

Abstract
Background
Since the causative gene linked to PARK8 parkinsonism was identified as LRRK2, LRRK2 gene mutations have been found to occur in about 4% of patients with hereditary Parkinson disease (PD); this percentage is even higher in certain populations. Moreover, no clear clinical differences between PARK8-linked parkinsonism and sporadic PD have been identified. Neuropathologic findings have been diverse in PARK8 parkinsonism, but few of the clinicopathologic examinations have been performed in the same family tree. We aimed to describe PET and neuropathologic findings in members of the same family tree with PARK8 parkinsonism.
Methods
We conducted PET of 2 subjects and neuropathologically examined 8 subjects in the same family from the Sagamihara district, the original source of PARK8-linked parkinsonism (I2020T mutation).
Results
The results of the PET scans were virtually identical to those seen in sporadic PD. The neuropathologic study results showed pure nigral degeneration with no Lewy bodies in 6 cases. One case, however, showed the presence of Lewy bodies and was similar neuropathologically to conventional PD with Lewy bodies. Another case had multiple system atrophy pathology.
Conclusions
Our study of PARK8-linked parkinsonism affecting several members of the same pedigree shows that the same gene mutation can induce diverse neuropathologies, even if the clinical picture and PET findings are virtually identical.
Keywords: nigral degeneration, PARK8, PET
Introduction
Parkinson disease (PD), a common neurodegenerative disorder, is becoming increasingly prevalent as the population ages. Of the various approaches used to identify factors that lead to PD, genetic analyses have been extremely important for etiologic studies of PD, including investigations of the causal genes for familial parkinsonism belonging to the PARK series.
Since we first discovered the gene locus of PARK8 parkinsonism in a Japanese family from the Sagamihara district (the Sagamihara family) [1,2], and the causative gene named LRRK2 was also identified [3,4], the frequency of the LRRK2 gene in hereditary PD has been estimated to be approximately 4% and is even higher in populations including North African Arabs and Ashkenazi Jews [5]. PARK8-linked parkinsonism has drawn much attention in studies of Western familial parkinsonism. On the basis of its predicted amino acid sequence, the product of the LRRK2 gene, leucine-rich repeat kinase 2, may have multiple domains and be a multifunctional protein. The clinical picture of PARK8-linked parkinsonism is quite homogeneous and is identical to that of sporadic parkinsonism, but its neuropathology is heterogeneous. We previously reported that the gene mutation in LRRK2 in the Sagamihara family resulted in the protein modification Ile2020Thr (I2020T), and we briefly presented neuropathologic findings of 5 cases [6]. Here, we describe PET results from 2 members of this family and autopsy results from 8 members, and we discuss the multiplicity of neuropathologic features present.
Overview of the Clinical Features of the Sagamihara Family
The clinical characteristics of the Sagamihara family members were first described in 1978 [7]. It is difficult to distinguish this familial disease clinically from sporadic PD. Age at onset, initial symptoms, response to levodopa and dopamine agonists, and clinical course are the same as in sporadic PD. The mean age at onset is 56 years (range, 38–74 years), and the initial symptoms are mainly rest tremor and gait disturbance. Gait disturbance is more frequent and tremor is less frequent than in sporadic PD and other PARK8 pedigrees. Autonomic symptoms are milder. Advances in medical therapy have greatly improved the prognoses for recent generations and currently living patients with Sagamihara parkinsonism; most patients respond well to levodopa and have symptoms of long-term levodopa use such as motor fluctuations. However, apparent dementia, cognitive dysfunction, and depression are very rare. The mean duration of the disease is greater than 25 years, and most of the patients who have died were older than 70 years. The immediate causes of death were mainly infections such as aspiration pneumonia, cardiovascular disease, or malignant neoplasm.
In patients with Sagamihara parkinsonism, olfactory function varies, ranging from slightly impaired to anosmia. 123I-meta-iodobenzylguanidine (123I-MIBG) myocardial scintigraphy shows that the uptake ratio in most of these patients ranges from normal to slightly decreased (unpublished results). If either olfaction or the 123I-MIBG uptake ratio is decreased, the extent of the decrease is generally mild.
Methods
PET Scans
Two patients from the Sagamihara family who already had overt PARK8-linked parkinsonism underwent PET at the University of British Columbia Pacific Parkinson’s Research Centre. The PET procedures were identical to those previously reported [8] and used the following nuclides: 18F-6-fluoro-L-dopa to assess dopamine synthesis and storage, 11C-α-dihydrotetrabenazine (11C-DTBZ) for the vesicular monoamine transporter, 11C-d-threo-methylphenidate for the membrane dopamine transporter, and 11C-raclopride for postsynaptic striatal D2 receptor function. Methods for tracer chemistry, PET studies and image analyses, and statistical analysis also were identical to those previously reported [8].
