

Abstract
Background
Amyotrophic lateral sclerosis (ALS)–Plus syndromes meet clinical criteria for ALS but also include 1 or more additional features such as dementia, geographic clustering, extrapyramidal signs, objective sensory loss, autonomic dysfunction, cerebellar degeneration, or ocular motility disturbance.
Methods
We performed a whole-brain and spinal cord pathologic analysis in a patient with an ALS-Plus syndrome that included repetitive behaviors along with extrapyramidal and supranuclear ocular motility disturbances resembling the clinical phenotype of progressive supranuclear palsy.
Results
There was motoneuron cell loss and degeneration of the corticospinal tracts. Bunina bodies were present. TAR DNA-binding protein-43 pathology was diffuse. Significant tau pathology was absent.
Conclusions
TAR DNA-binding protein-43 disorders can produce a clinical spectrum of neurodegeneration that includes ALS, frontotemporal lobar degeneration, and ALS with frontotemporal lobar degeneration. The present case illustrates that isolated TAR DNA-binding protein-43 disorders can produce an ALS-Plus syndrome with extrapyramidal features and supranuclear gaze palsy resembling progressive supranuclear palsy.
The diagnosis of amyotrophic lateral sclerosis (ALS) requires the presence of progressive upper and lower motor neuron symptoms and signs. The additional features of sensory disturbance, autonomic dysfunction, extrapyramidal features, ocular motility abnormalities, dementia, cerebellar dysfunction and/or geographic clustering are considered to exclude the diagnosis of classic ALS and when present define the ALS-Plus syndrome. The following case is an example of ALS-Plus with neuropathology that is remarkably similar in distribution and extent to that which has been reported in sporadic ALS.
REPORT OF A CASE
A 66-year-old man had dysarthria for 1½ years and dysphagia for 1 year before seeking medical attention. Clinical examination revealed that he had anarthria and marked difficulty swallowing liquids and solids and had lost 17 lb (to convert pounds to kilograms, multiply by 0.45). He was drooling and had difficulty with excessive pharyngeal secretions. One year previously, uncontrollable laughing developed. Concurrent with this, he developed a personality change that was manifested as apathetic, disinterested behavior, which was unusual for him, and he began to obsessively pace, climb and descend stairs, and open and close windows and doors. Three months before being seen, he developed progressive hand weakness and intrinsic hand muscle atrophy, greater on the left side than the right side, and diffuse muscular twitching in the arms, legs, and trunk. Two months previously, he developed a slow, stiff, unsteady gait and began to fall backward. In addition, he noted difficulty in voluntarily closing his eyes and an inability to voluntarily gaze, which was greater in the vertical than the horizontal direction. He did not report dyspnea or orthopnea; however, he did have frequent sleep arousal and excessive daytime sleepiness. He did not have pain, tremor, sensory disturbance, or bowel or bladder problems. He denied other medical history. His mother had developed dementia at the age of 60 years, which manifested as personality change and unusual behavior that included physical violence. Family history was otherwise normal.
NEUROLOGIC EXAMINATION
The patient was a thin, ill-appearing man in no acute distress. Forced vital capacity was 60% of predicted. He was awake, alert, and anarthric and could appropriately respond to yes/no questions and follow commands. He was able to pick out the name of objects from a written list. His writing was illegible. He blinked to visual threat. Pupils were 3 to 4 mm in diameter and reactive to light and near vision. He had a prominent stare with eyelid retraction and a surprised look. He blinked infrequently. Horizontal and vertical pursuit movements were interrupted and slow. Horizontal and vertical saccades were slow and hypometric. Downgaze was more affected than upgaze. He was unable to suppress head turning with attempted lateral gaze. The vertical and horizontal oculocephalic reflex was normal. Square wave jerks on fixation were noted. His mouth was held open; there was weakness of the masseter and pterygoid muscles, with masseter atrophy. The face was masked with upper and lower facial diparesis and prominent facial reflexes. He was unable to voluntarily close his eyes on command. Mental fasciculations were present. The palate elevated poorly but was midline. The sternocleidomastoid muscles and tongue were weak, atrophic, and fasciculating. Rapid tongue movements were slowed. Power was Medical Research Council (MRC) grade 5 in the deltoid, bicep, and tricep muscles but MRC grade 4 in the right and left finger extensor and hand intrinsic muscles. There was atrophy of the hand intrinsic muscles. Leg power was MRC grade 5, he was able to rise from a chair without using his hands, and heel and toe walking was normal. Large-amplitude fasciculations were noted throughout the arms, hands, chest, thighs, calves, forearms, and abdominal wall. Tone was more rigid in the legs than the arms, without tremor or cogwheeling. Rapid movements were slowed in the hands and feet with reduced amplitude. Bradykinesia was present. Walking was slow with a narrow base, small steps, and absent arm swing. Turning was poor, with multiple steps. Retropulsion was prominent. There was no limb apraxia or alien hand. Sensory and cerebellar examinations yielded normal findings. There was jaw clonus. Reflexes were 3 of 4 in the arms and legs, greater on the left side than the right side, with bilateral Hoffman sign and crossed adduction. Cutaneous abdominal reflexes were absent. Toes were extensor (the so-called Babinski sign) bilaterally. Snout, glabellar, and palmomental reflexes were present.
