

Abstract
Background
Linear motifs are short modules of protein sequences that play a crucial role in mediating and regulating many protein–protein interactions. The function of linear motifs strongly depends on the context, e.g. functional instances mainly occur inside flexible regions that are accessible for interaction. Sometimes linear motifs appear as isolated islands of conservation in multiple sequence alignments. However, they also occur in larger blocks of sequence conservation, suggesting an active role for the neighbouring amino acids.
Results
The evolution of regions flanking 116 functional linear motif instances was studied. The conservation of the amino acid sequence and order/disorder tendency of those regions was related to presence/absence of the instance. For the majority of the analysed instances, the pairs of sequences conserving the linear motif were also observed to maintain a similar local structural tendency and/or to have higher local sequence conservation when compared to pairs of sequences where one is missing the linear motif. Furthermore, those instances have a higher chance to co–evolve with the neighbouring residues in comparison to the distant ones. Those findings are supported by examples where the regulation of the linear motif–mediated interaction has been shown to depend on the modifications (e.g. phosphorylation) at neighbouring positions or is thought to benefit from the binding versatility of disordered regions.
Conclusion
The results suggest that flanking regions are relevant for linear motif–mediated interactions, both at the structural and sequence level. More interestingly, they indicate that the prediction of linear motif instances can be enriched with contextual information by performing a sequence analysis similar to the one presented here. This can facilitate the understanding of the role of these predicted instances in determining the protein function inside the broader context of the cellular network where they arise.
Introduction
Linear motifs (LMs) are short stretches of amino acids that populate protein sequences and play fundamental roles in protein interaction networks [1]. Their lengths are typically between three and ten amino acids [2], [3]. LMs frequently show wide variation in residue conservation: some positions accept only one or few amino acids while others do not have any preference and function as spacers [4]. These sequence features give to LMs an evolutionary plasticity and an important role in the evolution of cellular networks by the addition of new functionality to proteins [1].
LMs are mainly found in intrinsically unstructured regions of proteins [5]. Disordered regions allow a thermodynamical control of the affinity and specificity of protein interactions. They favour transient, that is to say low affinity, and conditional interactions, often depending on a previous modification like a phosphorylation [6]. Hence the localisation of LMs in disordered regions suits dynamic regulation of protein networks, where a rapid but deterministic response is needed [7]. Indeed, LM–mediated interactions allow the emergence of several regulatory modes (i.e. sequential, mutually exclusive and cooperative) frequently observed in signalling, vesicular trafficking and transcription pathways [8].
Function of LMs strongly depends on the context. An instance of the KDEL motif, which is an endoplasmic reticulum retrieving signal, is likely to be functional only if present in protein sequences known to localise to the ER or Golgi apparatus. On one hand, the context defines the natural constraints that act on LMs and therefore provides “rules” that can be applied to evaluate the reliability of a newly predicted pattern or instance. For example the domain masking strategy, which is used to discard instances occurring in protein regions inaccessible for interaction like globular domains or coiled coils [3], [9], [10], [11].
On the other hand, the context can also give detailed information about the mode of action of LMs. The role of the local amino acid composition in determining specificity of LM interactions has been experimentally studied at the interactome level [12], [13], [14]. At the structural level, unstructured regions flanking LMs have been observed to undergo disorder to order transition upon binding [15], forming either  -helices [16] or additional
-helices [16] or additional 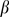 strands that join a
strands that join a 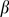 sheet of the partner [17]. This coincides with the observation that two thirds of LMs bind to their partners by mutual fit, meaning that they acquire a fixed structure upon binding to a well structured template [1]. Furthermore, a recent survey of 3D structures of protein–peptide complexes has estimated that neighbouring residues account for 20% of the global binding energy of peptide–mediated interactions. They are thought to improve the interaction affinity with the native partner or to impede non–native interactions [18].
sheet of the partner [17]. This coincides with the observation that two thirds of LMs bind to their partners by mutual fit, meaning that they acquire a fixed structure upon binding to a well structured template [1]. Furthermore, a recent survey of 3D structures of protein–peptide complexes has estimated that neighbouring residues account for 20% of the global binding energy of peptide–mediated interactions. They are thought to improve the interaction affinity with the native partner or to impede non–native interactions [18].
The evolutionary context of LMs has also been studied and used in predictive methods. Convergent evolution of LMs is at the basis of discovery algorithms like SLiMFinder [19] and DILIMOT [20], which search for over–represented motifs in unrelated proteins with a common functional attribute. Additionally, conservation of LMs in closely and distantly related proteins has been used to improve the identification of functional instances of known LM patterns [11], [21], [22], [23]. Methods for de novo discovery, have also benefited from the evolutionary signal provided by analysing patterns of conservation. SLiMFinder uses global or local sequence conservation to improve confidence in motif predictions [9], [24]; DILIMOT takes into account conservation of the motif in orthologs as part of the scoring scheme [10].
It is clear that LM predictions from the current generation of predictors require experimental validation to be considered genuine. The methods are often working at the limits of signal to noise and are dependent on the information content of the bioinformatics databases being used for LM prediction [3], [25], [26]. Nevertheless, LM prediction methods could be valuable tools for the study of high dimensional systems like the protein signalling networks. Therefore it is necessary to move from the identification of a LM in a protein towards the prediction of the role of that instance inside the functional framework of the protein, e.g. its network of interactors.
This work addresses the study of LM context from an evolutionary point of view. Conservation patterns of regions flanking 116 LM functional instances were examined in relation to the presence/absence of the LM inside protein families. Both sequence identity and structural tendency of the LM context was analysed. Notwithstanding the difficulty of assessing the generality of the results, due to the fragmentary knowledge about the complete set of cellular LMs, distinct evolutionary patterns were identified. For the majority of the studied instances, conservation of the local amino acid sequence and/or the local structural tendency was found to be differentially distributed between sequence pairs with and without the motif. These findings are supported by examples where the regulation of the LM mediated interaction has been shown to depend on the modifications at neighbouring positions or is thought to benefit from the binding versatility of disordered regions. Taken together, the results of the present study suggest that it is possible to enrich the identification of a LM instance with regulatory information by analysing the conservation pattern of its flanking regions.