Neuropathologic Examinations
Neuropathologic examinations were performed using conventional methods. Each brain was fixed in 10% formalin solution, and representative portions were embedded in paraffin wax. Thin sections (6-μm) were stained with hematoxylin-eosin, Klüver-Barrera, and Bodian methods. For immunohistochemistry, antibodies against phosphorylated α-synuclein (gifted by Prof. Takeshi Iwatsubo, Tokyo University), tau-2 (×1,000; MP Biomedicals, Solon, Ohio), and ubiquitin (×150; Dako, Glostrup, Denmark) were used and were detected with a labeled streptavidin-biotin kit (LSAB kit; Dako).
Subjects
This study was approved by the institutional review boards of the participating institutions. The subjects examined for this study were all from the Sagamihara family; their positions in the family tree are shown in Figure 1. Cases 2 and 7 are from a small pedigree that branched off from the main family tree sometime around 1600 to 1700 AD. Recent haplotype analysis of PD cases from the Sagamihara area has shown that small pedigrees exist isolated from the Sagamihara family pedigree [6,7]. All subjects were genetically determined to have the I2020T amino acid change in LRRK2. Cases 1 and 2 are living patients who were examined with PET. Cases 3 through 10 were deceased patients; we performed neuropathologic examination on paraffin sections of the representative parts of the 10% formalin-fixed brains. Detailed descriptions of the 10 case patients studied are provided in the Supplemental Materials (Appendix).
Fig. 1.
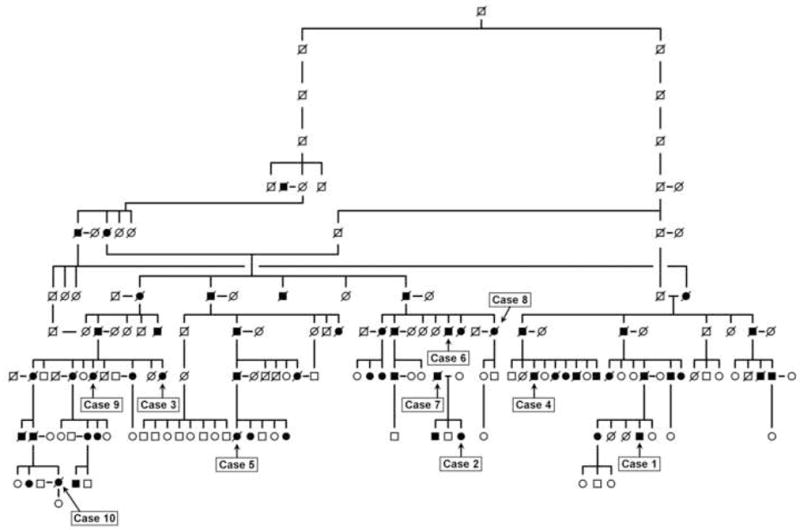
Pedigree of Sagamihara family with PARK8-linked parkinsonism. Filled symbols indicate affected members. Slanted lines indicate deceased members. Cases 1 to 10 indicate fully examined members reported in this study. Cases 2 and 7 are from a small family pedigree that had branched off from the main family sometime between 1600 and 1700 AD (based on the haplotype analysis).
Results
PET Scans
As shown in Table 1, the uptake of 11C-DTBZ, 11C-d-threo-methylphenidate, and 18F-6-fluoro-L-dopa for cases 1 and 2 was measured and compared with normal and sporadic PD control subjects [8]. Uptake of all 3 tracers of presynaptic dopamine function was decreased to levels more than 2 SD below the control mean compared with the control subjects. As in sporadic PD, uptake in the putamen was more severely affected than in the caudate nucleus.
Table 1.
Results of PET Studies in Cases 1 and 2 Compared with Normal and Sporadic PD Controls
| PET results* |
|||||
|---|---|---|---|---|---|
| Measure | Location | Normal controls† (n=33) | sPD controls† (n=67) | Sagamihara family members |
|
| Case 1 | Case 2 | ||||
| Kocc, min−1 | Caudate nucleus | 0.0116±0.0010 | 0.0085±0.0017 (74%) | 0.0055 (47%) | 0.0070 (60%) |
| Putamen | 0.0104±0.0011 | 0.0046±0.0017 (44%) | 0.0027 (26%) | 0.0036 (35%) | |
| BP (DTBZ) | Caudate nucleus | 0.967±0.008 | 0.539 (56%) | 0.41 (42%) | 0.44 (45%) |
| Putamen | 0.979±0.009 | 0.353 (36%) | 0.20 (20%) | 0.21 (21%) | |
| BP (MP) | Caudate nucleus | 1.460±0.002 | 0.848±0.252 (58%) | 0.43 (37%)‡ | 0.45 (39%)‡ |
| Putamen | 1.319±0.002 | 0.490±0.236 (37%) | 0.23 (26%)‡ | 0.25 (27%)‡ | |
| BP (RAC) | Caudate nucleus | 2.28±0.24§ | 2.08±0.32// | 2.89 | 2.59 |
| Putamen | 2.27±0.27 | 2.39±0.4 | 3.11 | 2.84 | |
BP, binding potential; DTBZ, 11C-α-dihydrotetrabenazine; Kocc, uptake rate constant of 18F-6-fluoro-L-dopa; MP, 11C-d-threo-methylphenidate; PD, Parkinson disease; RAC, 11C-raclopride; sPD, sporadic PD.