CLINICAL AND LABORATORY INVESTIGATIONS
Magnetic resonance images of the brain demonstrated diffuse atrophy with increased signal in the corticospinal tracts. Nerve conduction was normal. An electromyogram demonstrated acute and ongoing chronic partial denervation in multiple muscles of the left arm and leg, the tongue, the thoracic paraspinal region, and the sternocleidomastoid. Complete blood cell count was normal, as were electrolyte, anti-GM1 antibody, and creatine phosphokinase concentrations. Liver function studies, thyroid function studies, serum heavy metal screening, serum protein electrophoresis, and urine protein electrophoresis yielded normal results.
CLINICAL COURSE
The patient refused riluzole therapy. A gastrostomy tube was inserted 2 weeks after presentation. One month later, worsening dyspnea and orthopnea developed, and forced vital capacity decreased to 38% of predicted. Noninvasive positive-pressure ventilatory assistance was instituted. The patient declined long-term mechanical ventilatory assistance, and hospice care was instituted. Two months after the initial presentation, the patient died of respiratory failure.
POSTMORTEM EXAMINATION
NEUROPATHOLOGIC FINDINGS
The brain, spinal cord, and spinal roots were macroscopically normal. The substantia nigra was normally pigmented. The cervical and thoracic spinal cord demonstrated corticospinal tract degeneration with myelin staining. Axonal spheroids positive for α-synuclein antibody indicative of interrupted axonal transport were seen in the corticospinal tract. There was no other pathologic immunoreactivity for α-synuclein or Lewy body pathology elsewhere in the brain. Mild to moderate loss of motoneurons in the anterior horns was noted. Bunina bodies as well as TAR DNA-binding protein-43 (TDP-43)–positive cytoplasmic skein and Lewy body–like and punctate cytoplasmic inclusions were noted in some remaining motoneurons. Neuronal cytoplasmic TDP-43 inclusions and glial cytoplasmic inclusions were present in the motor cortex. Extensive neuronal and glial TDP-43 inclusions were also noted in the midbrain, hippocampus, globus pallidus, hypoglossal nucleus, substantial nigra, and caudate/putamen area.(Figure 1) A semiquantitiative pathologic scale (0, none; 0.25, rare; 1, mild; 2, moderate; and 3, severe) was used to score TDP-43 pathology in each brain region (Figure 2). Tau immunohistochemistry showed isolated and rare neurofibrillary tangles, which were limited to the entorhinal cortex (Braak and Braak stage I), likely the result of normal aging.
Figure 1.

TAR DNA-binding protein-43 pathology in the amygdala and midbrain (substantia nigra). A, Representative section of the amygdala stained with TDP-43 shows severe pathologic findings with abundant neuronal and some glial cytoplasmic inclusions. Note the characteristic nuclear clearing of the involved cells in contrast with the normal nuclear staining of a spared cell (arrow). This pattern was present in other brain areas such as the basal ganglia. B, In the substantia nigra, there were occasional round dense cytoplasmic inclusions (Lewy body–like inclusions) as well as glial cytoplasmic inclusions, as shown here in high power, adjacent to the affected dopaminergic neuron (arrowhead).
Figure 2.