Methods
Dataset
The analysis was done using the MAFFT [27] alignments of 75 protein families containing 85 protein sequences that have 116 non–redundant LM instances linked to experimental evidence in the ELM database [3]. Protein families were taken from the TreeFam4.0 database [28]. The 40% of the families in the dataset include proteins of metazoans (vertebrates and invertebrates) and plants (A. thaliana) or yeast (S. cerevisiae and S. pombe); 42% contain vertebrate and invertebrate sequences; the remaining 18% have only vertebrate proteins.
The presence/absence of each instance was determined in the sequences belonging to the protein family by looking for the regular expression of the corresponding LM, as defined in the ELM resource [3]. Sequence pairs in the protein family were assigned to one of the following sets: the presence set (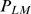 ), when both sequences have a match to the regular expression in the same position of the annotated ELM instance; the absence set (
), when both sequences have a match to the regular expression in the same position of the annotated ELM instance; the absence set (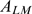 ), when the instance is missing in one of the sequences. Only protein sequences having a sub–sequence aligned to the region corresponding to the ELM instance were considered. This classification assumes that a LM instance is functional if it appears in a position that, according to the alignment, corresponds to that of the annotated ELM instance. Moreover, it depends on the adequacy of the ELM regular expression and might overestimate the size of the
), when the instance is missing in one of the sequences. Only protein sequences having a sub–sequence aligned to the region corresponding to the ELM instance were considered. This classification assumes that a LM instance is functional if it appears in a position that, according to the alignment, corresponds to that of the annotated ELM instance. Moreover, it depends on the adequacy of the ELM regular expression and might overestimate the size of the 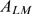 set. Sequence pairs where the instance is absent in both sequences were not considered, since any interpretation about their differences would imply making assumptions about the gain or loss of the instances during the evolution of the protein family.
set. Sequence pairs where the instance is absent in both sequences were not considered, since any interpretation about their differences would imply making assumptions about the gain or loss of the instances during the evolution of the protein family.
To perform comparisons between LMs located in similar structural contexts, each instance was assigned to a structural class. The structural class was defined in terms of disorder/order at two levels: protein family and module, where module is defined as an independent unit within the protein sequence with globular or disorder tendency. This classification was done in a semi–automated way, using the IUPred disorder predictor [29] and the SMART module research tool [30] and averaging the results over all the homologous sequences. Proteins were classified as disordered, when more than 70% of their residues are disordered (conservative IUPred threshold of 0.4); globular, when more than 70% of the residues belong to one or more SMART globular modules; mixed, for the proteins that could not be clearly allocated to any of the previous classes. Modules were similarly defined as disordered or globular. The final dataset has instances in all of the 6 structural classes resulting from the combination of protein and module class (see Text S1 for the complete dataset).
Local structure and sequence conservation metrics
Differences between sequences were studied in terms of conservation of the local structural tendency and the amino acid sequence at both local and global level. The conservation of the local structure was calculated for each sequence pair 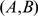 as:
as:
where 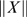 indicates the absolute value of
indicates the absolute value of 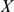 ;
; 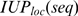 is the IUPred value averaged over the amino acids located 15 positions to the left and right of the LM in sequence
is the IUPred value averaged over the amino acids located 15 positions to the left and right of the LM in sequence 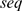 ;
; 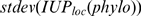 is the standard deviation of
is the standard deviation of 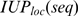 for all the sequences in the protein family. Therefore,
for all the sequences in the protein family. Therefore, 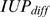 indicates whether the difference of the local tendency to disorder/order between A and B is higher or lower than the variability inside the whole protein family. Normalisation by standard deviation permits the comparison among instances belonging to different protein families, which have different IUPred variabilities. The
indicates whether the difference of the local tendency to disorder/order between A and B is higher or lower than the variability inside the whole protein family. Normalisation by standard deviation permits the comparison among instances belonging to different protein families, which have different IUPred variabilities. The 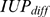 varies between −1 and infinity, with negative or small positive values indicating conservation of the local structural tendency around the LM instance.
varies between −1 and infinity, with negative or small positive values indicating conservation of the local structural tendency around the LM instance.
The protein sequence conservation between each pair 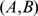 was calculated as the full-length sequence identity according to the multiple sequence alignment (
was calculated as the full-length sequence identity according to the multiple sequence alignment (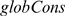 ) and as the sequence identity of the amino acids in the 15 positions flanking the LM instance both sides (
) and as the sequence identity of the amino acids in the 15 positions flanking the LM instance both sides (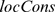 ).
).
The definition of 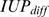 and
and 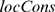 depends on the alignment quality of the flanking regions. Acknowledging the poor performance of multiple alignment programs in disordered regions [31], those values were calculated only when the 15 residue windows surrounding the instance contained at least 75% of non–gap positions; in other words, when there was enough information to estimate average conservation values.
depends on the alignment quality of the flanking regions. Acknowledging the poor performance of multiple alignment programs in disordered regions [31], those values were calculated only when the 15 residue windows surrounding the instance contained at least 75% of non–gap positions; in other words, when there was enough information to estimate average conservation values.
Frequency profiles and correlation between 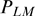
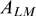 sets
sets
The distribution of the 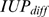 values as a function of the
values as a function of the 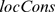 or
or 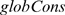 was represented as frequency profiles. Those profiles are no more than two-dimensional histograms which represent the number of pairs falling in a given range of the
was represented as frequency profiles. Those profiles are no more than two-dimensional histograms which represent the number of pairs falling in a given range of the 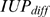 and a given range of
and a given range of 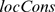 or
or 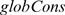 . Counts were normalised to avoid biases due to the different sizes of the protein families. Frequency profiles were calculated for the
. Counts were normalised to avoid biases due to the different sizes of the protein families. Frequency profiles were calculated for the 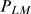 and
and 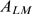 sets of each instance. Almost half of the instances (53 out of the 116) have a sufficient number of sequence pairs to allow this statistical representation.
sets of each instance. Almost half of the instances (53 out of the 116) have a sufficient number of sequence pairs to allow this statistical representation.
In order to compare the similarity between the 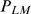 and
and 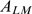 profiles, their correlation was estimated using the Spearman coefficient. The Spearman coefficient ranges between 1, high correlation, and −1 complete anticorrelation. In the context of the present study, a correlation of 1 would indicate that the
profiles, their correlation was estimated using the Spearman coefficient. The Spearman coefficient ranges between 1, high correlation, and −1 complete anticorrelation. In the context of the present study, a correlation of 1 would indicate that the 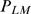 and
and 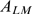 sets cover the same
sets cover the same 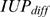 and
and 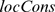 /
/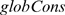 ranges. A correlation of −1 would imply that those ranges are completely disjoint and diametrically opposed (e.g. high
ranges. A correlation of −1 would imply that those ranges are completely disjoint and diametrically opposed (e.g. high 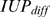 and low
and low 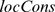 for
for 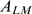 while low
while low 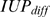 and high
and high 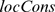 for
for 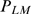 ). Small positive or negative values indicate that the
). Small positive or negative values indicate that the 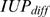 and
and 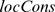 /
/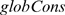 ranges of the
ranges of the 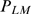 and
and  sets tend to be disjoint but not opposite.
sets tend to be disjoint but not opposite.