Values shown are mean±SD or value (% of normal) for each tracer.
Data from Adams et al (8), except for BP (RAC), which are unpublished data (normal controls) and from de la Fuente-Fernandez R, Sossi V, Huang Z, Furtado S, Lu J-Q, Calne DB, et al. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain 2004;127:2747–54 (sPD controls).
Age-adjusted.
N=10. Mean±SD age, 51.3±16.4 y.
N=8. Mean±SD age, 64.4±11.2 y.
11C-raclopride binding in case 1 and in the putamen of case 2 was increased to more than 2 SD greater than the control mean for normal subjects, as has been previously described in sporadic PD. Values were higher in the putamen than the caudate nucleus, whereas in healthy controls, the caudate nucleus and putamen values were identical (putamen:caudate ratio, 1.00±0.03).
In sporadic PD, the uptake ratio for 123I-MIBG cardiac scintigraphy was decreased compared with controls, which was considered to reflect loss of peripheral adrenergic neurons [9]. The results of 123I-MIBG cardiac scintigraphy were normal in case 1 but lower than normal subjects in case 2 (data not shown).
Neuropathologic Findings
Neuropathologic examinations were performed for the 8 deceased subjects described above (Table 2). In all, 6 cases (cases 3, 4, 5, 7, 8, and 9) showed mild depigmentation in the substantia nigra. Histologically, mild degeneration and loss of melanin-containing neurons in the pars compacta of the substantia nigra were noted. More integrated neuronal loss and gliosis were observed distinctly in the pars reticulata of the substantia nigra in all 6 cases; these findings were different from those seen in sporadic PD (Fig. 2 A and B) [10]. However, pigmented neurons in the locus ceruleus showed normal to minimal degeneration and loss in all 6 cases (Fig. 2 C and D). Lewy bodies were not found in the substantia nigra, the locus ceruleus, the dorsal nucleus of the vagal nerve, or any other regions of the cerebrum and brain stem in these 6 cases. Immunohistochemical staining with antibodies against tau, ubiquitin, and phosphorylated α-synuclein showed no positive staining. Moreover, no pertinent neuropathologic lesions were observed in the basal ganglia, cerebral cortex, or cerebellum.
Table 2.
Neuropathologic Findings in 8 Deceased Members of the Sagamihara Family
| Neuropathologic findings |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Neuronal loss |
Gliosis |
||||||||||
| Case no. | Disease duration, y | SN | LC | N.X | SN | LC | N.X | LB | NFT | GCI | Ub |
| 3 | 17 | + | − | − | ++ | − | − | − | − | − | − |
| 4 | 15 | + | − | − | ++ | − | − | − | − | − | − |
| 5 | 13 | ++ | + | − | + | − | − | − | − | − | − |
| 6* | 8 | ++ | ± | − | ++ | − | − | − | − | ++ | − |
| 7 | 21 | + | − | − | + | − | − | − | − | − | − |
| 8 | 32 | + | − | − | + | − | − | − | − | − | − |
| 9 | 33 | + | − | − | + | − | − | − | − | − | − |
| 10 | 20 | + | − | − | + | − | − | ++ | − | − | − |
GCI, glial cytoplasmic inclusions; LB, Lewy bodies; LC, locus ceruleus; NFT, neurofibrillary tangles; N.X, dorsal nucleus of vagus nerve; SN, substantia nigra; Ub, ubiquitin; −, absence of feature; ±, mild; +, moderate; ++, severe.
Neuropathologic diagnosis was multiple system atrophy parkinsonism.
Fig. 2.
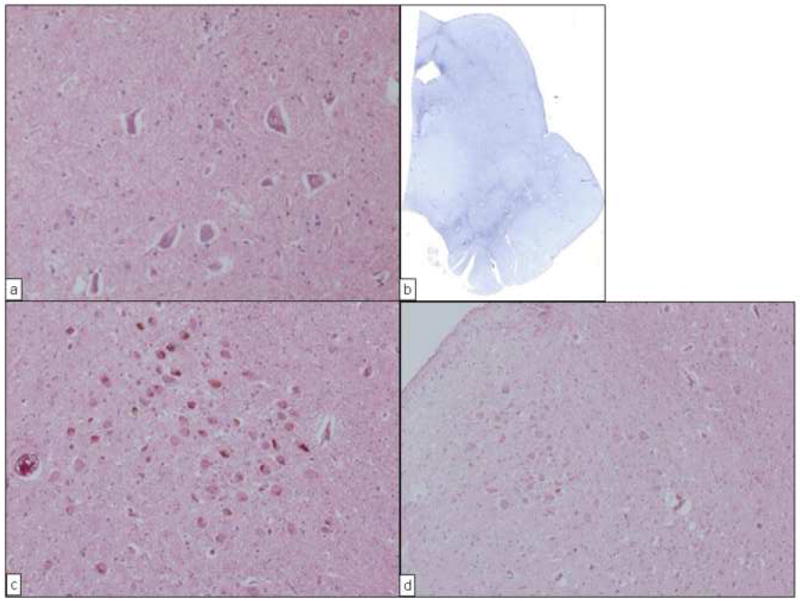
Neuropathologic findings from 2 subjects (cases 3 and 7) with PARK8 parkinsonism in the current study. A, View of the substantia nigra in case 3. Neuronal cell loss and gliosis were mild compared with that seen in sporadic Parkinson disease. (Hematoxylin-eosin; original magnification, ×200.) B, Scanning view of the midbrain in case 3. Marked gliosis was seen in the area of the substantia nigra, especially the pars reticulata. (Holzer stain.) C, View of the locus ceruleus in case 3. Neuronal cells are well preserved. (Hematoxylin-eosin; original magnification, ×100.) D, View of the locus ceruleus in case 7. Neuronal cells are well preserved. (Hematoxylin-eosin; original magnification, ×100.)