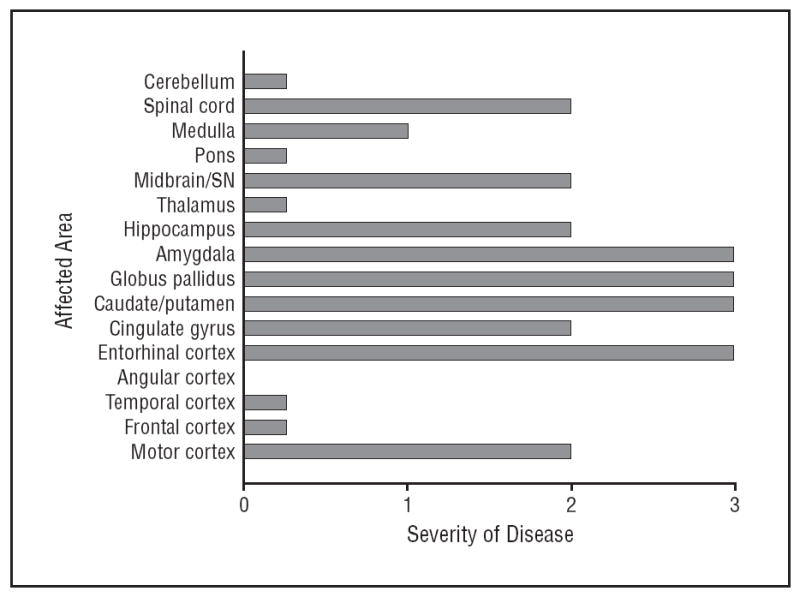
Extent and distribution of TAR DNA-binding protein-43 pathology. SN indicates substantia nigra. For disease severity, 1 is mild; 2, moderate; and 3, severe.
GENETIC FINDINGS
Mutations in the progranulin (GRN) (OMIM 138945) and TDP-43 (TARDBP) (OMIM 605078) genes were excluded using DNA sequencing. The DNA was extracted from brain tissue using an automated extractor according to the manufacturer’s protocol (M48 workstation; Qiagen GmbH, Hilden, Germany). DNA sequencing of the GRN and TAR-DBP genes was performed as previously described.1-3
COMMENT
This 66-year-old man had a rapidly progressive upper and lower motoneuron disorder culminating in death after 20 months as a result of neuromuscular respiratory failure. Symptoms began in the bulbar musculature and progressed and spread to include limb and respiratory muscles. Supranuclear ocular motility and extrapyramidal abnormalities developed without sensory or autonomic dysfunction. He manifested an apathetic, disinterested personality with motor stereotypies. Mental status examination was hampered by anarthria and limb weakness; however, severe memory or language impairment was not noted. Magnetic resonance images demonstrated increased signal in the corticospinal tract. An electromyelogram demonstrated evidence of a diffuse lower motoneuron process.
The patient met El Escorial criteria for clinically definite ALS.4 The extrapyramidal and ocular motility findings are reminiscent of progressive supranuclear palsy (PSP).5 Hyperintensity of the corticospinal tracts on magnetic resonance images is an occasional finding in ALS and has been reported in atypical primary lateral sclerosis.6,7 The personality change and motor stereotypies are characteristic of frontotemporal lobar degeneration (FTLD) and lesions of the orbitofrontal, ventral striatal, basal forebrain, and anterior temporal connections.8-11 Frontotemporal lobar degeneration may occur in ALS and in PSP.12-14 The ALS-Plus syndromes meet clinical criteria for ALS but also include 1 or more other features such as dementia, geographic clustering, extrapyramidal signs, objective sensory loss, autonomic dysfunction, cerebellar degeneration, or ocular motility disturbance. The combination of ALS, FTLD, and PSP-like features are consistent with the diagnosis of an ALS-Plus syndrome.15
The neuropathologic findings demonstrating Bunina bodies, loss of motoneurons, and corticospinal tract degeneration are consistent with the diagnosis of ALS. There was also diffuse TDP-43 pathology. TAR DNA-binding protein-43 has been identified as a major disease-associated protein in FTLD characterized by ubiquitin inclusions and in sporadic ALS with and without FTLD.16-20 In ALS, TDP-43 pathology is often diffusely distributed.21 Thus, it is not surprising that the present case of ALS-Plus syndrome was associated with TDP-43 proteinopathy in the brain and spinal cord motor regions and in other brain regions closely associated with behavioral change and extrapyramidal disorders.