Statistical coupling analysis
Positional coupling [32] between each non–wildcard position of the LM instance and each one of the residues of the module (globular or disordered) was calculated. The method could be applied for the instances located in modules whose multiple sequence alignment is diverse, such that the frequencies of amino acids at some positions are near to their mean values in all proteins, i.e. those positions are poorly conserved. Only positions in the module with coupling values that emerge from noise were considered. Noise threshold was set to two standard deviations above the mean coupling value of all the residues in the module.
Coupled positions were classified as neighbouring, when located within 15 positions both sides of the LM instance, and as distant for all the others. For the instances located towards the limits of the module, the partial window (i.e. less than 15 residues) was considered. In other words, the module boundaries were taken into account when defining neighbouring residues.
Assuming that the probability of coupling is equal for any residue in the protein sequence, the number of coupled positions was weighted by the total number of potentially coupled positions: 30 for the neighbouring residues and the length of the module minus the length of the instance region (15+ motif length +15) for the distant ones. This weighted value is defined as the frequency of coupling.
Results
LM presence and the conservation of the local structural tendency
This section explores the relationship between LM presence and the conservation of the structural tendency in the regions flanking the motif. Figure 1 shows the 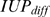 distribution for the pairs of the
distribution for the pairs of the 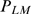 and the
and the 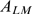 sets averaged over all the instances. Even if there is a non–negligible overlap between the two distributions, negative
sets averaged over all the instances. Even if there is a non–negligible overlap between the two distributions, negative 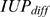 values, that indicate conservation of the local structural tendency, are significantly more frequent in
values, that indicate conservation of the local structural tendency, are significantly more frequent in 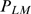 than in
than in 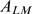 sequence pairs (Kolmogorov-Smirnov test: difference = 0.423, p-value
sequence pairs (Kolmogorov-Smirnov test: difference = 0.423, p-value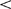 0.00001). This difference is lost for higher
0.00001). This difference is lost for higher 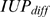 values.
values.
Figure 1. Frequency distribution of IU Pdiff for the PLM and ALM sets.

Frequency is calculated per instance as the proportion of sequence pairs falling in a given IU Pdiff range. Error bars indicate the standard deviation of the frequency when averaging over all the instances in that range. Significant difference (p-value<0.00001) between PLM and ALM distributions is marked by the asterisk.
When the analysis is repeated comparing the 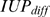 distributions of
distributions of 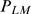 and
and 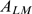 sets of each instance, inside each protein family, analogous results are obtained. For all the structural classes the mean
sets of each instance, inside each protein family, analogous results are obtained. For all the structural classes the mean 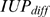 for the
for the 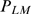 set is lower than that of the
set is lower than that of the 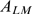 set, as shown in Table 1. Additionally, comparison of the two
set, as shown in Table 1. Additionally, comparison of the two 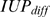 distributions gives statistically significant differences for 57 out of 116 instances (Kolmogorov-Smirnov test: differences between 0.303 and 0.791, p-values
distributions gives statistically significant differences for 57 out of 116 instances (Kolmogorov-Smirnov test: differences between 0.303 and 0.791, p-values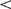 0.05, see complete results in Table S1). This means that, for almost 50% of the instances the
0.05, see complete results in Table S1). This means that, for almost 50% of the instances the 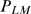 and
and 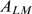 sets have different local structural tendencies that can be quantified and used to statistically differentiate between those sequence pair sets.
sets have different local structural tendencies that can be quantified and used to statistically differentiate between those sequence pair sets.
Table 1. IU Pdiff ranges and mean IU Pdiff for the PLM and ALM sets per structural class.
| protein class | module class | numbera | min | max | mean | |||
| Plm | Alm | Plm | Alm | Plm | Alm | |||
| DIS | DIS GLOB | 41 | −0.9 | −0.8 | 3.4 | 4.9 | 0.6 | 1.2 |
| 4 | −1.0 | −1.0 | 1.8 | 3.5 | 0.0 | 0.6 | ||
| GLOB | DIS GLOB | 16 | −0.9 | −0.9 | 3.9 | 6.9 | 0.6 | 1.6 |
| 14 | −1.0 | −0.8 | 2.1 | 5.2 | 0.1 | 1.2 | ||
| MIXED | DIS GLOB | 32 | −0.9 | −0.9 | 3.2 | 6.0 | 0.5 | 1.8 |
| 9 | −1.0 | −0.9 | 2.1 | 4.6 | 0.2 | 1.3 | ||
IU Pdiff values are averaged over all the instances belonging to the same structural class.
number of instances per structural class.
For the remaining instances the 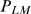 and
and 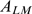 sets have the same
sets have the same 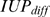 ranges. These instances suggest that, sometimes, the local structure is conserved even if the LM is lost. This is not surprising if considering that the LM is a module evolving inside a higher order unit (e.g. the protein sequence) composed of several other functional modules. Disambiguation of the selective pressure imposed by the LM, based exclusively in its local structure conservation, will be difficult in these cases. Consequently it is worth analysing the conservation of the local structural tendency in relation to the evolution of the rest of the protein modules.
ranges. These instances suggest that, sometimes, the local structure is conserved even if the LM is lost. This is not surprising if considering that the LM is a module evolving inside a higher order unit (e.g. the protein sequence) composed of several other functional modules. Disambiguation of the selective pressure imposed by the LM, based exclusively in its local structure conservation, will be difficult in these cases. Consequently it is worth analysing the conservation of the local structural tendency in relation to the evolution of the rest of the protein modules.
LM evolution and the relationship between local structural tendency and sequence conservation
In order to explore how the conservation of the local structure, in terms of disorder/order, is related to the evolution of the protein sequence, the distribution of 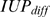 was analysed as a function of the global and local sequence conservation. Frequency profiles of the combined distribution of
was analysed as a function of the global and local sequence conservation. Frequency profiles of the combined distribution of 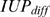 versus the local and global sequence conservation (
versus the local and global sequence conservation (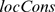 and
and 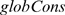 ) were calculated for both the
) were calculated for both the 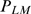 and
and 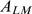 sets of each instance.
sets of each instance.