Neuropathologically, case 6 showed features seen in multiple system atrophy parkinsonism (MSA-P) (Fig. 3A–C). Immunostaining with antibody to phosphorylated α-synuclein showed many glial cytoplasmic inclusions (GCIs). The GCI-positive cells were more extensively distributed in this case than in conventional cases of MSA-P [11].
Fig. 3.
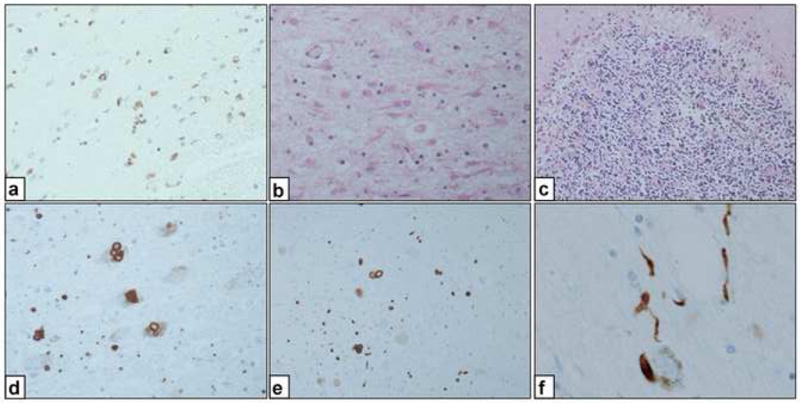
Neuropathologic findings from case 6 (A–C) and case 10 (D–F) in the current study. A, View of the putamen (case 6). Many oligodendroglia with positively stained cytoplasm are seen. (Anti-α-synuclein antibody; original magnification, ×200.) B, View of the cerebellum (case 6). Diffuse loss of Purkinje cells and granular neurons with gliosis in the white matter. (Hematoxylin-eosin; original magnification, ×100.) C, View of the putamen (case 6). Many glial cytoplasmic inclusions and protoplasmic astrocytes are seen. (Hematoxylin-eosin; original magnification, ×200.) D, View of the locus ceruleus (case 10) showing Lewy bodies. (Anti-α-synuclein antibody; original magnification, ×200.) E, View of the dorsal vagus nerve (case 10) showing Lewy bodies. (Anti-α-synuclein antibody; original magnification, ×200.) F, View of the locus ceruleus (case 10); some Lewy neurites also were observed. (Anti-α-synuclein antibody; original magnification, ×200.)
In case 10, hematoxylin and eosin staining showed Lewy bodies. When stained with anti-phosphorylated-synuclein antibody, many Lewy bodies and Lewy neurites were observed in the substantia nigra, locus ceruleus, dorsal motor nucleus of the vagal nerve, and raphe nuclei (Fig. 3D–F). The neuronal loss in the affected nuclei was mild given the long clinical course. Neuronal loss in the locus ceruleus was extremely mild. When these regions were stained with anti-ubiquitin antibody or anti-tau antibody, the positively stained regions were also positive with anti-synuclein antibody, as previously described in sporadic PD (Table 2) [12,13].
Discussion
The clinical features of Sagamihara family parkinsonism are very similar to those seen in sporadic cases of PD, with all cardinal features of this illness. These patients have good response to levodopa and dopamine agonists; however, later on in the course of the illness, complications of dopaminergic therapy develop, as in cases of sporadic PD.
PET findings for patients with PARK8 mutations generally differ little from those with idiopathic PD. The PET findings from our 2 cases do not conflict with those of sporadic PD. The I2020T mutant line has a long and benign clinical course. The PET results seem to be consistent with this clinical course. The data from olfactory examinations and MIBG cardiac scintigraphy vary from normal to moderately impaired (data not shown), but the presence of dysfunction is not significantly associated with the clinical course of PARK8-linked parkinsonism with the I2020T mutation. Recently, Goldstein et al [14] reported that 1 PARK8 patient, a carrier of the T2356I mutation, showed cardiac sympathetic denervation. 123I-MIBG scans have demonstrated that a few members of the Sagamihara family have moderate cardiac sympathetic denervation. However, they do not have any orthostatic hypertension or other signs of autonomic system dysfunction. Common neuropathologic features of parkinsonism in the Sagamihara family are 1) neuronal cell loss and gliosis in the reticular zone of the substantia nigra, 2) loss of only neuronal cells in the compact zone of the substantia nigra, and 3) very mild neuronal cell loss in the locus ceruleus.
Other than the Sagamihara family, few cases of PARK8 parkinsonism have been investigated neuropathologically. A previous study reported neuropathologic findings on Family A, which consists of Canadian siblings with a mean onset age of 53 years [15]. The clinical features of the disease in 13 siblings across 4 generations were reported: the presentation was basically the same as in conventional PD. However, muscle atrophy was observed in 2 cases and dementia in 2 cases. The causative LRRK2 gene mutation was identified and produces the amino acid change Tyr1699Cys. Neuropathologically, atypical nigral degradation accompanying ubiquitin-positive inclusions was observed in 2 cases, 1 case showed neuronal changes similar to those seen in Alzheimer disease, and 1 case showed mild motor neuron degradation.
The Western Nebraska Family D comprises 190 family siblings, of whom 22 were affected with PD [16,17]. The mean age at onset was 65 years, and the affected members showed the 4 major signs related to PD. They responded well to levodopa therapy. The causative LRRK2 mutation has been identified as Arg1441Cys. Autopsies have been conducted on 4 subjects. Lewy bodies were restricted to the brain stem in case 1. Case 2 showed an extensive distribution of Lewy bodies, and that patient was diagnosed pathologically as having diffuse Lewy body disease despite lack of dementia on clinical examination. Case 3 showed neurofibrillary tangles without Lewy bodies, and the diagnosis was neurodegenerative tauopathy, which is similar to progressive supranuclear palsy. However, the patient did not clinically fulfill the criteria for diagnosis of progressive supranuclear palsy. Case 4 showed only nigral degeneration and no Lewy bodies or neurofibrillary tangles, which is similar to the Sagamihara family members.
In another report, many Lewy bodies were observed in various areas in the central nervous system in patients with the Gly2019Ser (G2019S) mutation of LRRK2, but they did not show apparent dementia [18]. Another report concerned the neuropathology of the G2019S mutation: family SK had slowly progressive parkinsonism. Neuropathologic examination in 1 case revealed no Lewy bodies but did show tau-immunoreactive neurofibrillary tangles and an argyrophilic grain disease–like pathology [19]. The authors believed that the Lewy body pathology and tau pathology were distinct pathologic processes, but that the pathologic end points may share the same primary cause. Therefore, these different pathologies may be incidental to other coexisting insults, or perhaps they represent independent events; this dilemma remains to be resolved. However, as of yet, no interaction is known to exist between LRRK2, tau, and α-synuclein.
Recently, 2 cases of PARK8 associated with the G2019S LRRK2 mutation showed mild neuronal loss in the substantia nigra and had neither Lewy bodies nor tau- or ubiquitin-positive cytoplasmic inclusions [20,21]. These cases suggest that the underlying pathogenic mechanism may be an upstream pathway for other proteins implicated in the pathogenesis of neurodegeneration. In another case with the G2019S mutation, pathologic examination showed frontotemporal lobar degeneration with ubiquitin-immunoreactive neuronal inclusions [22]. The authors thought that the comorbid condition in this case may be coincidental, although patients with LRRK2 mutations may display atypical features.
Neuropathologic findings were reported for a case with an Ile1371Val mutation of LRRK2 [23]. This case showed typical ubiquitin-and α-synuclein-positive Lewy body pathology. Although this case showed sporadic PD pathology, it would be desirable to accumulate neuropathologic findings from other cases.
Pure nigral degeneration without Lewy bodies is a core neuropathologic change in Sagamihara family (I2020T) parkinsonism. Of the 8 cases studied here, 2 (1 with MSA-P and 1 with Lewy body–positive PD) showed different core images. In the case with Lewy bodies and an approximately 16-year history of highly advanced clinical symptoms, the neuronal loss was less marked than would be expected given the long duration of the disease and the advanced stage. These findings may indicate that the subject had PARK8-linked parkinsonism, rather than sporadic PD. Also, compared with conventional cases of MSA-P, the case in the current study with MSA-P showed an extensive distribution of numerous GCI lesions, as well as mild neuronal loss and astrogliosis in the olivopontocerebellar afferent system, which suggests early-stage pathologic changes in MSA-P. These pathologic findings from cases 6 and 10 suggest that, neuropathologically, the Sagamihara family principally shows pure nigral degeneration, but once the α-synuclein aggregation process starts, phosphorylation may occur rapidly.
The mutations of LRRK2 currently identified to be in the kinase-motif domain of the protein are G2019S, I2020T, and Ile2012Thr (I2012T). Neuropathologic characteristics linked to the I2012T mutation have not been reported. The G2019S mutation may induce the accumulation of α-synuclein and the acceleration of phosphorylation. The I2020T mutation might not be related to the accumulation of α-synuclein, but once accumulation starts, phosphorylation of α-synuclein may occur. To clarify the mechanism, the function of the mitogen-activated protein kinase kinase kinase (MAPKKK) domain should be investigated; however, MAPKKK activity may not be related to the neuropathologic features. As for the I2020T mutation, the reports on MAPKKK activity vary. The Sagamihara family examined in the present study showed various neuropathologic findings, some of which sometimes promote phosphorylation when mutations are induced in MAPKKK, but at other times decrease phosphorylation.
The neuropathology of PARK8 is modified by point mutation loci and is characterized by polymorphism in the siblings. This suggests that the LRRK2 mutations may be involved in the mechanism responsible for the onset of not only PD but also other neurodegenerative diseases. Antibodies for labeling the MAPKKK domain currently are being produced. We will use these antibodies to analyze the role of PARK8 in the progression of neurodegeneration in PD.
Acknowledgments
This work was partially supported by a Grant-in-Aid from the Research Committee of CNS Degenerative Disease, the Ministry of Health, Labor and Welfare of Japan. A.J.S. was supported by a CIHR Team grant in Parkinson’s Disease, a Michael Smith Foundation for Health Research Research Unit award, and the Canada Research Chairs program. A.J.S. and Z.K.W. were supported by a Pacific Alzheimer’s Research Foundation Centre grant. Z.K.W. was also supported by a Morris K. Udall NIH/NINDS grant awarded to Mayo Clinic Jacksonville.
We thank Prof. Isao Okayasu, Masafuchi Ryo, and Yutaka Ogino for material preparation and comments on the manuscript, Mrs. Hiroko Kawashima for preparing this manuscript, and Mrs. Etsuko Tadokoro for immunostaining. We thank Dr. Thomas J. Ruth and the UBC-TRIUMF PET program for conducting the PET studies. We are also grateful to our patients of the Sagamihara family.
Abbreviations
- 11C-DTBZ
11C-α-dihydrotetrabenazine
- GCI
glial cytoplasmic inclusion
- HY
Hoehn and Yahr scale
- 123I-MIBG
123I-meta-iodobenzylguanidine
- MAPKKK
mitogen-activated protein kinase kinase kinase
- MSA-P
multiple system atrophy parkinsonism
- PD
Parkinson disease
Appendix
Case 1
A male member of the Sagamihara parkinsonism family noticed a gait disturbance, particularly in the right leg, at age 40 years. He was admitted to Sagamihara National Hospital (SNH) at age 49 years. Tremors in his left hand and leg, rigidity in all extremities, and akinesia were noted. He was diagnosed as having PD and treated with levodopa, which controlled his symptoms well. In 2004, the PD was characterized as PARK8. He is now 63 years old. Levodopa and a dopamine agonist (pramipexole) control his parkinsonism at Hoehn and Yahr (HY) stage 3, but he shows mild wearing-off phenomenon and overt start hesitation. 123I-MIBG cardiac scintigraphy performed in 2006 indicated that his uptake ratio was in the normal range. His Unified Parkinson Disease Rating Scale total score is now 29 (parts II and III, 25).
Case 2
A female member of a small pedigree of the Sagamihara family had tremor in the left hand beginning at age 60 years. She noticed lumbar pain at that time and was admitted to the orthopedics division of SNH. Lumbago was treated by medication but her symptoms did not improve, so she consulted our clinic. The 4 cardinal symptoms of parkinsonism were noted, and PARK8-linked parkinsonism was diagnosed genetically. After beginning therapy with levodopa and dopamine agonist, her symptoms became well controlled at HY stage 3. 123I-MIBG cardiac scintigraphy showed a slight decrease in the uptake ratio in the early and delayed phases. Her Unified Parkinson Disease Rating Scale total score is now 12 (parts II and III, 6).
Case 3
A woman in the Sagamihara family noted a hauling gait in the right leg when she was 52 years old. When her symptoms worsened, she consulted SNH—the diagnosis was PD. Administration of trihexyphenidyl improved her symptoms. After that, she was given amantadine hydrochloride, levodopa, and bromocriptine, each of which was effective. However, after approximately 6 years, wearing-off and on-off phenomena began to appear. Medical therapy maintained her condition at HY stage 3 for approximately 15 years. Other than dementia and constipation, no autonomic symptoms were seen. At age 67 years, liver dysfunction of an unknown nature developed, which caused an advance to HY stage 4. She died of pneumonia at age 69 years; the entire course of the disease lasted 17 years.
Case 4
At age 60 years, this man experienced abnormal gait with reduced stride initially affecting primarily the right leg. After akinesia had gradually spread to all extremities, he consulted SNH. We noted the 4 major PD signs and diagnosed PD. Administration of amantadine hydrochloride and levodopa alleviated his symptoms. However, the tremor, rigidity, akinesia, and postural instability gradually worsened. Treatment with the dopamine agonist selegiline maintained his HY stage at 3. Approximately 13 years after the onset of PD, his falling tendency worsened, which compelled him to use a wheelchair. However, apparent autonomic symptoms and dementia were never seen. He died of pneumonia at age 75 years; the duration of PD was 15 years.
Case 5
Onset of PD, initiated by a gait disturbance and tremors, occurred in this woman at age 43 years, at which time she consulted SNH. Administration of amantadine hydrochloride and levodopa relieved her symptoms, and maintained her condition at HY stage 3. After that, bromocriptine and pergolide were given, which enabled her to work. Nine years after the onset, akinesia was acutely exacerbated by severe diarrhea, and her HY stage advanced to stage 5. She was able to communicate clearly without dementia throughout the 13-year course of the disease. She died after a bout of pneumonia at the age of 56 years.
Case 6
A 74-year-old man began having hand tremors and bradykinesia. The diagnosis was PD. Although he was older, we noted no decline in cognitive function. He responded well to levodopa and his HY stage remained at 3 for several years. Four years after the onset of PD, a dopamine agonist was added to his regimen to relieve the persistent akinesia. However, his symptoms were not relieved, and the disease advanced to stage 4. When he was 82 years old, he had repeated bouts of hypothermia during the winter and died, 8 years after the onset of PD. No cognitive impairment or autonomic disturbance ever occurred.
Case 7
A 56-year-old man noted left hand tremor at rest and visited SNH. Akinesia and rigidity were also observed. Treatment was started with amantadine hydrochloride, which produced a good response and maintained his condition at HY stage 2. Later, levodopa and bromocriptine were added. He responded well to these therapies as well, without adverse effects. At age 66 years, he noticed wearing-off phenomenon and dyskinesia. Despite the use of medication, his disease advanced to HY stage 3 during his “on” periods and to stage 4 during his “off” periods. His clinical course was free of cognitive dysfunction or apparent autonomic symptoms. Gastric cancer later developed, and he died at age 77 years, 21 years after the onset of PD.
Case 8
The onset of PD, initiated by tremor, occurred when this woman was 46 years old. At age 48 years, body bradykinesia appeared; she visited SNH, and PD was diagnosed. Treatment was started with trihexyphenidyl, and her HY stage improved to stage 2. Amantadine hydrochloride, levodopa, and bromocriptine were added to alleviate individual symptoms. She responded to those therapies as well, but her clinical state advanced to HY stage 3. Although she was free of apparent cognitive dysfunction and autonomic symptoms, difficulty in swallowing developed from about the age of 75 years, and pneumonia began to occur frequently. She died of pneumonia at age 78 years. The entire course of the disease lasted 32 years.
Case 9
In this woman, PD was initiated by a shuffling walk when she was 55 years old. At about age 59 years, she began to have a stooped posture. She visited SNH and was diagnosed as having PD. Treatment began with trihexyphenidyl, which improved her symptoms. Later, levodopa and bromocriptine were added, and her symptoms improved again. Clinically, retropulsion and falling occurred and her clinical course progressed to HY stage 3. When she was 77 years old, dehydration aggravated her akinesia, and the disease advanced to HY stage 4. At the age of 80 years, a tracheotomy was performed for pneumonia, and her clinical course advanced to HY stage 5. However, she was able to communicate clearly, and no dementia was noted. She died of pneumonia at age 88 years.
Case 10
The onset of PD occurred in this woman when she was 48 years old, initiated by difficulty moving the left leg and tremor in the left hand. She presented with the 4 major signs related to PD. Treatment was started with trihexyphenidyl. To control the symptoms, we prescribed amantadine hydrochloride, levodopa, and a dopamine agonist. She responded well to these therapies, and her clinical state was controlled at HY stage 3. Nine years after the onset of PD, exacerbated wearing-off phenomenon occurred and she became depressed. Her cognitive function was normal. Sixteen years after disease onset, her medication gradually became ineffective for unknown reasons, and her disease advanced to HY stage 4 during “on” periods and to HY stage 5 during “off” periods. Because of frequent pneumonia, a tracheotomy was required, along with the temporary use of a respirator. After the tracheotomy, she required tube feeding. The wearing-off phenomenon was complicated by dyskinesia, and her symptoms became difficult to control with medications. Her disease advanced to HY stage 5, and she died at age 68 years. The entire course of the disease lasted 20 years.
Footnotes
Disclosure: ZKW has a potential financial interest associated with technology entitled “Identification of Mutations in PARK8, a Locus for Familial Parkinson’s Disease,” Mayo Clinic case #2004-185. A patent has been issued for this technology, and it has been licensed to a commercial entity. No royalties have accrued to ZKW; however, Mayo Clinic has received royalties of greater than $10,000, the federal threshold for significant financial interest, from the licensing of this technology.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2–q13.1. Ann Neurol. 2002;51:296–301. doi: 10.1002/ana.10113. [DOI] [PubMed] [Google Scholar]
- 2.Hasegawa K, Funayama M, Matsuura N, Furusawa H, Sakai F, Kowa H, et al. Analysis of alpha-synuclein, parkin, tau, and UCH-L1 in a Japanese family with autosomal dominant parkinsonism. Eur Neurol. 2001;46:20–4. doi: 10.1159/000050751. [DOI] [PubMed] [Google Scholar]
- 3.Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 4.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, et al. International LRRK2 Consortium. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7:583–90. doi: 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Funayama M, Hasegawa K, Ohta E, Kawashima N, Komiyama M, Kowa H, et al. An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann Neurol. 2005;57:918–21. doi: 10.1002/ana.20484. [DOI] [PubMed] [Google Scholar]
- 7.Nukada H, Kowa H, Saitoh T, Tazaki Y, Miura S. A big family of paralysis agitans [translated by the authors] [Japanese] Rinsho Shinkeigaku. 1978;18:627–34. [PubMed] [Google Scholar]
- 8.Adams JR, van Netten H, Schulzer M, Mak E, Mckenzie J, Strongosky A, et al. PET in LRRK2 mutations: comparison to sporadic Parkinson’s disease and evidence for presymptomatic compensation. Brain. 2005;128:2777–85. doi: 10.1093/brain/awh607. [DOI] [PubMed] [Google Scholar]
- 9.Orimo S, Ozawa E, Nakade S, Sugimoto T, Mizusawa H. 123I-metaiodobenzylguanidine myocardial scintigraphy in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1999;67:189–94. doi: 10.1136/jnnp.67.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowe JS, Leigh N. Disorders of movement and system degenerations. In: Graham DI, Lantos PL, editors. Greenfield’s neuropathology. 7. Vol. 2. New York: Arnold; 2002. pp. 325–430. [Google Scholar]
- 11.Inoue M, Yagishita S, Ryo M, Hasegawa K, Amano N, Matsushita M. The distribution and dynamic density of oligodendroglial cytoplasmic inclusions (GCIs) in multiple system atrophy: a correlation between the density of GCIs and the degree of involvement of striatonigral and olivopontocerebellar systems. Acta Neuropathol (Berl) 1997;93:585–91. doi: 10.1007/s004010050655. [DOI] [PubMed] [Google Scholar]
- 12.Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol. 2003;62:389–97. doi: 10.1093/jnen/62.4.389. [DOI] [PubMed] [Google Scholar]
- 13.Saito Y, Ruberu NN, Sawabe M, Arai T, Kazama H, Hosoi T, et al. Lewy body-related α-synucleinopathy in aging. J Neuropathol Exp Neurol. 2004;63:742–9. doi: 10.1093/jnen/63.7.742. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein DS, Imrich R, Peckham E, Holmes C, Lopez G, Crews C, et al. Neurocirculatory and nigrostriatal abnormalities in Parkinson disease from LRRK2 mutation. Neurology. 2007;69:1580–4. doi: 10.1212/01.wnl.0000268696.57912.64. [DOI] [PubMed] [Google Scholar]
- 15.Wszolek ZK, Vieregge P, Uitti RJ, Gasser T, Yasuhara O, McGeer P, et al. German-Canadian family (family A) with parkinsonism, amyotrophy, and dementia: longitudinal observations. Parkinsonism Relat Disord. 1997;3:125–39. doi: 10.1016/s1353-8020(97)00013-8. [DOI] [PubMed] [Google Scholar]
- 16.Wszolek ZK, Pfeiffer B, Fulgham JR, Parisi JE, Thompson BM, Uitti RJ, et al. Western Nebraska family (family D) with autosomal dominant parkinsonism. Neurology. 1995;45:502–5. doi: 10.1212/wnl.45.3.502. [DOI] [PubMed] [Google Scholar]
- 17.Wszolek ZK, Pfeiffer RF, Tsuboi Y, Uitti RJ, McComb RD, Stoessl AJ, et al. Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology. 2004;62:1619–22. doi: 10.1212/01.wnl.0000125015.06989.db. [DOI] [PubMed] [Google Scholar]
- 18.Ross OA, Toft M, Whittle AJ, Johnson JL, Papapetropoulos S, Mash DC, et al. LRRK2 and Lewy body disease. Ann Neurol. 2006;59:388–93. doi: 10.1002/ana.20731. [DOI] [PubMed] [Google Scholar]
- 19.Rajput A, Dickson DW, Robinson CA, Ross OA, Dachsel JC, Lincoln SJ, et al. Parkinsonism, LRRK2 G2019S, and tau neuropathology. Neurology. 2006;67:1506–8. doi: 10.1212/01.wnl.0000240220.33950.0c. [DOI] [PubMed] [Google Scholar]
- 20.Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ, et al. Biochemical and pathological characterization of LRRK2. Ann Neurol. 2006;59:315–22. doi: 10.1002/ana.20791. [DOI] [PubMed] [Google Scholar]
- 21.Gaig C, Marti MJ, Ezquerra M, Rey MJ, Cardozo A, Tolosa E. G2019S LRRK2 mutation causing Parkinson’s disease without Lewy bodies. J Neurol Neurosurg Psychiatry. 2007;78:626–8. doi: 10.1136/jnnp.2006.107904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dachsel JC, Ross OA, Mata IF, Kachergus J, Toft M, Cannon A, et al. LRRK2 G2019S substitution in frontotemporal lobar degeneration with ubiquitin-immunoreactive neuronal inclusions. Acta Neuropathol (Berl) 2007;113:601–6. doi: 10.1007/s00401-006-0178-1. [DOI] [PubMed] [Google Scholar]
- 23.Giordana MT, D’Agostino C, Albani G, Mauro A, Di Fonzo A, Antonini A, et al. Neuropathology of Parkinson’s disease associated with the LRRK2 Ile1371Val mutation. Mov Disord. 2007;22:275–8. doi: 10.1002/mds.21281. [DOI] [PubMed] [Google Scholar]