Sporadic PSP is characterized by the presence of tau-positive inclusions and may demonstrate corticospinal tract degeneration in atypical cases.7 Mutations in the GRN gene have been reported to cause diffuse TDP-43 proteinopathy manifesting as FTLD, associated in some patients with atypical parkinsonism including features of PSP or corticobasoganglionic degeneration without ALS.1,22,23 A search for GRN mutations in 272 patients with sporadic ALS, 40 with familial ALS, and 49 with ALS-FTLD yielded missense mutations of uncertain significance in 1 patient with limb-onset sporadic ALS and 1 with ALS-FTLD.24 It is notable that our patient’s mother had dementia suggestive of FTLD. A GRN mutation was not identified in our patient. Although he did not have the clinical features of corticobasoganglionic degeneration, a unique predominantly glial TDP-43 pathology with staining of astrocytic plaquelike structures and coiled bodies with biochemical changes of TDP-43 similar to that seen in FTLD with ubiquitin inclusions has been reported in up to 15.4% of patients with corticobasoganglionic degeneration.25 Both TDP-43 and tau pathologic features are present in Pacific Island geographic clusters of ALS, that is, parkinsonism-dementia complex.26 Mutations in the TDP-43 gene have been reported in both familial and sporadic ALS.1-3,27,28 However, our patient was not found to carry a mutation of the TDP-43 gene.
This case report suggests the possibility that the extent and distribution of TDP-43 proteinopathy may determine clinical features. Confirming this will require pathologic analysis of a large number of ALS-Plus cases. While the most frequent manifestation may be ALS with or without cognitive impairment or dementia, the present case suggests that isolated TDP-43 proteinopathy can also produce an extrapyramidal disorder with supra-nuclear gaze paresis like that seen in PSP. Further studies are needed to determine whether isolated TDP-43 pathology can produce ALS-Plus syndromes with other features. This case also highlights the clinical overlap between tau and TDP-43 proteinopathies. Frontotemporal dementia occurs in PSP, FTLD, and corticobasoganglionic degeneration and also in ALS with or without FTLD. While supranuclear gaze abnormalities have often been clinically linked to presumed tau pathology, this case and previous reports of atypical PSP in patients with GRN mutations suggest that such assumptions may be flawed and perhaps particularly so when there is a clinical or pathologic indication of associated upper or lower motoneuron degeneration.
Footnotes
Author Contributions: Dr McCluskey had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: McCluskey and Siderowf. Acquisition of data: McCluskey, Martinez-Lage, Van Deerlin, Yuan, and Clay. Analysis and interpretation of data: McCluskey, Elman, Martinez-Lage, Van Deerlin, and Trojanowski. Drafting of the manuscript: McCluskey and Trojanowski. Critical revision of the manuscript for important intellectual content: McCluskey, Elman, Martinez-Lage, Van Deerlin, Yuan, Clay, Siderowf, and Trojanowski. Administrative, technical, and material support: Martinez-Lage, Van Deerlin, Clay, and Trojanowski. Study supervision: McCluskey, Siderowf, and Trojanowski.
Financial Disclosure: None reported.
References
- 1.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 2.Van Deerlin VM, Wood EM, Moore P, et al. Clinical, genetic, and pathologic characteristics of patients with frontotemporal dementia and progranulin mutations. Arch Neurol. 2007;64(8):1148–1153. doi: 10.1001/archneur.64.8.1148. [DOI] [PubMed] [Google Scholar]
- 3.Van Deerlin VM, Leverenz JB, Bekris LM, et al. TARDP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histological analysis. Lancet Neurol. 2008;7(5):409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brooks BR, Miller RG, Swash M, Munsat TL. World Federation of Neurology Research Group on Motoneuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motoneuron Disord. 2000;1(5):293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 5.Ward C. Characteristics and symptom management of progressive supra-nuclear palsy: a multidisciplinary approach. J Neurosci Nurs. 2006;38(4):242–247. doi: 10.1097/01376517-200608000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Kriaa S, Zbidi M, Hafsa C, Golli M, Gannouni A. MRI in amyotrophic lateral sclerosis: hyperintensity of the corticospinal tract. Neurol Clin Neurophysiol. 2003;2005:3. [PubMed] [Google Scholar]
- 7.Josephs KA, Katsuse O, Beccano-Kelly DA, et al. Atypical progressive supra-nuclear palsy with corticospinal tract degeneration. J Neuropathol Exp Neurol. 2006;65(4):396–405. doi: 10.1097/01.jnen.0000218446.38158.61. [DOI] [PubMed] [Google Scholar]
- 8.Libon DJ, Xie SX, Moore P, et al. Patterns of neuropsychological impairment in frontotemporal dementia. Neurology. 2007;68(5):369–375. doi: 10.1212/01.wnl.0000252820.81313.9b. [DOI] [PubMed] [Google Scholar]
- 9.Nyatsanza S, Shetty T, Gregory C, Lough S, Dawson K, Hodges JR. A study of stereotypic behaviours in Alzheimer’s disease and frontal and temporal variant frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2003;74(10):1398–1402. doi: 10.1136/jnnp.74.10.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bathgate D, Snowden JS, Varma A, Blackshaw A, Neary D. Behaviour in frontotemporal dementia, Alzheimer’s disease and vascular dementia. Acta Neurol Scand. 2001;103(6):367–378. doi: 10.1034/j.1600-0404.2001.2000236.x. [DOI] [PubMed] [Google Scholar]
- 11.Ames D, Cummings JL, Wirshing WC, Quinn B, Mahler M. Repetitive and compulsive behavior in frontal lobe degenerations. J Neuropsychiatry Clin Neurosci. 1994;6(2):100–113. doi: 10.1176/jnp.6.2.100. [DOI] [PubMed] [Google Scholar]
- 12.Kurz AF. Uncommon neurodegenerative causes of dementia. Int Psychogeriatr. 2005;17(suppl 1):S35–S49. doi: 10.1017/s1041610205001936. [DOI] [PubMed] [Google Scholar]
- 13.Rampello L, Buttà V, Raffaele R, et al. Progressive supranuclear palsy: a systematic review. Neurobiol Dis. 2005;20(2):179–186. doi: 10.1016/j.nbd.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 14.Kertesz A, Munoz D. Relationship between frontotemporal dementia and corti-cobasal degeneration/progressive supranuclear palsy. Dement Geriatr Cogn Disord. 2004;17(4):282–286. doi: 10.1159/000077155. [DOI] [PubMed] [Google Scholar]
- 15.Zoccolella S, Palagano G, Fraddosio A, et al. ALS-plus: 5 cases of concomitant amyotrophic lateral sclerosis and parkinsonism. Neurol Sci. 2002;23(suppl 2):S123–S124. doi: 10.1007/s100720200100. [DOI] [PubMed] [Google Scholar]
- 16.Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 17.Kwong LK, Neumann M, Sampathu DM, Lee VM, Trojanowski JQ. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motoneuron disease. Acta Neuropathol. 2007;114(1):63–70. doi: 10.1007/s00401-007-0226-5. [DOI] [PubMed] [Google Scholar]
- 18.Neumann M, Kwong LK, Sampathu DM, Trojanowski JQ, Lee VM. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch Neurol. 2007;64(10):1388–1394. doi: 10.1001/archneur.64.10.1388. [DOI] [PubMed] [Google Scholar]
- 19.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. see comment in Science 2006;314(5796):42-43. [DOI] [PubMed] [Google Scholar]
- 20.Mackenzie IR, Bigio EH, Ince PG, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61(5):427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 21.Geser F, Brandmeir NJ, Kwong LK, et al. ALS is a multisystem TDP-43 proteinopathy. Arch Neurol. 2008;65(5):636–664. doi: 10.1001/archneur.65.5.636. [DOI] [PubMed] [Google Scholar]
- 22.Gass J, Cannon A, Mackenzie IR, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15(20):2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- 23.Josephs KA, Ahmed Z, Katsuse O, et al. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol. 2007;66(2):142–151. doi: 10.1097/nen.0b013e31803020cf. [DOI] [PubMed] [Google Scholar]
- 24.Schymick JC, Yang Y, Andersen PM, et al. Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J Neurol Neurosurg Psychiatry. 2007;78(7):754–756. doi: 10.1136/jnnp.2006.109553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uryu K, Nakashima-Yasuda H, Forman M, et al. Concomitant TDP-43 pathology in Alzheimer’s disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol. 2008;67(6):555–564. doi: 10.1097/NEN.0b013e31817713b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geser F, Winton MJ, Kwong LK, et al. Pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol. 2008;115(1):133–145. doi: 10.1007/s00401-007-0257-y. [DOI] [PubMed] [Google Scholar]
- 27.Kabashi E, Valdmanis PN, Dion P, et al. TARDP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 28.Gitcho MA, Baloh RH, Chakraverty S, et al. TDP-43 A315T mutation in familial motoneuron disease. Ann Neurol. 2008;63(4):535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]