Figure 2 presents the frequency profile of 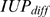 versus
versus 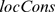 and
and 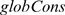 . Since they represent the distribution of the above variables for the
. Since they represent the distribution of the above variables for the 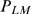 and
and 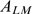 sets averaged over all the instances, those profiles do not allow a comparative analysis between
sets averaged over all the instances, those profiles do not allow a comparative analysis between 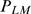 and
and 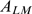 sets or sequence conservation variables. Differences among protein families due to dissimilar evolutionary rates are not averaged out. The structural composition of proteins belonging to different structural classes (disordered, globular, mixed) might add further disparity, since sequences with long disordered regions tend to have heterogeneous evolutionary rates [33].
sets or sequence conservation variables. Differences among protein families due to dissimilar evolutionary rates are not averaged out. The structural composition of proteins belonging to different structural classes (disordered, globular, mixed) might add further disparity, since sequences with long disordered regions tend to have heterogeneous evolutionary rates [33].
Figure 2. Frequency profiles for the PLM and ALM sets.

Distribution of IU Pdiff as a function of sequence conservation: locCons (A,B) and globCons (C,D). Colour represents the frequency of sequence pairs whose local structure and sequence conservation values fall in a given range of IU Pdiff and locCons/globCons, averaged over all the instances.
Nevertheless those profiles provide an idea about the general trends of the relationship between 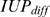 and sequence conservation. As expected, the
and sequence conservation. As expected, the 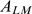 sets cover mainly low sequence conservation values (Figure 2B and D). Indeed, even if low sequence similarity does not necessarily imply the loss of the LM, closely related protein sequences are more likely to have similar LM instances than distantly related or paralogous sequences [1], [4]. Instead, the frequency profiles of the
sets cover mainly low sequence conservation values (Figure 2B and D). Indeed, even if low sequence similarity does not necessarily imply the loss of the LM, closely related protein sequences are more likely to have similar LM instances than distantly related or paralogous sequences [1], [4]. Instead, the frequency profiles of the 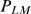 sets exhibit an additional feature: low
sets exhibit an additional feature: low 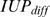 values are frequent in both high and low sequence conservation values (Figure 2A and C). In other words, conservation of the amino acid sequence is not required for the maintenance of the disorder tendency around the LM.
values are frequent in both high and low sequence conservation values (Figure 2A and C). In other words, conservation of the amino acid sequence is not required for the maintenance of the disorder tendency around the LM.
The above result suggests that structural and sequence conservation, intended as sequence identity, are not redundant and both might provide information about the LM evolution. Indeed the IUPred method predicts disordered/ordered regions by estimating the total pair wise interresidue interaction energy [29] and therefore there is no a priori reason why the conservation of the local structural tendency should imply the conservation of the exact amino acid sequence. To further explore this, the frequency profiles of the 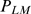 and
and 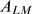 sets of each instance were obtained and their Spearman correlation coefficient calculated separately. The analysis per instance has the additional advantage of discarding artificial differences between
sets of each instance were obtained and their Spearman correlation coefficient calculated separately. The analysis per instance has the additional advantage of discarding artificial differences between 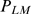 and
and 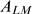 caused by dissimilar evolutionary rates among the protein families.
caused by dissimilar evolutionary rates among the protein families.
All the structural classes have low mean correlation coefficients indicating that, on average, the 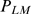 and
and 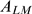 frequency profiles of each instance can be discriminated; correlation values range from 0.11 to 0.34 for
frequency profiles of each instance can be discriminated; correlation values range from 0.11 to 0.34 for 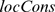 and from 0.02 to 0.22 for
and from 0.02 to 0.22 for 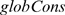 depending on the structural class (see Table S2). The low number of instances per structural class, makes any comparative statistical analysis unreliable, e.g. between structural classes or conservation variables. Nevertheless, having a closer look at the results per instance (Table 2), three groups with distinct behaviour can be identified. Examples of instances belonging to each one of those groups are presented in Figure 3. Those trends do not change when the
depending on the structural class (see Table S2). The low number of instances per structural class, makes any comparative statistical analysis unreliable, e.g. between structural classes or conservation variables. Nevertheless, having a closer look at the results per instance (Table 2), three groups with distinct behaviour can be identified. Examples of instances belonging to each one of those groups are presented in Figure 3. Those trends do not change when the 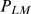 set is enlarged by considering subsequences that partially match the ELM regular expression as LM instances (see Table S3 for further details).
set is enlarged by considering subsequences that partially match the ELM regular expression as LM instances (see Table S3 for further details).
Table 2. Spearman correlation coefficient between the PLM and ALM frequency profiles.
| structural classa | TreeFam id | UniProt id | ELM id | Start | locCons corr | globCons corr |
| TF106427 | P29374 | LIG_RB | 957 | −0.14 | 0.12 | |
| TF106496 | P25054 | TRG_NES_CRM1_1 | 163 | −0.09 | −0.05 | |
| TF316358 | P10636 | LIG_SH3_1 | 565 | −0.05 | 0.42 | |
| TF300785 | P51531 | LIG_RB | 1294 | −0.01 | 0.12 | |
| TF314303 | O15147 | LIG_SH3_5 | 389 | −0.01 | 0.16 | |
| TF325994 | P35568 | LIG_14-3-3_3 | 267 | 0.12 | 0.29 | |
| TF331759 | O60315 | LIG_CtBP | 785 | 0.16 | 0.20 | |
| TF323952 | P17535 | LIG_COP1 | 241 | 0.16 | 0.33 | |
| DIS DIS | TF325994 | P35568 | LIG_14-3-3_3 | 371 | 0.29 | 0.42 |
| TF318445 | O35973 | TRG_NES_CRM1_1 | 488 | 0.32 | 0.50 | |
| TF325994 | P35570 | LIG_SH2_GRB2 | 896 | 0.45 | 0.52 | |
| TF101166 | P05205 | LIG_RB | 61 | 0.53 | 0.10 | |
| TF320471 | P35712 | LIG_CtBP | 424 | 0.36 | 0.15 | |
| TF313876 | Q91VZ6 | LIG_Clathr_ClatBox_l | 192 | 0.31 | 0.16 | |
| TF325994 | P35570 | LIG_SH2_PTP2 | 1179 | 0.23 | 0.21 | |
| TF331759 | O60315 | LIG_CtBP | 859 | 0.34 | 0.31 | |
| TF323952 | P05412 | MOD_PIKK_l | 246 | 0.55 | 0.52 | |
| TF105306 | Q00987 | MOD_PIKK_l | 392 | −0.02 | 0.02 | |
| DIS GLOB | TF323952 | P05412 | LIG_MAPK_1 | 32 | 0.55 | 0.27 |
| TF314861 | Q05140 | LIG_PIP2_ANTH_1 | 28 | 0.51 | 0.36 | |
| TF325994 | P35570 | MOD_CK2_1 | 96 | 0.48 | 0.39 | |
| TF335892 | P04235 | TRG_LysEnd_APsAcLL_l | 138 | 0.18 | 0.28 | |
| TF300460 | Q04656 | TRG_LysEnd_APsAcLL_l | 1483 | 0.38 | 0.00 | |
| TF105137 | Q02750 | LIG_MAPK_1 | 3 | 0.34 | 0.11 | |
| TF300618 | P27797 | TRG_ER_KDEL_l | 414 | 0.53 | 0.22 | |
| GLOB DIS | TF105135 | P45985 | LIG_MAPK_1 | 40 | 0.36 | 0.28 |
| TF105115 | Q99683 | LIG_14-3-3_1 | 963 | 0.33 | 0.31 | |
| TF300540 | P04040 | TRG_PTS1 | 523 | 0.38 | 0.36 | |
| TF105044 | P36604 | TRG_ER_KDEL_1 | 660 | 0.45 | 0.42 | |
| TF106381 | P09103 | TRG_ER_KDEL_l | 506 | 0.52 | 0.48 | |
| TF105042 | P17156 | LIG_TPR | 630 | 0.65 | 0.52 | |
| TF335892 | P19377 | MOD_TYR_ITAM | 146 | −0.05 | −0.08 | |
| TF101211 | Q8AY27 | MOD_PIKK_l | 2 | 0.03 | 0.00 | |
| TF101004 | P24385 | LIG_RB | 5 | 0.22 | 0.10 | |
| GLOB GLOB | TF105115 | Q99683 | LIG_RB | 916 | 0.19 | 0.17 |
| TF105122 | P28562 | LIG_MAPK_2 | 339 | 0.44 | 0.25 | |
| TF315491 | P27918 | MOD_CMANNOS | 318 | 0.37 | 0.27 | |
| TF105331 | Q96GD4 | LIG_APCC_Dbox_l | 314 | 0.65 | 0.53 | |
| TF316520 | O00268 | LIG_HP1_1 | 762 | −0.07 | 0.01 | |
| TF101065 | Q12834 | LIG_APCC_KENbox_2 | 96 | −0.01 | 0.04 | |
| TF313542 | P49418 | LIG_AP2alpha_l | 324 | 0.03 | 0.15 | |
| TF300772 | P49736 | MOD_PIKK_1 | 105 | 0.09 | 0.16 | |
| MIXED DIS | TF105351 | P35465 | LIG_SH3_2 | 13 | 0.11 | −0.06 |
| TF332149 | O75074 | TRG_LysEnd_GGAAcLL_l | 764 | 0.35 | 0.01 | |
| TF106101 | P04637 | TRG_NES_CRM1_1 | 339 | 0.04 | 0.01 | |
| TF318574 | Q9UJY5 | TRG_LysEnd_GGAAcLL_2 | 355 | 0.34 | 0.03 | |
| TF101089 | P53350 | LIG_APCC_Dbox_l | 336 | 0.33 | 0.14 | |
| TF105722 | P35251 | LIG_RB | 662 | −0.11 | −0.06 | |
| TF300901 | P23396 | LIG_MAPK_2 | 77 | 0.24 | 0.41 | |
| MIXED GLOB | TF333209 | P54274 | MOD_PIKK_1 | 216 | −0.08 | −0.10 |
| TF318283 | P46061 | MOD_SUMO | 525 | 0.10 | −0.07 | |
| TF101066 | Q8UWJ8 | LIG_CYCLIN_l | 445 | 0.31 | 0.19 | |
| TF330851 | P10912 | LIG_SH2_STATB | 566 | 0.30 | 0.21 |
Spearman correlation coefficient calculated between the PLM and ALM frequency profiles of each instance. Correlation of the frequency profiles of IU Pdiff versus locCons and IU Pdiff versus globCons are indicated as locCons corr and globCons corr respectively. Correlation of 1 would indicate that the PLM and ALM sets cover the same IU Pdiff and locCons/globCons ranges. A correlation of −1 would imply that those ranges are completely disjoint and diametrically opposed (e.g. high IU Pdiff and low locCons for ALM while low IU Pdiff and high locCons for PLM). Small positive or negative values indicate that the ranges tend to be disjoint but not opposite. Instances in bold have PLM and ALM sets with significantly different IU Pdiff distributions (p-values<0.05).
protein and module structural classes.
Figure 3. Examples of evolutionary patterns of the regions flanking LM.

IU Pdiff versus locCons and globCons for the sequence pairs in PLM (black dots) and ALM (blue asterisks) sets per instance. Three groups with distinct evolutionary behaviour can be identified: instances whose PLM and ALM frequency profiles of IU Pdiff versus locCons are less correlated than the corresponding IU Pdiff versus globCons profiles (A); instances where the contrary is true (B); instances that, additionally, have a significantly different IU Pdiff distribution (C,D).
The first group consists of those instances whose 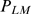 and
and 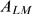 frequency profiles of
frequency profiles of 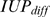 versus
versus 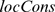 are less correlated than the corresponding
are less correlated than the corresponding 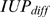 versus
versus 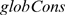 profiles (Figure 3A). This indicates that variations in the local protein sequence are more connected to the LM presence/absence than the modifications happening in the rest of the protein. The 37% of the instances in Table 2 have this kind of behaviour, especially those ones located in disordered modules of disordered proteins (8 out of 13).
profiles (Figure 3A). This indicates that variations in the local protein sequence are more connected to the LM presence/absence than the modifications happening in the rest of the protein. The 37% of the instances in Table 2 have this kind of behaviour, especially those ones located in disordered modules of disordered proteins (8 out of 13).
The second group is formed of instances where the contrary is true, meaning that the LM presence/absence is better distinguished by the global conservation (Figure 3B). In those cases, the main selective pressure on the LM presence might be coming from the protein sequence as a whole unit. Not surprisingly all of the 8 instances located in globular proteins (both in disordered and globular modules) belong to this group.
A third group of instances appears when merging the results of the previous section, that is to say, considering those instances whose 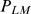 and
and 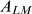 sets have significantly different
sets have significantly different 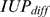 distributions (in bold in Table 2, Figure 3C and D). In these cases, the presence or absence of the LM is correlated with changes in both the local structural tendency and the sequence conservation. Those instances reach, on average, lower correlation values independently from the conservation variable (0.18 for the
distributions (in bold in Table 2, Figure 3C and D). In these cases, the presence or absence of the LM is correlated with changes in both the local structural tendency and the sequence conservation. Those instances reach, on average, lower correlation values independently from the conservation variable (0.18 for the 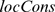 and 0.15 for the
and 0.15 for the 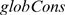 ) than the instances with no significant
) than the instances with no significant 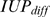 distinction between
distinction between 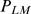 and
and 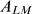 (0.30 for
(0.30 for 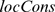 and 0.26 for
and 0.26 for 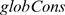 ). This last group of instances is the best evidence in favour of the hypothesis proposed above, about the additive value of the structural and sequence conservation information in the analysis of LM evolution.
). This last group of instances is the best evidence in favour of the hypothesis proposed above, about the additive value of the structural and sequence conservation information in the analysis of LM evolution.
Co-evolution of the LM and their flanking regions
To get additional evidence about the co–evolution between LMs and their flanking regions, the statistical coupling [32] was used as an independent method. This method has been used to identify clusters of positions that statistically co–vary with one another and therefore are likely to co–evolve and to be functionally related [34]. In this case only pair coupling between the non–wildcard positions of the LM instance and all the other residues in the corresponding module was considered. The frequency of coupling with neighbouring and distant residues was calculated and compared in terms of the sequence conservation that best describes the LM evolution, that is to say the variable that gives the lowest correlation in Table 2.
For the instances that have lower 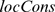 correlation (e.g. Figure 3A), the frequency of neighbouring coupling is significantly higher (Kolmogorov-Smirnov test: difference = 0.576, p-value
correlation (e.g. Figure 3A), the frequency of neighbouring coupling is significantly higher (Kolmogorov-Smirnov test: difference = 0.576, p-value 0.005) than the frequency of distant coupling (Figure 4A). In other words, the instances whose evolution is better described by the local sequence conservation combined with the
0.005) than the frequency of distant coupling (Figure 4A). In other words, the instances whose evolution is better described by the local sequence conservation combined with the 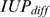 have a higher chance of correlated amino acid changes with neighbouring rather than with distant residues in the module. Conversely, for the instances where the global sequence conservation is the better descriptor (e.g. Figure 3B), the coupling between non–wildcard positions and neighbouring or distant positions is equally frequent (Figure 4B).
have a higher chance of correlated amino acid changes with neighbouring rather than with distant residues in the module. Conversely, for the instances where the global sequence conservation is the better descriptor (e.g. Figure 3B), the coupling between non–wildcard positions and neighbouring or distant positions is equally frequent (Figure 4B).
Figure 4. Frequency of coupling between LM and neighbouring or distant residues.

Box plots show the distribution of the frequency of coupling for instances in Table 2. A. Distribution for instances whose presence/absence is better described by the local rather than the global sequence conservation (i.e. locCons correlation<globCons correlation) B. Distribution for instances with globCons correlation<locCons correlation.
Discussion
This study presents evidence for the concerted evolution of LMs and their flanking regions. Although the current knowledge of the complete set of cellular LMs is fragmentary and it is not possible to assess the representativity of the analysed dataset, there are clear trends that are worth considering. LMs are known to be evolutionarily labile modules, which can be easily lost by point mutation [4]. Nonetheless, the results of the present study show that LMs, in some cases, determine the conservation of the structural tendency and/or the sequence of the neighbouring amino acids. Here those findings are discussed in the light of the protein interactions mediated by LMs.
In the first section of the Results it was shown that, for some instances, the conservation of the LM is associated with the maintenance of the structural tendency of the surrounding residues. What is the meaning of this conservation? As mentioned in the Introduction, two thirds of the LM–mediated interactions lead to the formation of secondary structure elements ( –helices or
–helices or 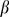 –strands) [1]. If the LM functionality is to be maintained, the structural properties of the neighbouring amino acids that allow such disorder/order transition are likely to be conserved. This local propensity would be reflected by the corresponding IUPred values and hence the low
–strands) [1]. If the LM functionality is to be maintained, the structural properties of the neighbouring amino acids that allow such disorder/order transition are likely to be conserved. This local propensity would be reflected by the corresponding IUPred values and hence the low 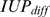 observed in the
observed in the 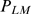 sets would indicate the conservation of such propensity.
sets would indicate the conservation of such propensity.
However, the conservation of the local structural tendency could also indicate the maintenance of the local disorder. Several studies on protein–protein interactions have drawn attention to the importance of intrinsic disorder in the formation of protein complexes [6], [35], [36], [37]. If the local disorder provides the flexibility required to bind different patterns, it is not surprising to observe the conservation of this structural tendency in the regions involved in such interactions. Previous work by [38] has connected the conservation of predicted disordered regions in eukaryotic proteins with DNA/RNA binding domains. The conservation of disorder around LMs would extend this result to a broader set of biological processes.
The instances of the molecular hub p53 exemplify the double meaning of the structural conservation measured by the IU Pdiff. For three out of four of the p53 instances in the dataset (TRG_NES_CRM1_1, 339–352; MOD_SUMO, 385–388; MOD_PIKK_1, 12–18), the presence of the instance coincides with the conservation of the local structural tendency. They belong to the group of instances that have a significantly different distribution of the 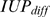 between
between 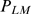 and
and 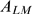 sets (p-value
sets (p-value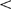 0.05). Those instances are located in the C and N terminal regions of P53, which are disordered modules known to bind different partners by acquiring different conformations [39]. Additionally, the MOD_SUMO and the MOD_PIKK_1 (but not the TRG_NES_CRM1_1) occur in predicted α–MoREs, disordered regions having propensities to form α–helix upon molecular recognition [16].
0.05). Those instances are located in the C and N terminal regions of P53, which are disordered modules known to bind different partners by acquiring different conformations [39]. Additionally, the MOD_SUMO and the MOD_PIKK_1 (but not the TRG_NES_CRM1_1) occur in predicted α–MoREs, disordered regions having propensities to form α–helix upon molecular recognition [16].
A more detailed study of the structural conservation as function of the different types of mutual fit interaction (i.e. α–helix formation, 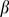 augmentation or irregular topology) may be interesting. It would shade light on the specific requirements of each conformation. This would require the definition of a more elaborated metric for the local structure conservation than the
augmentation or irregular topology) may be interesting. It would shade light on the specific requirements of each conformation. This would require the definition of a more elaborated metric for the local structure conservation than the 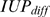 . However, independently from its specific meaning, the structural tendency conservation around the LM suggests the occurrence of overlapping interaction surfaces. Those clustered overlaps are likely to entail different regulatory mechanisms for the spatial or temporal isolation of the mutually exclusive interactions.
. However, independently from its specific meaning, the structural tendency conservation around the LM suggests the occurrence of overlapping interaction surfaces. Those clustered overlaps are likely to entail different regulatory mechanisms for the spatial or temporal isolation of the mutually exclusive interactions.
In the second and third part of the Results it was shown that the presence of some LM instances is accompanied by the conservation of the amino acids flanking the motif. This is the case for 42% of the instances in Table 2 that have 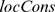 correlation values lower that 0.20 between the
correlation values lower that 0.20 between the 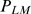 and
and 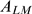 sets. The local sequence conservation could be explained in some cases by the conservation of the local structural tendency (instances in bold in Table 2, Figure 3C and D). Still, as shown in the Results (Figure 2), sequence identity does not seem to be a requirement for the maintenance of the local order/disorder tendency. Indeed, it has been recently demonstrated by nuclear magnetic resonance spectroscopy that intrinsically disordered regions can maintain their dynamic behaviour despite low sequence similarity [40]. Yet there must be a functional meaning for the local sequence conservation associated with these instances, especially considering that it allows to discriminate sequences with and without the motif (
sets. The local sequence conservation could be explained in some cases by the conservation of the local structural tendency (instances in bold in Table 2, Figure 3C and D). Still, as shown in the Results (Figure 2), sequence identity does not seem to be a requirement for the maintenance of the local order/disorder tendency. Indeed, it has been recently demonstrated by nuclear magnetic resonance spectroscopy that intrinsically disordered regions can maintain their dynamic behaviour despite low sequence similarity [40]. Yet there must be a functional meaning for the local sequence conservation associated with these instances, especially considering that it allows to discriminate sequences with and without the motif (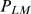 and
and 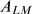 sets), even when local structural tendencies between those sequences are not significantly different (e.g. Figure 3A and B). Furthermore, these instances have higher chance of co–evolving with the neighbouring residues in comparison to the distant ones (Figure 4A).
sets), even when local structural tendencies between those sequences are not significantly different (e.g. Figure 3A and B). Furthermore, these instances have higher chance of co–evolving with the neighbouring residues in comparison to the distant ones (Figure 4A).
It is likely that the flanking regions of those instances are related with the regulation of the LM or with the regulation of another interaction, which is functionally connected to the one mediated by the motif. This is the case of the LIG_AP2alpha_1 in positions 324–328 of amphiphysin (P49418, 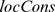 correlation 0.03), which is involved in clathrin coated vesicle formation. Phosphorylation of amphiphysin by Cdk5 in S276, S285 and T310 has been shown to directly regulate the intramolecular interaction in amphiphysin, which in turn regulates dynamin-dependent endocytosis [41], [42]. Likewise, other instances with
correlation 0.03), which is involved in clathrin coated vesicle formation. Phosphorylation of amphiphysin by Cdk5 in S276, S285 and T310 has been shown to directly regulate the intramolecular interaction in amphiphysin, which in turn regulates dynamin-dependent endocytosis [41], [42]. Likewise, other instances with 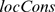 correlation between −0.05 and 0.16 (LIG_SH3_1 P10636 565–572, LIG_COP1 P17535 241–248) have experimentally verified phosphorylation sites in their flanking regions: T561 for P17535 and S251, S255 and S259 for P17535 [25]. Those phosphorylation site are likely to regulate the local protein conformation and activity, as recently shown in a phosphoproteomic analysis of the mouse brain cytosol [43].
correlation between −0.05 and 0.16 (LIG_SH3_1 P10636 565–572, LIG_COP1 P17535 241–248) have experimentally verified phosphorylation sites in their flanking regions: T561 for P17535 and S251, S255 and S259 for P17535 [25]. Those phosphorylation site are likely to regulate the local protein conformation and activity, as recently shown in a phosphoproteomic analysis of the mouse brain cytosol [43].
Finally, it is opportune to consider how current LM prediction methods can benefit from these results. A simple sequence analysis, similar to the one described here, would allow the identification of flanking regions with relevant conservation patterns, adding contextual information to already predicted LM instances. This can lead to a more detailed understanding of the role of LMs in determining the protein function. Indeed we consider that the LM field is ready – and has the potential – to go one step further from the timeless binary interactions towards the construction of more dynamic and realistic protein networks.
Supporting Information
Dataset of functional instances. List of the 116 instances, classified per structural class with phylogeny, sequence and motif identifiers.
(0.00 MB TXT)
Comparison of the IUPdiff distribution between the PLM and ALM sets. Kolmogorov-Smirnov test comparing the IUPdiff distribution of the PLM and ALM sets of each instance. The difference is the Kolmogorov-Smirnov statistic calculated from the cumulative distributions of the compared samples.
(0.03 MB PDF)
Mean and standard deviation of the correlation between PLM and ALM frequency profiles. Spearman correlation coefficient calculated between the PLM and ALM frequency profiles of each instance. Correlation of the frequency profiles of IUPdiff versus locCons and IUPdiff versus globCons are indicated as locCons corr and globCons corr respectively.
(0.02 MB PDF)
Effect of the stringency of the regular expression matching on the correlation between the PLM and ALM frequency profiles. Spearman correlation coefficient calculated between the PLM and ALM frequency profiles of each instance. Correlation of the frequency profiles of IUPdiff versus locCons and IUPdiff versus globCons are indicated as locCons corr and globCons corr respectively. Percentages indicate the stringency used to define a match to the ELM regular expression: 100% stringency supposes that a LM is present only if there is a perfect match to the ELM regular expression in the same position of the annotated instance; lower percentages consider that a LM is present also in case of partial match to the regular expression. Correlation values in bold show the biggest difference (more than 0.05) with the corresponding 100% stringency correlation value. Missing values can not be calculated due insufficient number of sequence pairs in the ALM set.
(0.05 MB PDF)
Acknowledgments
The authors would like to thank Steve W. Lockless and Rama Ranganathan for providing the code for the SCA implementation, Aidan Budd and Daniel Castaño for fruitful discussion at the beginning of the project, Niall Haslam for critical reading of the manuscript and Norman Davey for pointing out at “flanking”.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was partially supported by the EU EMBRACE (LHSG-CT-2004-512091) grant. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Diella F, Haslam N, Chica C, Budd A, Michael S, et al. Understanding eukaryoticlinear motifs and their role in cell signaling and regulation. Front Biosci. 2008;13:6580–603. doi: 10.2741/3175. [DOI] [PubMed] [Google Scholar]
- 2.Sigrist C, Cerutti L, Hulo N, Gattiker A, Falquet L, et al. PROSITE: a documented database using patterns and profiles as motif descriptors. Brief Bioinform. 2002;3(3):265–74. doi: 10.1093/bib/3.3.265. [DOI] [PubMed] [Google Scholar]
- 3.Puntervoll P, Linding RC, Chabanis-Davidson GS, Mattingsdal M, et al. ELM server: A new resource for investigating short functional sites in modular eukaryotic proteins. Nucleic Acids Res. 2003;31(13):3625–30. doi: 10.1093/nar/gkg545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neduva V, Russell R. Linear motifs: evolutionary interaction switches. FEBS Lett. 2005;579(15):3342–3345. doi: 10.1016/j.febslet.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Fuxreiter M, Tompa P, Simon I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics. 2007;23(8):950–6. doi: 10.1093/bioinformatics/btm035. [DOI] [PubMed] [Google Scholar]
- 6.Wright P, Dyson H. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293(2):321–31. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 7.Gibson T. Cell regulation: determined to signal discrete cooperation. Trends Biochem Sci. 2009 doi: 10.1016/j.tibs.2009.06.007. (in press) [DOI] [PubMed] [Google Scholar]
- 8.Seet B, Dikic I, Zhou M, Pawson T. Reading protein modifications with interaction domains. Nat Rev Mol Cell Biol. 2006;7(7):473–83. doi: 10.1038/nrm1960. [DOI] [PubMed] [Google Scholar]
- 9.Davey N, Shields D, Edwards R. SLiMDisc: short, linear motif discovery, correcting for common evolutionary descent. Nucleic Acids Res. 2006;34(12):3546–54. doi: 10.1093/nar/gkl486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neduva V, Linding R, Su-Angrand I, Stark A, de F Masi, et al. Systematic discovery of new recognition peptides mediating protein interaction networks. PLoS Biol. 2006;3(12):e405. doi: 10.1371/journal.pbio.0030405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dinkel H, Sticht H. A computational strategy for the prediction of functional linear peptide motifs in proteins. Bioinformatics. 2007;23(24):3297–303. doi: 10.1093/bioinformatics/btm524. [DOI] [PubMed] [Google Scholar]
- 12.Landgraf C, Panni S, Montecchi-Palazzi L, Castagnoli L, Schneider-Mergener J, et al. Protein interaction networks by proteome peptide scanning. PLoS Biol. 2004;2(1):e14. doi: 10.1371/journal.pbio.0020014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stiffler M, Chen J, Grantcharova V, Lei Y, Fuchs D, et al. PDZ domain binding selectivity is optimized across the mouse proteome. Science. 2007;317(5836):364–9. doi: 10.1126/science.1144592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zarrinpar A, Park S, Lim W. Optimization of specificity in a cellular protein interaction network by negative selection. Nature. 2003;426(6967):676–80. doi: 10.1038/nature02178. [DOI] [PubMed] [Google Scholar]
- 15.Mohan A, Oldfield C, Radivojac P, Vacic V, Cortese M, et al. Analysis of molecular recognition features (MoRFs). J Mol Biol. 2006;362(5):1043–59. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- 16.Oldfield C, Cheng Y, Cortese M, Romero P, Uversky V, et al. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry. 2005;44(37):12454–70. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- 17.Remaut H, Waksman G. Protein–protein interaction through beta–strand addition. Trends Biochem Sci. 2006;31:436–444. doi: 10.1016/j.tibs.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 18.Stein A, Aloy P. Contextual specificity in peptide-mediated protein interactions. PLoS ONE. 2008;3(7):e2524. doi: 10.1371/journal.pone.0002524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards R, Davey N, Shields D. SLiMFinder: A probabilistic method for identifying over-represented, convergently evolved, short linear motifs in proteins. PLoS ONE. 2007;2(10):e967. doi: 10.1371/journal.pone.0000967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neduva V, Russell R. DILIMOT: discovery of linear motifs in proteins. Nucleic Acids Res. 2006;34(Web Server issue):W350–5. doi: 10.1093/nar/gkl159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chica C, Labarga A, Gould C, López R, Gibson T. A tree-based conservation scoring method for short linear motifs in multiple alignments of protein sequences. BMC Bioinformatics. 2008;9:229. doi: 10.1186/1471-2105-9-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balla S, Thapar V, Verma S, Luong T, Faghri T, et al. Minimotif Miner: a tool for investigating protein function. Nat Methods. 2006;3(3):175–7. doi: 10.1038/nmeth856. [DOI] [PubMed] [Google Scholar]
- 23.Gutman R, Berezin C, Wollman R, Rosenberg Y, Ben-Tal N. QuasiMotiFinder: protein annotation by searching for evolutionarily conservedmotif-like patterns. Nucleic Acid Res. 2005;33(Web Server issue):W255–61. doi: 10.1093/nar/gki496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davey N, Shields D, Edwards R. Masking residues using context-specific evolutionary conservation significantly improves short linear motif discovery. Bioinformatics. 2009;25(4):443–450. doi: 10.1093/bioinformatics/btn664. [DOI] [PubMed] [Google Scholar]
- 25.Diella F, Gould C, Chica C, Via A, Gibson T. Phospho.ELM: a database of phosphorylation sites–update 2008. Nucleic Acids Res. 2008;36:D240–4. doi: 10.1093/nar/gkm772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Obenauer J, Cantley L, Yaffe M. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31(13):3635–41. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002;30(14):3059–66. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Coghlan A, