be as free of exogenous DNA as possible. For example, during the manufacturer’s validation of the ID kit no non-specific peaks were generated (13). To reduce the possibility of including spurious alleles in profiles when detected and to facilitate complex data interpretation, samples should be amplified at least twice and only alleles that repeat assigned to the consensus profile, also termed the composite profile (7-11,14,16,19). In Gill’s study of 1225 comparisons of 50 amplifications examined in pairs, only four samples (0.3%) produced the same spurious allele twice (7).
Replication of each sample dictates the use of a PCR STR system that simultaneously amplifies all of the loci required for comparison to a database system. This conserves the sample by avoiding dilution of the DNA extract which occurs if 2 kits targeting fewer loci were used. When utilizing the ID kit with our optimized parameters, stochastic effects were noted for samples amplified with less than 100 pg in our study. Over 800 samples were examined to derive interpretation protocols that would account for the stochastic effects observed and, when followed, would generate 100% correct allelic assignments.
Following review of our validation studies for LT-DNA testing, our quality control, testing, and interpretation protocols were approved by the DNA Subcommittee of the New York State Forensic Science Commission, and subsequently, by the entire commission, in December 2005. In addition, these studies have since been reviewed and approved by Federal Bureau of Investigation-trained DNA auditors during routine external audits. The validation studies demonstrate that by employing these protocols, LT-DNA testing is reliable and robust.
Materials and methods
Personnel, workspace, equipment, and consumables’ preparation
Laboratory personnel donning double gloves, hair coverings, face masks, booties, eye protection, and laboratory coats worked exclusively in rooms dedicated to LT-DNA evidence examination or pre-amplification testing. Hoods were specifically allocated for each procedure (22). Gloves were changed between examination of individual items, between assays, and often several times during preparation of an assay. Workbenches, tools, and instruments were cleaned with 10% bleach followed by water and 70% ethanol before and after each procedure and between each item of evidence examined. A thorough weekly cleaning of the entire laboratory space and equipment was performed.
All consumables and water were irradiated in a Stratalinker® UV Crosslinker 2400 (Stratagene, La Jolla, CA, USA) prior to use (23). The strength of the UV light in the Stratalinker was tested weekly (25). Pipets were calibrated biannually and only aerosol-resistant filter tips were used. For manual processes, only 1 sample tube was opened at a time and either a clean cap opener or paper tissue was used. Tissue culture technique was employed in order to avoid crossing over an open tube or plate (21). To ensure that the sensitivity of the capillary electrophoresis (CE) machines was maintained, the heights of the 200 bp internal GeneScan® 500 LIZ® Size Standard (Applied Biosystems) injected with each amplification negative were monitored. Prior to using each electrophoresis capillary with samples, GeneScan® 500 LIZ® Size Standard was injected to confirm that the capillaries were free of exogenous DNA and functioning.
Moreover, analysts completed additional training and competency testing for the described procedures, and periodically performed LT-DNA testing on proficiency tests. For proficiency test purposes, if high amounts of DNA were recovered, extracts were diluted and the target amount of LT-DNA, 100 pg, processed.
Quality control of reagents
All reagents used in evidence examination, extraction, quantitation, amplification, and separation were tested with their respective assay to ensure they were functioning and free of contaminating DNA. The sensitivity of the ID kit was verified for each new kit or every 4 months with amplification of 100 pg, 25 pg, and 6.25 pg of DNA. The number of alleles determined for each concentration of DNA was compared with the expected results described in this study. If the anticipated results were not generated with a certain kit, it was not used.
Samples
Several sample sources were used in these studies. These included control DNA as indicated, buccal swabs and semen swabs from known donors, previously processed blood stored on stain cards, human bones, and items handled by known individuals. The DNA profiles of all of these sources were previously determined; thus, the profiles generated from this study could be compared and verified. Negative controls which consisted of irradiated (23) UltraPure DNase/RNase-Free Distilled Water (GIBCO Invitrogen, Carlsbad, CA, USA) were processed in the same or a more sensitive manner than their corresponding samples.
Recovery of DNA from touched evidence
Samples were swabbed with the New York City Office of Chief Medical Examiner’s swab (patent pending) pre-moistened with 0.01% sodium dodecyl sulfate (SDS). Swabbing was performed with a light touch and, if applicable, with the grain of the item. If needed, more than one swab per item or section of an item was used. DNA was extracted from swabs within 1-2 days of collection.
Extraction
Samples were incubated in 0.01% SDS and 0.72 mg/mL proteinase K at 56°C for 30 minutes with shaking at 1400 rpm, then at 99°C for 10 minutes without shaking. Following centrifugation, the digest was purified and concentrated with a Microcon® 100 (Millipore, Billerica, MA, USA) pretreated with 1 ng of poly-A RNA and eluted with 20 µL of water (26). Extracts were quantitated and amplified within 1-3 days of extraction. Some samples were extracted using Chelex resin (Bio-Rad, Hercules, CA, USA) (27).
Quantitation
Two microliters of sample were quantitated on the Rotor-Gene Q 3000® (Qiagen, Valencia, CA, USA) using an Alu-based real time PCR assay based on the method described by Nicklas and Buel (28), with the exception of the addition of 0.3 µL of 100X SYBR green I (Molecular Probes) and 0.525 mg/mL BSA in a 25 µL reaction volume. The concentrations of 2 known sources of human genomic DNA (Promega Corporation, Madison, WI, USA) used for the standards and calibrators were verified in triplicate 3 times for a total of 9 tests using a Synergy HT Multi-Mode Microplate Reader (BioTeq Instruments, Winooski, VT, USA). The human genomic DNA was stored at -20°C in single use aliquots. The dynamic range of the assay extends from 0.39 pg/µL to 1600 pg/µL and measurements are within 30% of their expected value. The no template control threshold was set at 0.1 pg/µL. In addition, for concordance studies for this quantitation assay, samples were measured with the Quantiblot slot-blot method (Applied Biosystems) according to the manufacturer’s protocol.
Amplification
Samples as indicated and negative controls were amplified in triplicate using the AmpFℓSTR® Identifiler PCR Amplification Kit according to the manufacturer’s recommendations with the exception of a 2-minute annealing time and a half-reaction volume with 2.5 U of TAQ for 31 cycles (14). All amplifications were carried out in the AB GeneAmp® PCR System 9700 thermal cycler (Applied Biosystems) within thin-walled 0.2 mL AB MicroAmp® (Applied Biosystems). AmpFℓSTR® Control DNA 9947A (Applied Biosystems) was amplified with the target amount, 100 pg of DNA.
Separation
Five or 6 μL of each PCR product and 0.375 µL of GeneScan® 500 LIZ® Size Standard (Applied Biosystems) were prepared with HIDI formamide (Applied Biosystems) for a total volume of 50 µL and injected at either 1 kV for 22 seconds (low), 3 kV for 20 seconds (normal), or 6 kV for 30 seconds (high) on the ABI Prism® 3100 Genetic Analyzer (Applied Biosystems). All 150 pg, 100 pg, and 75 pg samples were injected low; 50 pg, 25 pg, and 20 pg samples were injected normal; and 12.5 pg and 6.25 pg samples were injected high, unless otherwise indicated. For the low and the normal injections, 0.5 µL of allelic ladder and 1 µL of a 1/10 dilution of the 9947A positive control with 0.375 µL of LIZ were prepared with HIDI formamide in a total volume of 16 µL. For the high injection, 0.3 µL of allelic ladder and 1 µL of a 1/20 dilution of 9947A positive control were prepared in the same fashion. Data were collected using non-variable binning. In addition to injecting each sample replicate, as indicated, 5 µL of each of the 3 replicate PCR products were combined and mixed to form a pooled sample. Five microliters of this pooled sample was also injected.
Analysis
Data were analyzed with GeneScan® and Genotyper® Analysis software (Applied Biosystems) with a peak height threshold of 75 relative fluorescence units (RFU), a 251-baseline window, and a general filter of 10%. This filter removes labels from peaks that are less than 10% of the height of the highest peak at each locus. Stutter filters provided by the manufacturer were as follows: 6% (TPOX and THO1), 9% (D7S820 and CSF1PO), 10% (D13S317 and D5S818), 11% (D3S1358, vWA, and FGA), 12% (D8S1179), 13% (D21S11 and D16S539), 15% (D2S1338), 16% (D18S51), and 17% (D19S433). For single-source samples, stutter peaks were also removed if they were less than 20% of the height of the main peak. The consensus profile, also termed the composite profile, was generated including all alleles that were labeled in at least 2 of the 3 replicates.
Results were expressed as the percentage of alleles determined compared with the expected 32 alleles or the number of complete database loci determined. The locations searched in the US database include all those typed in the ID kit with the exception of D19S433, D2S1338, and Amelogenin. Loci were considered to be complete and not partial when they contained a heterozygote pair or a true homozygote allele. For samples amplified with less than 20 pg of DNA, when only 1 peak was observed at a locus that locus was always considered a potential false homozygote. The designation “Z” was used to indicate the possible presence of a second allele.
Results were also expressed as peak height ratios (PHR), defined as the smaller peak/larger peak, with perfect balance resulting in a peak height ratio of 1. Furthermore, stutter rates were calculated as follows: (number of alleles observed in the stutter position/number of true alleles detected) × 100. Unless otherwise indicated, when evaluating stutter, samples were analyzed without the application of the loci specific stutter filters or the 10% general filter. In order to avoid miscalculating ratios for both intralocus peak balance and stutter, only loci with alleles below the CE instrument’s saturation threshold were considered.
Results
Quantitation
The quantitative real time PCR assay used in our LT-DNA studies proved to be accurate and reliable. Over 5 assays, measurements of 28 samples of 50 pg/µL of standard DNA varied by a factor of 0.3 or less from the expected value. The standards included 9947A control DNA, Quantiblot standard A, and 2 sources of genomic DNA from Promega whose concentrations were verified previously with a spectrophotometer in triplicate 3 times for a total of 9 tests.
To determine the range of this precision, a buccal swab sample, previously measured in triplicate 3 times, was diluted by factors of 2 from 1000 pg/µL to 0.12 pg/µL and these concentrations were quantitated in triplicate. Measured values for the 1000 pg/µL, 500 pg/µL, and 250 pg/µL samples were within 33%, 36%, and 30% of their respective values. With less DNA input, measurements were more accurate, varying 21% and 11% from their expected values for the 125 pg/µL and 62.5 pg/µL samples, respectively. Considering that at most 5 µL of template volume may be added to our amplification reaction, the concentrations of DNA surrounding the range of LT-DNA testing, 31.25 pg/µL to 0.98 pg/µL, were also precise, measuring within 12 ± 5% of their expected values (Figure 1). This precision extended down to 0.24 pg/µL with a variance of 12%. However, the average measured value for 0.12 pg/µL was slightly more variable, within 45% of the expected amount.
Figure 1.

Sensitivity of the quantitative real time polymerase chain reaction assay within the low template DNA range. A buccal swab previously measured in triplicate 3 times was diluted from 1000 pg/µL to 0.12 pg/µL, as indicated. Results are depicted for samples diluted from 31.25 pg/µL to 0.98 pg/µL. Data for 1000 pg/µL to 62.5 pg/µL and 0.49 pg/µL to 0.12 pg/µL are not shown. Results are expressed as the mean ± standard deviation, where n = 3. The average value for each measured amount of DNA is labeled.
Furthermore, the reproducibility of the test was confirmed with 15 buccal samples measured in triplicate 3 times which varied at most 9.8% within an experiment and 11.17% between experiments (data not shown). Additional measurements were made of 15 blood and 15 semen samples in duplicate. Five of each of these samples were amplified with the same template amount and yielded comparable peak heights (data not shown).
Initial design
The duplicate and the triplicate amplification approaches were compared using 37.5 pg of template DNA distributed over 2 or 3 amplifications (data not shown). Using 3 amplifications, despite the lower amount of DNA amplified in each reaction, resulted in more confirmed alleles (29). Furthermore, in 33% of 24 buccal samples amplified with either 25 pg, 12.5 pg, or 6.25 pg, each peak of a heterozygote pair was observed as a single peak in 2 amplifications, while in the third amplification, both peaks were present. Employing the consensus approach with 3 replicates, rather than 2, would confirm both alleles. With both methodologies, however, spurious alleles were resolved. As demonstrated in the experiments below, the triplicate approach provides additional information that can be used for mixture and homozygote assessment. For example, in 17% of the 24 samples referred to above, the same single peak of a heterozygote pair was apparent in 2 amplifications whereas a third amplification revealed the other peak.
Reproducibility and precision for amplification and separation
In order to determine the precision and reproducibility of multiple injections of the allelic ladder, 16 preparations of allelic ladders were injected twice on the same day on 2 different instruments at 1 kV 22 seconds (low), 3 kV 20 seconds (normal), and 6 kV 30 seconds (high) parameters. The standard deviation of the average base pair size of the largest and the smallest alleles at each locus was on average between 0.04 and 0.07 bp for all conditions. Additionally, data collected for 24 ladders injected at high parameters on different days and instruments exhibited, on average, a variation of 0.47 bp. Yet, within an injection or between 2 injections performed on the same day, the precision was within 0.07 bp as above. For each study, all alleles were assigned correctly (data not shown).
The reproducibility of the modified PCR conditions was verified with amplifications of 9947A control DNA, as well as non-probative DNA extracts. Over a period of 4 months, 24 separate amplification products of 100 pg of 9947A control DNA injected with high, normal, and low injection parameters were evaluated. All samples generated the correct allelic determinations with no instances of allelic drop-in or dropout. Within each injection condition, the peak heights of alleles for each locus were comparable (data not shown).
Furthermore, 9947A control DNA and 5 different buccal sample extracts were amplified in triplicate on different days using 100 pg, 75 pg, 50 pg, 25 pg, 12.5 pg, and 6.25 pg of DNA. All 108 PCR products were injected on 2 different instruments at 3 kV for 20 seconds (data not shown). All samples generated the correct allelic determinations. Since the single-source 100 pg and 75 pg samples displayed saturated loci at this injection parameter, they were also injected with 1 kV and 22 seconds. Conversely, peaks below threshold were apparent with 12.5 pg and 6.25 pg samples, and thus they were injected with 6 kV and 30 seconds, resulting in at least a 2-fold increase in peak heights and one more called locus. This CE strategy of using different injection parameters based upon starting DNA amounts was adopted for all subsequent validation experiments.
Figure 2 illustrates the average number, over all replicates, of complete loci which are defined as either a heterozygote pair or a true homozygote. For all concentrations and samples, the amplification success for the replicates was consistent, as the average standard deviation for the number of loci detected was 1.03 loci on one instrument and 1.02 loci on another instrument. Larger amounts of template DNA produced smaller variances. Amplification of 12.5 pg and 6.25 pg of DNA had to be treated as potentially false homozygotes; therefore, fewer loci were deemed complete.
Figure 2.

Reproducibility of single amplifications with enhanced polymerase chain reaction conditions. Five different buccal extracts and 9947A control DNA were amplified in triplicate over a range of concentrations from 100 pg to 6.25 pg on different days. Individual replicates were evaluated for the number of complete loci defined as a heterozygote pair or a true homozygote. For samples with less than 20 pg of DNA, loci with single alleles are considered false homozygotes and are not complete. Results are expressed as the average ± standard deviation, where n = 3 for a total of 108 amplifications. Results for 9947A control DNA are represented by the diagonally striped columns and the 5 buccal extracts by the dotted, open, hashed, lined, and closed columns.
Sensitivity and allelic dropout
Amplification of LT-DNA samples typically produces stochastic phenomena such as allelic dropout and drop-in, intralocus peak imbalance, and increased stutter. For the following studies, these effects were evident with 100 pg of DNA or less and were magnified with amounts below 50 pg. 9947A control DNA samples and 5 buccal samples were amplified in triplicate with starting quantities of 100 pg, 75 pg, 50 pg, 25 pg, 20 pg, 12.5 pg, and 6.25 pg and were injected on 2 different instruments. Control sample 9947A was also amplified at 150 pg, but not at 20 pg. At least 92% of the expected alleles were determined in the composite or consensus profile for all samples amplified with 25 pg or more. Amplification of 12.5 pg and 6.25 pg generated 77% and 51% of the expected alleles, respectively (Figure 3). Most importantly, correct alleles were assigned for all samples at all loci.
Figure 3.

Percentage of alleles determined in the composite profile. 9947A control DNA and 5 different buccal sample extracts were amplified in triplicate at varying concentrations from 150 pg to 6.25 pg of DNA on different days. The number of alleles that repeated in 2 of the 3 replicate amplifications of each sample was compared with the total expected number of alleles, 32. Data are expressed as the average percentage ± standard deviation of the population, where n = 6 confirmed samples for 100 pg, 75 pg, 50 pg, 25 pg, 12.5 pg, and 6.25 pg; n = 2 for 20 pg; and n = 1 for 150 pg for a total of 117 amplifications.
Although allelic dropout occurred with smaller amounts of DNA, injecting these samples with high parameters yielded robust peak heights, often above 1000 RFUs (Figure 4). Since less sensitive injection conditions were required for greater amounts of DNA injected, peak heights within some loci did not vary significantly among concentrations. For example, peak heights for 75 pg samples were only 2% taller than those of 12.5 pg samples, although for single cell amounts, 6.25 pg, the difference was 30%. What was more striking to note was that for all concentrations, the peak heights of alleles at some loci, such as THO1, D16S539, and D2S1338, were considerably shorter than alleles labeled with the same color at other loci. It follows that different loci displayed varying degrees of allelic dropout. For the 25 pg samples, the larger loci, CSF1PO, D2S1338, D18S51, and FGA did not display either one or both alleles in 28% of the amplification replicates. Two loci, THO1 and D16S539, also showed high degrees of allelic dropout, 26.6% and 33.3%, respectively, regardless of their position as compared with 9.6% for the remaining loci. Regarding only the dropout rate for the second allele of a heterozygous pair, this was 2.2 times higher for THO1, D16S539, and the largest loci than for the others. These sensitivity titrations were used to formulate interpretation protocols ensuring proper recognition of allelic dropout and minimizing the risk of calling a false homozygote.
Figure 4.

Locus to locus peak height comparisons. Data were evaluated from 3 separate buccal samples amplified in triplicate on different days from 100 pg to 6.25 pg, as indicated. Values are expressed as the average peak height of heterozygote alleles for each locus + standard deviation of the population, where n = 9. Quantities amplified are represented as follows: 100 pg (solid), 75 pg (diagonal), 50 pg (open), 25 pg (horizontal stripes), 12.5 pg (vertical stripes), and 6.25 pg (dots). Letters in parenthesis represent dye colors. Only heterozygous loci were included.
Stochastic effects, spurious alleles, and stutter
Using the results of the sample amplifications described above, heterozygote peak imbalance was examined. One representative data set, shown in Figure 5, depicts 9947A control DNA amplified in triplicate from 150 pg to 6.25 pg and separated consecutively on 2 different CE instruments. Amplification of 100 to 150 pg of DNA did not demonstrate significant peak imbalance as PHRs were on average within 77 ± 18%. Specifically, the PHRs for 150 pg peaks were 70% to 99%. One hundred pictogram samples exhibited slightly more imbalance as the PHRs extended from 23% to 98%. Heterozygote pairs of 50 pg to 6.25 pg of DNA varied more, on average 62 ± 22%. Although the average imbalance for template amounts of 50 pg, 25 pg, 12.5 pg, and 6.25 pg was relatively similar – 66%, 60%, 57%, and 59%, respectively, the peaks varied more for the lower DNA inputs. The range of imbalance observed was from 7.5% to 98%. These PHRs were consistent between the 2 CE instruments. In summary, it was found that no sample amplified with more than 20 pg, where the second peak was less than 30% of the main peak, was actually a heterozygote. Although peak height ratios varied widely, the average value, including the standard deviation, was not below 30% (Figure 5). Only for samples amplified with less than 20 pg, was a true heterozygous peak observed to be less than 30% of the main peak, and this occurred infrequently with a rate of 8%.
Figure 5.

Peak imbalance within heterozygote loci. 9947A control DNA amplified in triplicate with 150-6.25 pg of DNA, as indicated, was injected consecutively onto 2 capillary electrophoresis (CE) instruments, CE 1 (closed circles) and CE 2 (closed squares). The average peak height ratio (PHR)±standard deviation, expressed as a percentage, was calculated for each concentration, where n = 30 loci for 150 pg, 100 pg, and 25 pg; n = 29 loci for 50 pg; n = 19 loci for 12.5 pg: and n = 8 loci for 6.25 pg.
Another concern arising from enhanced amplification protocols is the detection of extraneous sources of DNA. Data from 12 extraction negative controls and 10 amplification negative controls that had been amplified in duplicate or triplicate for a total of 57 replicates and were injected at various parameters, were examined for spurious alleles. Drop-ins occurred in 8% of the amplification negatives and 11% extraction negatives. However, implementation of the consensus approach addressed this concern as no alleles repeated. Regarding these non-repeating spurious alleles, the average number per replicate for all 57 amplifications was 0.7 ± 0.28. The average number of drop-ins over all 3 replicates for 22 controls was 0.9 ± 1.4. Considering only the 11 control samples that displayed drop-ins, the average number over all replicates was 1.8 ± 1.6.
For samples containing DNA, spurious alleles were detected at similar rates if peaks in the -4 stutter position were considered separately. The data set from the sensitivity study of 6 different samples amplified in triplicate at a range of concentrations from 100 pg to 6.25 pg for a total of 38 samples and 114 amplifications was examined. A foreign allele was detected in 28% of these amplifications. The majority of these peaks were in the -4 stutter position, totaling 18.4% of the replicates. The remaining foreign alleles, 9.6% of the total amplifications, were either in the +4 stutter position (6.1%) or in neither of these positions (3.5%).
Amplification of LT-DNA with additional cycles generated increased stutter. Using the same PCR conditions with the exception of the cycle number, the rate of stutter was compared between the 28-cycle high template DNA (HT-DNA) system and the 31-cycle LT-DNA system. Samples were analyzed without the locus-specific stutter filters or the general 10% filter. As shown in Table 1 and described by other authors (7), the occurrence of stutter increases for low amounts of DNA. Using our system, an increase of a factor of 2 was observed. However, the average height of the stutter peak relative to the main peak did not differ significantly. For example, only 3% of the LT-DNA stutter peaks observed were above 20%.
Table 1.
Comparison of the rate of stutter among high template DNA and 2 quantitative categories of low template DNA samples*
| Sample | Total amplifications examined | Alleles detected | No (%) of observed stutter peaks | No (%) of stutter not removed by filters | No (%) of stutter ≥20% |
|---|---|---|---|---|---|
| 28 cycles: 500 pg | 30 | 845 | 238 (28.2) | 0 (0.0) | 0 (0.0) |
| 31 cycles: 100 pg | 61 | 1664 | 974 (58.5) | 53 (5.4) | 8 (0.8) |
| 31 cycles: 20, 25, and 50 pg | 24 | 586 | 300 (51.2) | 32 (10.7) | 9 (3.0) |
*Data sets amplified with 500 pg for 28 cycles, 100 pg for 31 cycles, and 20 pg, 25 pg, or 50 pg with 31 cycles were examined for the presence of peaks in the stutter position. The rates of stutter were expressed as percentages and defined as follows: the percentage of alleles with stutter (the number of observed stutter peaks/the total number of true alleles detected over all loci) × 100, the percentage of stutter peaks not removed (number of stutter peaks above the locus specific filter thresholds and the 10% general filter/number of observed stutter peaks) × 100, and the percentage of stutter ≥20% (number of stutter peaks that were above or equal to 20% of the height of the main peak/number of observed stutter peaks) × 100.
Figures 6 and 7 depict the average and standard deviations of stutter percentages, as well as the maximum and minimum stutter values, for each locus for 2 quantitative categories of low template DNA. Although stutter averages do not vary between the amounts of DNA amplified, the range and the highest percentage of stutter were larger for some loci amplified below 50 pg. The tallest stutter peak for some loci, such as D3, could be artificially inflated by a dye artifact at that position which could contribute to the height. Nevertheless, these peaks should be noted as they would not be filtered by the software. Plus 4 stutter was rare, but was observed particularly with the 100 pg samples. Not considering the observed stutter, but rather all loci that could potentially stutter, the frequency of stutter approaches that observed with 28-cycle amplification of 100 pg of DNA (15). Specifically, 3.1% of the potential stutter events in samples amplified with 100 pg exceeded either the software cut-off values or the 10% general filter, the only filters applied to mixed samples. For the samples amplified from 50 pg or less, this value increased to 5.4%. These values varied by 1% if only the locus specific filters were applied.
Figure 6.

Locus-specific stutter percentages for 100 pg samples. Sixty one amplifications of 100 pg with Identifiler® for 31 cycles were analyzed without application of the locus-specific stutter filters or the 10% general filter. The following loci were examined: D8S1179 (n = 103), D21S11 (n = 93), D7S820 (n = 45), CSF1PO (n = 34), D3S1358 (n = 94), THO1 (n = 30), D13S316 (n = 93), D16S539 (n = 67), D2S1338 (n = 62), D19S433 (n = 81), vWA (n = 77), TPOX (n = 11), D18S51 (n = 69), D5S818 (n = 73), and FGA (n = 42). Results are expressed as the average percentage stutter represented by a closed square ± standard deviation. Maximum and minimum percentage stutter values were also designated by closed triangles and closed diamonds, respectively. Asterisk indicates that the maximum stutter value observed at D18S51 (60%) was outside the range of the graph.
Figure 7.

Locus-specific stutter percentages for 50 pg, 25 pg, and 20 pg samples. Twenty-four amplifications of 50 pg, 25 pg, and 20 pg with Identifiler® for 31 cycles were analyzed without application of the locus-specific stutter filters or the 10% general filter. The following loci were examined: D8S1179 (n = 37), D21S11 (n = 27), D7S820 (n = 13), CSF1PO (n = 19), D3S1358 (n = 32), THO1 (n = 10), D13S316 (n = 16), D16S539 (n = 25), D2S1338 (n = 18), D19S433 (n = 26), vWA (n = 21), TPOX (n = 6), D18S51 (n = 20), D5S818 (n = 18), and FGA (n = 12). Results are expressed as the average percentage stutter represented by a closed square ± standard deviation. Maximum and minimum percentage stutter values were also designated by closed triangles and closed diamonds, respectively. Asterisk indicates that the maximum stutter values observed at D7S820 (40%), D3S1358 (49%), and D16S539 (39%) were outside the range of the graph.
Evaluation of Identifiler® using the NIST SRM DNA Panel and non-probative case work samples
Ten samples from the National Institute of Standards and Technology standard reference material (NIST SRM) DNA Panel and 68 non-probative case work samples (15 buccal samples, 15 blood samples, 15 semen samples, and 23 bone samples) were amplified repeatedly and injected on 2 different instruments at the appropriate parameters. Initially, 1 allele at vWA was missing in one NIST standard sample. Accordingly, the sample was re-injected with high parameters and the allele was detected (this allele was also confirmed by re-amplification). The optimum amount of DNA, 100 pg, was amplified with the exception of 6 bone samples for which the amount of template DNA available for each amplification replicate was 10 pg, 20 pg, 25 pg, 30 pg, 40 pg, or 80 pg. On average, the composite profiles revealed that full profiles or 100% of the database loci were generated for the NIST, buccal, and semen samples. For the blood and the bone samples, 99.16% and 99.4% of the loci, respectively, were determined. No drop-in alleles were apparent in the composite profiles. Moreover, peak heights for each color, injection parameter, and instrument were consistent. Furthermore, 109 non-probative casework samples from touched or handled items or single fingerprints were also examined (30). At least 40 pg of DNA was recovered from 42% of these samples, with four containing sufficient DNA for HT-DNA testing. Thirty nine of the remaining samples were amplified with at least 10 pg of DNA in triplicate. There were 61.5% of the LT-DNA samples amplified which yielded database eligible profiles, of which 48.7% were single source and 12.8% were major contributors to mixtures (Table 2).
Table 2.
Profiles generated from DNA recovered from touched items*
| Sample group | No (%) of samples amplified | No (%) of database eligible samples in each group | No (%) of database eligible samples of total amplified (n = 39) |
|---|---|---|---|
| 10 pg – 19 pg – single source | 9 (23.1) | 2 (22.2) | 2 (5.1) |
| 10 pg – 19 pg – non-deducible mixtures | 1 (2.6) | N/A | N/A |
| 20 pg – 100 pg – single source | 18 (46.1) | 17 (94.4) | 17 (43.6) |
| 20 pg – 100 pg – deducible mixtures | 7 (17.9) | 5 (71.4) | 5 (12.8) |
| 20 pg – 100 pg – non-deducible mixtures | 4 (10.3) | N/A | N/A |
| Total | 39 (100.0) | N/A | 24 (61.5) |
*DNA in the low template DNA range was recovered from 39 samples from touched or handled items or single fingerprints. Sample groups were defined according to the amount of DNA amplified in each of 3 replicates and whether the resulting profiles were from 1 source or were mixtures wherein the major component could or could not be resolved. Data are expressed as the number or percentage of either samples amplified or database eligible profiles which contain at least 6 complete loci and 10 loci overall.
All consensus profile results from amplifications with known samples and 9947A control DNA described in this paper demonstrate concordance with the results from amplification of high template amounts of DNA of these same samples with standard protocols. Following validation approval, consensus profiles from forensic casework were also examined and were similarly deemed concordant if profiles from LT-DNA samples amplified with enhanced conditions were consistent with profiles from other HT-DNA samples in the case amplified with standard protocols. All sample comparisons (n = 319) were concordant.
Mixture studies
Four studies were performed to examine the behavior of contributors to LT-DNA mixtures. Dropout rates for a certain concentration of each contributor were higher than for the same concentration of DNA in a single-source sample. Mixtures were resolved according to the interpretation protocols developed from this study as described in the discussion section of this paper. Examination of the pooled sample revealed that the peak height ratios of heterozygote pairs were 25% more balanced than those of individual replicates for 25 pg samples. Accordingly, the pooled sample results better approximated the known mixture ratios, and thus were used to confirm allelic assignments. Moreover, if there appeared to be inconsistencies between the replicates and the pooled sample did not confirm the calls from the individual replicates, the locus was deemed inconclusive. However, the pooled samples were never used independently as an interpretation tool, as they sometimes contained alleles that did not repeat and thus a drop-in allele could potentially be assigned.
The first mixture study consisted of one series of Chelex 100 extracted samples mixed in ratios of 5:1, 3:1, and 1:1 with 100 pg, 50 pg, and 25 pg of total DNA. The second series of mixtures contained 100 pg of DNA mixed at 50:1, 20:1, 10:1, 5:1, 3:1, 1:1, 1:3, 1:5, 1:10, 1:20, and 1:50 ratios. For the third and forth series, samples extracted with the high sensitivity method were mixed at 49:1, 19:1, 9:1, 4:1, 2:1, 1:1, 1:2, 1:4, 1:9, 1:19, and 1:49 ratios with 100 pg of DNA. For all studies, mixtures were amplified in triplicate. Table 3 illustrates the number of complete and partial loci that were assigned to the major components of 100 pg mixtures from all of the series. At least 10 complete loci were deduced from mixtures with ratios from 1:50 to 1:4. Mixtures with the 1:3 and 1:2 ratios were partially resolved, with on average 3.7 complete and 6.4 partial loci. The 50 pg mixtures generated 11 and 8 complete loci from 1:5 and 1:3 mixtures, respectively. Although the 25 pg 1:5 and 1:3 mixtures yielded on average only 4 or 3 complete loci, 10 or 9 partial loci were deduced, respectively (data not shown).
Table 3.
Resolution of major components in 100 pg mixtures*
| Sample | Complete loci | Partial loci | Database loci | Inconclusive loci |
|---|---|---|---|---|
| 1:50a | 11 | 2 | 13 | 0 |
| 1:50b | 11 | 2 | 13 | 0 |
| 1:49c | 12 | 0 | 13 | 0 |
| 1:49d | 12 | 1 | 13 | 0 |
| 1:49e | 11 | 2 | 13 | 0 |
| 1:49f | 10 | 3 | 13 | 0 |
| 1:20a | 11 | 2 | 13 | 0 |
| 1:20b | 11 | 2 | 13 | 0 |
| 1:19c | 10 | 3 | 13 | 0 |
| 1:19d | 12 | 1 | 13 | 0 |
| 1:19e | 11 | 2 | 13 | 0 |
| 1:19f | 10 | 3 | 13 | 0 |
| 1:10a | 10 | 3 | 13 | 0 |
| 1:10b | 10 | 3 | 13 | 0 |
| 1:9c | 9 | 4 | 13 | 0 |
| 1:9d | 12 | 1 | 13 | 0 |
| 1:9e | 11 | 2 | 13 | 0 |
| 1:9f | 10 | 3 | 13 | 0 |
| 1:5a | 10 | 3 | 13 | 0 |
| 1:5b | 8 | 5 | 13 | 0 |
| 1:5g | 12 | 1 | 13 | 0 |
| 1:4c | 5 | 8 | –† | 7 |
| 1:4d | 10 | 1 | 13 | 0 |
| 1:4e | 6 | 1 | 9 | 4 |
| 1:4f | 5 | 8 | –† | 2 |
| 1:3a | 5 | 3 | –† | 5 |
| 1:3b | 4 | 7 | –† | 5 |
| 1:3g | 8 | 5 | 13 | 0 |
| 1:2c | 1 | 0 | –† | 9 |
| 1:2d | 3 | 10 | –† | 4 |
| 1:2e | 3 | 10 | –† | 4 |
| 1:2f | 2 | 10 | –† | 9 |
*100 pg mixtures of 6 buccal samples were prepared in ratios of 1:50 to 1:1 as indicated (1:1 mixtures not shown). Designations of “a,” “b,” “c,” “d,” “e,” “f,” and “g” correspond to the combination of samples used to prepare the mixtures. All mixture samples were amplified in triplicate.
†Deduced profiles had fewer than 6 complete loci.
All of these deductions were correct; however, the minor components could not be reliably deduced. Rather, peaks attributable to the minor component were apparent and some repeated for all concentrations and ratios tested. For example, the median number of repeating minor peaks detected in 100 pg mixed samples with 1:50-1:49, 1:20-1:19, 1:10-1:9, 1:5-1:4, and 1:3-1:2 ratios were 1, 3, 8, 13, and 13, respectively (Table 4). Similar results were observed for 50 pg and 25 pg mixtures, with a median of 12.5 minor repeating alleles observed (data not shown). Additionally, evaluation of all 4 sets of mixtures or 126 amplifications showed 176 repeating peaks. Of these, all but 2 peaks (1.1%) were either attributable to the major or minor components or were in the stutter position.
Table 4.
Detection of repeating minor component alleles in 100 pg mixtures*
| LOCI | D8 | D21 | D7 | CSF | D3 | THO1 | D13 | D16 | D2 | D19 | vWA | TPOX | D18 | Amel | D5 | FGA | Total repeating alleles |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1:50a | ● | ○ | N/A | ○ | ● | ○ | ○ | N/A | 2 | ||||||||
| 1:50b | N/A | ○ | ● | ○ | ● | N/A | N/A | ○ | 2 | ||||||||
| 1:49c | N/A | ● | ○ | 1 | |||||||||||||
| 1:49d | ○ | 0 | |||||||||||||||
| 1:49e | N/A | ○ | N/A | ● | N/A | N/A | 1 | ||||||||||
| 1:49f | ○ | N/A | ○ | N/A | N/A | N/A | ○ | ○ | N/A | N/A | 0 | ||||||
| 1:20a | ○ | ● | ○ | ● | ● | N/A | ● | ● | N/A | 5 | |||||||
| 1:20b | ○ | ● | ○ | N/A | ● | ● | ○ | N/A | N/A | ○ | 3 | ||||||
| 1:19c | ○ | ○ | ○ | 0 | |||||||||||||
| 1:19d | ○ | ● | ○ | ● | ○ | ○ | ● | ○ | ● | 4 | |||||||
| 1:19e | N/A | ○ | ○ | ● | ● | N/A | ○ | N/A | ○ | ○ | ● | N/A | 3 | ||||
| 1:19f | N/A | N/A | N/A | ○ | N/A | N/A | 0 | ||||||||||
| 1:10a | ● | ● | ● | ○ | ● | ● | N/A | ● | ● | ● | ● | N/A | ○ | 9 | |||
| 1:10b | ○ | ● | ● | ● | ● | ● | ○ | N/A | ○ | ● | ○ | ○ | N/A | N/A | ● | 7 | |
| 1:9c | ○ | ○ | ○ | ○ | ● | 1 | |||||||||||
| 1:9d | ● | ○ | ● | ○ | ● | ○ | ○ | ○ | ● | ● | ● | ○ | ● | 9 | |||
| 1:9e | ○ | N/A | ● | ○ | ● | ● | N/A | ● | ○ | N/A | ● | ● | ● | ● | N/A | 8 | |
| 1:9f | ○ | ● | ○ | ● | ○ | ○ | N/A | N/A | N/A | ○ | ● | N/A | N/A | 8 | |||
| 1:5a | ● | ● | ● | ● | ● | ● | ● | N/A | ○ | ● | ● | ● | ● | ● | N/A | ● | 13 |
| 1:5b | ● | ● | ● | ● | ● | ● | ● | N/A | ● | ● | ● | ● | ● | N/A | N/A | ● | 13 |
| 1:5g | ● | ● | ● | N/A | ● | ● | ● | ● | ● | ● | N/A | ● | ● | ● | ● | 13 | |
| 1:4c | ● | ● | ● | ● | ● | ● | ○ | ● | ● | ○ | ● | ● | ● | 9 | |||
| 1:4d | ● | ● | ● | ○ | ● | ● | ● | ● | ○ | ● | ● | ● | ● | ● | ● | ● | 14 |
| 1:4e | ● | N/A | ● | ○ | ● | ● | N/A | ● | ● | N/A | ● | ○ | ● | ● | ● | N/A | 10 |
| 1:4f | ● | ● | ● | ● | N/A | N/A | ● | N/A | ● | ● | ● | N/A | N/A | 8 | |||
| 1:3a | ● | ● | ● | ● | ● | ● | ● | N/A | ● | ● | ● | ● | ● | ● | N/A | ● | 14 |
| 1:3b | ● | ● | ● | ● | ● | ● | ● | N/A | ● | ● | ● | ● | ● | N/A | N/A | ● | 13 |
| 1:3g | ● | ● | ● | N/A | ● | ● | ● | ● | ● | ● | N/A | ● | ● | ● | ● | 13 | |
| 1:2c | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● | 13 |
| 1:2d | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● | 16 |
| 1:2e | ● | N/A | ● | ● | ● | ● | N/A | ● | ○ | N/A | ● | ○ | ● | ● | ● | N/A | 9 |
| 1:2f | ● | ● | ● | ● | ● | ● | N/A | N/A | ● | N/A | ● | ● | ● | ● | N/A | N/A | 11 |
*100 pg mixtures of 6 buccal samples were prepared in ratios of 1:50 to 1:1 as indicated (1:1 mixtures not shown). Designations of “a,” “b,” “c,” “d,” “e,” “f,” and “g” correspond to the combination of samples used to prepare the mixtures. All mixture samples were amplified in triplicate. Loci for each sample concentration were assigned a closed circle if a minor component allele repeated in 2 of the 3 amplification replicates, or an open circle if a minor allele was observed in only one replicate. “N/A” was assigned to loci for which the mixture components shared genotypes.
Discussion
Quantitation accuracy
Since the amount of template amplified is directly correlated with STR performance, the reliability of the quantitation system is essential to effectively triage a sample for enhanced PCR conditions. Equipped with a quantitative assay measuring within 30% of the actual amount of DNA, reproducibility studies demonstrated that our amplification protocols produce predictable results. However, based on the fact that our quantitation assay targets a 124 bp fragment of DNA, approximating the size of the smallest loci amplified with the ID system, amplification success for a degraded forensic sample would not be as accurately assessed. Additional screening assays to identify degraded DNA are warranted in order to better triage samples for amplification with mini-STRs (31). However, even with mini-STRs, the stochastic effects inherent to LT-DNA must still be considered.
Sample interpretation: allelic drop-ins
The first step in analysis is construction of the consensus profile, termed the composite profile, which contains only alleles that repeat in 2 of the 3 replicates. The advantage of 3 amplifications rather than 2 is correlated with the locus dropout rate and becomes obvious for lower amounts of DNA. If the dropout of the starting amount divided by 3 was very similar to the dropout rate divided by 2, then the success rate would be higher for the triplicate amplification method than for the duplicate method. Additional experiments to identify the exact threshold are in progress; however, given that many forensic touched samples are mixtures, a triplicate approach from the onset may prove most efficient.
Due to the increased sensitivity of the system, as is evidenced from our titration studies, drop-in alleles are expected and may originate from a very minor contributor to a sample. From our validation, these drop-in alleles did not display any repeated pattern; therefore, no conclusions could be drawn regarding the source of these alleles. The appearance of spurious alleles was random and they were only observed within a single sample tube and did not repeat in a control. Our data thus supports the assertion by Gill et al (7) that non-repeating peaks in the negative controls do not affect their corresponding samples, as they are likely to have originated from a single molecule, which, by definition, can only exist in a single tube. Yet, to account for the randomness of drop-ins, more controls are required and should be amplified in triplicate along with case samples to determine whether drop-ins are reproducible, indicating global contamination. These negative controls should also be injected at the same or more sensitive injection parameters than their associated case samples.
An approximate 1:3 ratio was maintained between the samples and the negative control replicates, considering the extraction, additional purification, and amplification controls, all of which were amplified in triplicate. A sample batch fails if one of the negative control sets displays more than 2 repeated alleles. In the event of only 1 or 2 repeated peaks in the negative control, the affected loci will be deemed inconclusive in the associated samples. As described in the results section, the validation showed that, even with protocols in place to minimize laboratory sources of drop-ins, we expect to observe some spurious alleles in our negative controls. Nevertheless, too many non-repeating peaks over 3 amplifications will also result in a failed batch, although these peaks may be isolated to that control tube and not indicate gross contamination.
Mixture assessment
If 3 confirmed alleles are present at only one locus in a sample where all other loci show 2 confirmed alleles at most, although this allele may be due to a second contributor to the sample, no conclusions can be drawn regarding the source of this extra allele. For example, the 1:50 and 1:49 mixtures displayed only 2 loci with repeating minor peaks that were significantly shorter than the main peaks. The major component peaks behaved in the same manner as single-source samples. Therefore, samples with less than 3 repeating peaks may be interpreted with the protocols for single-source samples with the following exception. Mixtures amplified from very small amounts of DNA, for example <20 pg, as observed within the touched items study, may not contain sufficient DNA to generate repeating peaks, but there may be inconsistencies among the replicates such that the possibility of a mixture should be considered. In these instances, the samples should be analyzed with mixture interpretation protocols as described below.
Stutter filters
The threshold where an analyst could remove stutter peaks from single-source samples, following locus-specific stutter filtering performed by the software, was set at 20%. Although occasionally higher stutter was observed, this value was chosen to prevent the removal of real allele peaks. Manual stutter removal was not permitted for mixed samples. Interestingly, the overwhelming majority of non-repeating foreign peaks observed in our samples were in the stutter position. There is the possibility that a non-repeating peak in the stutter position is both a drop-in peak and a stutter peak and that either one or both would not be detectable if they did not occur concurrently. Due to the low incidence of allelic drop-in observed in our negative controls and the expectation of a higher fidelity rate in samples rather than in negative controls, attributable to the competing template DNA, these peaks are likely stutters. Nevertheless, to take a conservative approach, and since the level of peak imbalance precluded adjusting the stutter filters further, these concerns were addressed with the interpretation protocols as discussed below.
Heterozygote assignment for single-source samples
A locus was considered to be heterozygous based on the 2 highest repeating peaks in 2 amplifications. Due to the wide range of peak height differences, the assignment was not limited by an imbalance threshold with a few exceptions. To accommodate the range of stutter seen, if a repeating allele was in the plus or minus 4 bp stutter position, and was less than 30% of the major peak height in 2 out of 3 amplifications, the possibility of a homozygote was considered, and a “Z” assigned to indicate the possible presence of a second allele. The peak was considered a true heterozygote peak if the potential stutter peak was larger than 50% of the main allele in the third amplification (Figure 8). Furthermore, if 2 repeating peaks were clearly major peaks, any additional minor repeating alleles were not considered in the profile. These peaks were often in the stutter position as observed in our evaluations of spurious alleles or could have been from a very minor contributor to the sample.
Figure 8.
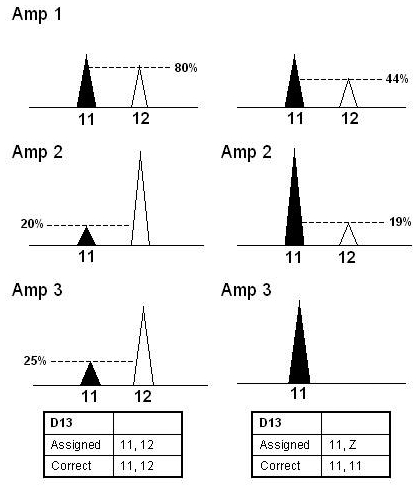
Examples of repeating peaks in stutter position for single-source samples. This schematic shows 2 scenarios where a repeating peak is in the plus or minus four stutter position. The peak height ratio should be evaluated to determine the genotype. If the second repeating peak is less than 30% of the major peak in 2 out of 3 amplifications, the possibility of a homozygote should be considered and a “Z” assigned. If however, the second repeating peak is greater than 50% in one of the 3 amplifications, a heterozygote pair can be assigned.
Homozygote assignment for single-source samples
Homozygote types have to be interpreted with caution to avoid assigning potential false homozygotes. As evidenced from the sensitivity studies, due to the enhanced peak heights resulting from high injection parameters, peak height thresholds were not effective tools to discern true homozygote alleles. Rather, homozygotes were correctly assigned according to the following protocols based on peak height imbalance as well as the locus and quantity specific dropout rates presented previously. An allele had to appear in all 3 amplifications to be considered a homozygote. The presence of an additional allele in one of the 3 amplifications can be indicative of allelic dropout in the other 2 replicates. However, if one peak was clearly the major peak and the minor peaks (even repeating minor peaks) were less than 30% of the major peak height in all 3 amplifications, the allele was considered a true homozygote. Alternatively, if the non-repeating minor drop-in was greater than 30% of the repeating peak, allelic drop out was suspected and the locus marked with a “Z”, to indicate the possible presence of a second allele (Figure 9).
Figure 9.
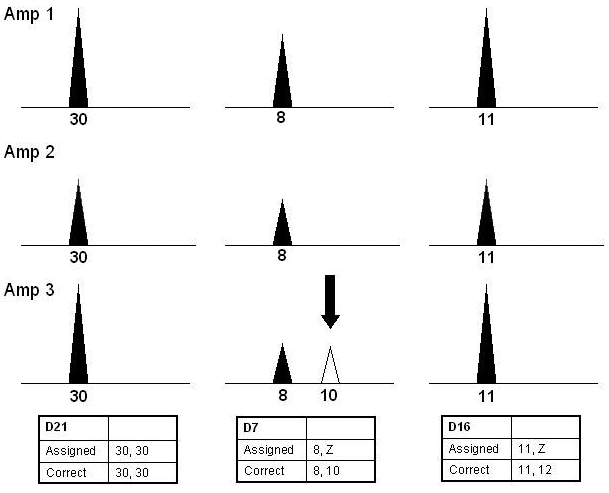
Example of replicate amplifications for potential false homozygotes. This schematic shows 3 scenarios of potentially homozygous loci. For samples amplified with more than 20 pg of DNA, a homozygote type may be assigned to a single peak appearing in all replicates at a robust locus such as D21S11 above. In a case of drop-in, such as the example at another robust locus, D7S820, a conservative approach is taken given that a second peak is greater than 30% of the height of the first. Depicted in the third example, allelic dropout is always considered for all high molecular weight loci (CSF, D2S1338, D18S51, and FGA), as well as the loci identified as less efficient (D16S539 and THO1). In these cases a “Z” is assigned to indicate that a second allele may be present.
Our validation demonstrated that the largest locus detected in each color, particularly for samples amplified in the lower LT-DNA range, as well as THO1 and D16S539, often appeared as false homozygote loci. To be more conservative, allelic dropout was therefore always considered for the last locus detected in each color and for THO1 and D16S539. In addition, due to the rate of dropout for samples with less than 20 pg, all apparently homozygous loci in samples amplified with this amount of DNA were assigned a “Z”. Care was also exercised if alleles in one of 3 amplifications were completely different from those in the first 2 amplifications. In this case, the locus was deemed inconclusive.
Evaluation of mixed samples
For mixed samples, only clear major components can be interpreted unless 1 of the DNA sources is known, for example an intimate sample. According to the mixture studies, mixtures with apparently equal ratios of contributors can only be used for direct comparison. Samples with varying ratios of contributors at several loci also may only be used for direct comparison. The number of contributors to a sample should also be considered for interpretation. A sample may have 3 or more contributors if 5 or more repeating alleles are present in at least 2 loci. If there is a clear major component, it may be possible to deduce the major contributor from a 3-person mixture. Often, this is not feasible at all of the loci. For example, some degraded samples appear to be 3-person mixtures at the small loci, but the minor component(s) are not visible at the large loci.
Heterozygote assignment of major components in mixtures
In these studies, alleles were correctly assigned to a major component if they appeared in all 3 amplifications and if they were the major peaks in 2 of the 3 amplifications. A heterozygote pair for the major component was called if 2 of the 3 amplifications showed a peak balance greater than or equal to 50% (Figure 10A). During the validation, examination of peak imbalance in single-source samples indicated that for more than 99% of the replicates and locations tested no more than 1 replicate for each locus showed peak imbalance greater than 50%. Therefore, if major peaks were within 50% of one another in at least 2 replicates, a heterozygote pair was likely, which proved to be empirically correct.
Figure 10.
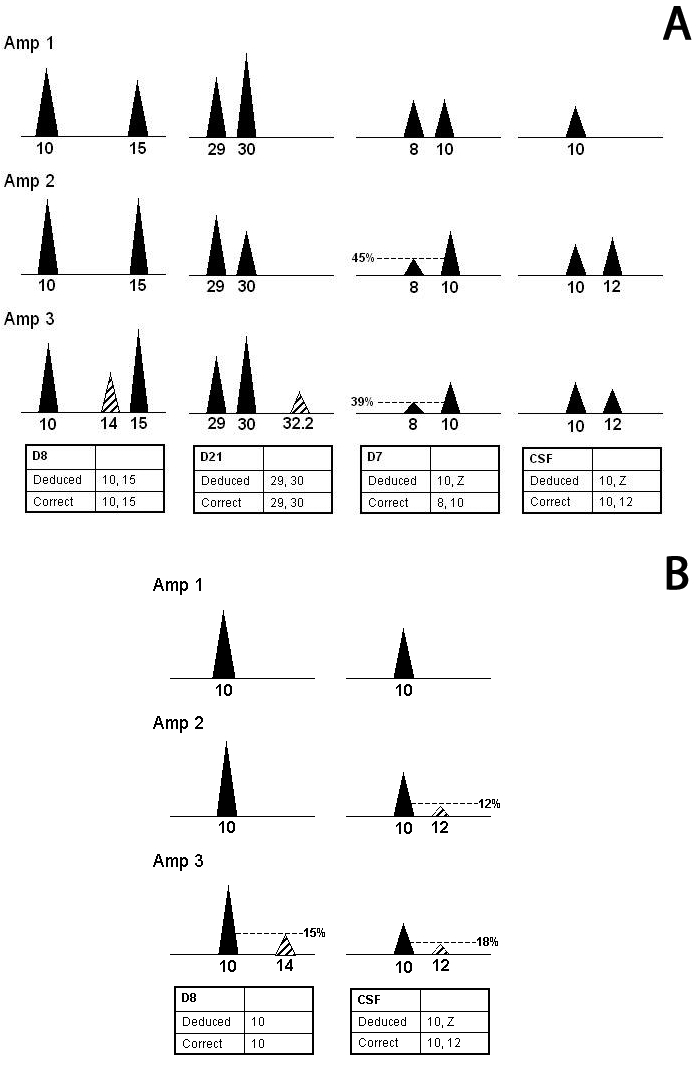
Deduction of major components of mixed samples. This schematic shows scenarios for assignment of major component genotypes. (A) Alleles can be assigned to a major component if they appear in all 3 amplifications, are the major peaks in 2 out of the 3, and the pair shows a peak balance ≥0.5. (B) Illustration of the possibility of a true homozygote if all additional peaks are less than 30% of the main peak and the locus is robust. However, for non-robust-loci, the possibility of a false homozygote must be considered and a “Z” is assigned. In the above examples, all peaks are within 50% of each other unless otherwise indicated. Striped peaks represent minor component alleles.
In order to ensure correct allelic pairing, care should be taken to not consider saturated peaks in ratio calculations. For degraded samples, high injection parameters may be required to detect long amplicons, whereas alleles at small loci may saturate the AB 3100 detector and are not suitable for ratio calculations. In these instances, one may combine injection results as long as data from overlapping loci are generated.
Regardless of the peak balance, if it was not apparent that a sample had a ratio of at least 1:3, then all loci that contained only 2 alleles were considered possible mixed loci. This was particularly relevant for the less robust loci and TPOX, a locus prone to primer binding site mutations (32) and applicable to mixtures containing a homozygote and a heterozygote which share the same allele. Note also that the contribution of the minor component to a peak shared with the major component is not subtracted prior to calculating the peak imbalance. Therefore, in a mixed sample, the peak imbalance may seem exaggerated, but this serves to ensure a more conservative approach.
Homozygote assignment of major component in mixtures
As evidenced in these studies, homozygote genotypes had to be deduced carefully. If 1 peak was clearly the major peak and the minor peaks (even if they repeated) were less than 30% of the major peak in all 3 amplifications, an allele was assigned as a true homozygote. In between cases, where the height of the second allele was between 30% and 50% of the height of the main peak, it was not clear whether the major contributor was heterozygous or homozygous. In this case, a major peak was assigned to the major component along with a “Z” (Figure 10B).
Based on validation data, as for single-source samples, if only one peak could be called, allelic dropout and a “Z” were considered for the high molecular weight or less efficient loci. This therefore affected CSF1PO, THO1, D16S539, D2S1338, D18S51, and FGA or the last evident locus of a particular color in 2 of 3 amplifications in a degraded sample. Additionally, a “Z” was always assigned at TPOX for the reason discussed above. As with single-source samples, for amplifications with less than 20 pg, all potential homozygotes at all loci were considered false and a “Z” was assigned.
Additional mixture interpretation issues
When alleles could not be assigned to the major component, the locus was not deduced and was deemed inconclusive. Minor components could not be reliably determined. The degree of allelic sharing within a mixture was not readily evident as replicate amplifications displayed shifts in ratios between various mixture alleles within the same locus. Nevertheless, minor peaks may be used for comparison. From our mixture studies, 98.9% of the non-repeating peaks were either attributable to the major or the minor components or were in stutter position and thus were likely not due to random events. However, for extreme mixtures often very few minor alleles are detected, and these samples may not be suitable for comparison to the minor component.
All mixtures, as well as single-source interpretation decision points and guidelines, were summarized in a flowchart (Figure 11). In addition to applying these protocols to the replicates, the pooled sample should be considered. Although the pooled sample is not evaluated independently, if it does not confirm the allelic assignments from the replicates, caution should be exercised.
Figure 11.
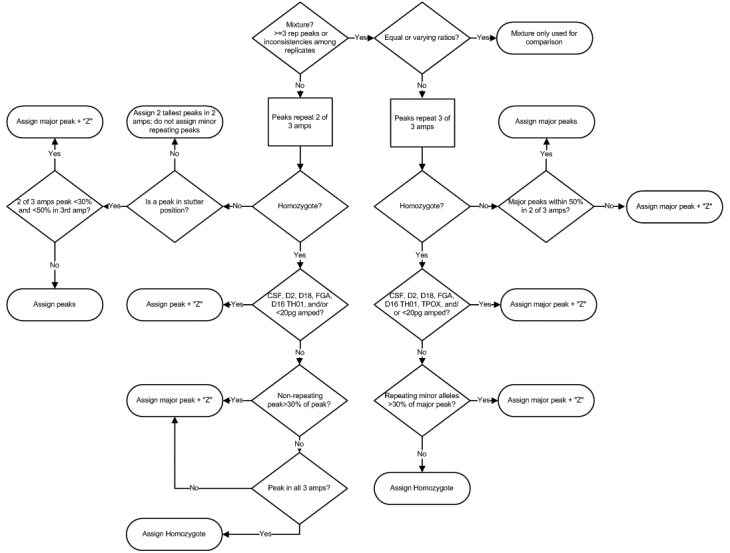
High sensitivity interpretation decision points. This flowchart summarizes the low template DNA interpretation process.
Profile comparison and statistical evaluation
For the purposes of statistical evaluation for single-source or deduced major component profiles when calculating the random match probability, only the assigned alleles that meet the interpretation requirements should be used. For loci assigned a “Z,” only one allele is considered and the random match probability is calculated as 2p. For mixtures where the profile of the major component cannot be determined, a known sample may be compared and qualitative conclusions can be drawn. If the known sample’s alleles are labeled at all loci where DNA alleles are repeating, the probability of exclusion of the individual from the mixture may be calculated using the Random Man Not Excluded method.
Since allelic dropout caused by stochastic effects is a common occurrence for LT-DNA samples, the absence of a comparison sample’s alleles from a mixture is not necessarily indicative of an exclusion. An analyst should consider whether the alleles detected are what one would expect to see had an individual contributed to the mixture. The amount of DNA amplified, the number of contributors to the mixture, the empirically defined loci characteristics, and the length of the repeats should be evaluated. The phenomena of allelic drop-in and dropout must be taken into account when evaluating the weight of the missing allele(s). Statistical methods to achieve this purpose are in the process of being refined and validated for forensic casework (33,34). In the absence of such validated statistical methods, qualitative results may still be provided. Once likelihood ratio methods are validated, they could also be applied to the scenarios described above.
Conclusion
Through extensive testing of low template single-source and mixed samples, we have developed quality control, testing, and interpretation protocols. These protocols were designed to address the concerns regarding the increased sensitivity of this system and the accompanying stochastic effects (20). Although not every situation can be anticipated, employing these conservative interpretation protocols and evaluating the sample as a whole including the pooled injection ensures that allelic assignments, when they can be made, are correct.
The detection of an individual’s alleles in an evidence profile, whether it be from skin cells recovered from an item or from a blood, saliva, or semen stain, simply indicates that their DNA may be present. No inferences about the means of deposition should or can be made. Conversely, in the case of handled items, the absence of an individual’s alleles does not indicate that this individual did not touch the object; rather the DNA might not have been detectable. Very partial profiles must be interpreted carefully and they may not be suitable for comparison. The weight of the evidence and the absence or presence of DNA is dependent upon the context of the case. It is for the finders of fact to assign the significance of these results.
These concerns do not impact the reliability of high sensitivity DNA testing and the interpretation protocols described in this paper. Our studies demonstrated that these methodologies generated robust and reliable results which may prove valuable depending upon the context of the case. Therefore, implementation of these methods, or others suitably verified, in conjunction with an appropriate quality control program, ensures that LT-DNA testing is fit for forensic purposes.
Acknowledgments
The authors thank Adele Mitchell for reviewing the manuscript.
References
- 1.Wickenheiser RA, Roney JM, Hummel KHJ, Szakacs NA, MacMillan CE, Kuperus WR. Unusual exhibit material yielding successful DNA profiles using PCR STR typing. In: Proceedings of the 11th Annual Symposium on Human Identification; 1999 Sep 29-Oct 3; Lake Buena Vista, FL, USA. Madison (WI): Promega Corporation; 1999. [Google Scholar]
- 2.Hummel KHJ, Wickenheiser RA, Roney JM, Szakacs NA, MacMillan CE, Kuperus WR. Update on unusual exhibit material yielding successful DNA profiles using PCR STR typing. In: Proceedings of the 4th Annual Cambridge Health Institute’s DNA Forensics Conference; 2000 May 31-Jun 2; Springfield, VA, USA. Madison (WI): Promega Corporation; 2000. [Google Scholar]
- 3.Szakacs NA, MacMillan CE, Roney JM, Wickenheiser RA, Hummel KHJ, Kuperus WR. Perspectives on DNA casework: unusual exhibits, mixture interpretation, and profiles from inhibited PCR reactions. In: Proceedings of the 11th Annual Symposium on Human Identification; 2000 Oct 10-13, Biloxi, MI, USA. Madison (WI): Promega Corporation; 2000. [Google Scholar]
- 4.Wickenheiser RA. Trace DNA: a review, discussion of theory, and application of the transfer of trace quantities of DNA through skin contact. J Forensic Sci. 2002;47:442–50. [PubMed] [Google Scholar]
- 5.Gill P. Application of low copy number DNA profiling. Croat Med J. 2001;42:229–32. [PubMed] [Google Scholar]
- 6.Sewell J, Quinones I, Ames C, Multaney B, Curtis S, Seeboruth H, et al. Recovery of DNA and fingerprints from touched documents. Forensic Sci Int; Genet. 2008;2:281–5. doi: 10.1016/j.fsigen.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Gill P, Whitaker J, Flaxman C, Brown N, Buckleton J. An investigation of the rigor of interpretation rules for STRs derived from less than 100 pg of DNA. Forensic Sci Int. 2000;112:17–40. doi: 10.1016/S0379-0738(00)00158-4. [DOI] [PubMed] [Google Scholar]
- 8.Kloosterman AD, Kersbergen P. Efficacy and limits of genotyping low copy number (LCN) DNA samples by multiplex PCR of STR loci. J Soc Biol. 2003;197:351–9. doi: 10.1051/jbio/2003197040351. [DOI] [PubMed] [Google Scholar]
- 9.Staiti N, Giuffre G, Di Martino D, Simone A, Sippelli G.Tuccari G, et alMolecular analysis of genomic low copy number DNA extracted from laser-microdissected cells. Int Congr Ser 20061288568–70. 10.1016/j.ics.2005.11.126 [DOI] [Google Scholar]
- 10.Anjos MJ, Andrade L, Carvalho M, Lopes V, Serra A, Oliveira C, et al. Low copy number: Interpretation of evidence results. Int Congr Ser. 2006;1288:616–8. doi: 10.1016/j.ics.2005.09.118. [DOI] [Google Scholar]
- 11.Roeder AD, Elsmore P, Greenhalgh M, McDonald A. Maximizing DNA profiling success from sub-optimal quantities of DNA: a staged approach. Forensic Sci Int; Genet. 2009;3:128–37. doi: 10.1016/j.fsigen.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Findlay I, Taylor A, Quirke P, Frazier R, Urquhart A. DNA fingerprinting from single cells. Nature. 1997;389:555–6. doi: 10.1038/39225. [DOI] [PubMed] [Google Scholar]
- 13.Applied Biosystems, Human Identification Department. AmpFℓSTR® Identifiler™ PCR amplification kit user’s manual. Foster City (CA): Applied Biosystems; 2001. [Google Scholar]
- 14.Bajda E, Shaler R, Prinz M, Caragine T. Modification of amplification and STR conditions to enhance sensitivity of the Identifiler® (AB) and PowerPlex® 16 (Promega) kits for low copy number DNA samples. In: Proceedings of the 15th Annual International Symposium on Human Identification; 2004 Oct 4-7; Phoenix, AZ, USA. Madison (WI): Promega Corporation; 2004. [Google Scholar]
- 15.Smith PJ, Ballantyne J. Simplified low-copy-number DNA analysis by post-PCR purification. J Forensic Sci. 2007;52:820–9. doi: 10.1111/j.1556-4029.2007.00470.x. [DOI] [PubMed] [Google Scholar]
- 16.Forster L, Thomson J, Kutranov S. Direct comparison of post-28-cycle PCR purification and modified capillary electrophoresis methods with the 34-cycle “low copy number” (LCN) method for analysis of trace forensic DNA samples. Forensic Sci Int; Genet. 2008;2:318–28. doi: 10.1016/j.fsigen.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Strom CM, Rechitsky S. Use of nested PCR to identify charred human remains and minute amounts of blood. J Forensic Sci. 1998;43:696–700. [PubMed] [Google Scholar]
- 18.Hanson EK, Ballantyne J. Whole genome amplification strategy for forensic genetic analysis using single or few cell equivalents of genomic DNA. Anal Biochem. 2005;346:246–57. doi: 10.1016/j.ab.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 19.Whitaker JP, Cotton EA, Gill P. A comparison of the characteristics of profiles produced with the AMPFlSTR SGM Plus multiplex system for both standard and low copy number (LCN) STR DNA analysis. Forensic Sci Int. 2001;123:215–23. doi: 10.1016/S0379-0738(01)00557-6. [DOI] [PubMed] [Google Scholar]
- 20.Budlowle B, Hobson D, Smerick J, Smith J. Low copy number-consideration and caution. In: Twelth International Symposium on Human Identification; 2001 Oct 9-12, Biloxi, MI, USA. Madison (WI): Promega Corporation; 2001. [Google Scholar]
- 21.Davis JM, editor. Basic cell culture: A practical approach. Oxford (UK): Oxford University Press; 1994. [Google Scholar]
- 22.Kwok S, Higuchi R. Avoiding false positives with PCR. Nature. 1989;339:237–8. doi: 10.1038/339237a0. [DOI] [PubMed] [Google Scholar]
- 23.Tamariz J, Voynarovska K, Prinz M, Caragine T. The application of ultraviolet irradiation to exogenous sources of DNA in plasticware and water for the amplification of low copy number DNA. J Forensic Sci. 2006;51:790–4. doi: 10.1111/j.1556-4029.2006.00172.x. [DOI] [PubMed] [Google Scholar]
- 24.Shaw K, Sesardic I, Bristol N, Ames C, Dagnall K, Ellis C, et al. Comparison of the effects of sterilisation techniques on subsequent DNA profiling. Int J Legal Med. 2008;122:29–33. doi: 10.1007/s00414-007-0159-5. [DOI] [PubMed] [Google Scholar]
- 25.Stratalinker® UV Crosslinker Instruction Manual, Model 2400 Catalog #400075 (120 V), #400076 (230 V) and #400676 (100 V). Revision #122003 IN #70034-06. La Jolla (CA): Stratagene Copyright; 2002. [Google Scholar]
- 26.Schiffner LA, Bajda EJ, Prinz M, Sebestyen J, Shaler R, Caragine TA. Optimization of a simple, automatable extraction method to recover sufficient DNA from low copy number DNA samples for generation of short tandem repeat profiles. Croat Med J. 2005;46:578–86. [PubMed] [Google Scholar]
- 27.Walsh PS, Metzger DA, Higuchi R. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques. 1991;10:506–13. [PubMed] [Google Scholar]
- 28.Nicklas JA, Buel E. Development of an Alu-based, real-time PCR method for quantitation of human DNA in forensic samples. J Forensic Sci. 2003;48:936–44. [PubMed] [Google Scholar]
- 29.Caragine T, Bajda E, Sebestyen J, Tamariz J, Prinz M. A High Sensitivity DNA testing methodology. problems and proposed solutions. In: The 4th European-American School in Forensic Genetics and Mayo Clinic Course in Advanced Molecular and Cellular Medicine, 2005; Dubrovnik, Croatia. Zagreb: Exto Produkcija; 2005. [Google Scholar]
- 30.Prinz M, Schiffner L, Sebestyen JA, Bajda E, Tamariz J, Shaler RC, et al. Maximization of STR DNA typing success for touched objects. Int Congr Ser. 2006;1288:651–3. doi: 10.1016/j.ics.2005.10.051. [DOI] [Google Scholar]
- 31.Dixon LA, Dobbins AE, Pulker HK, Butler JM, Vallone PM, Coble MD, et al. Analysis of artificially degraded DNA using STRs and SNPs – results of a collaborative European (EDNAP) exercise. Forensic Sci Int. 2006;164:33–44. doi: 10.1016/j.forsciint.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 32.Kline MC, Jenkins B, Rodgers S. Non amplification of a vWA allele. J Forensic Sci. 1998;43:250. [Google Scholar]
- 33.Gill P, Kirkham A, Curran J. LoComatioN: a software tool for the analysis of low copy number DNA profiles. Forensic Sci Int. 2007;166:128–38. doi: 10.1016/j.forsciint.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 34.Gill P, Curran J, Neumann C, Kirkham A, Clayton T, Whitaker J, et al. Interpretation of complex DNA profiles using empirical models and a method to measure their robustness. Forensic Sci Int; Genet. 2008;2:91–103. doi: 10.1016/j.fsigen.2007.10.160. [DOI] [PubMed] [Google Scholar]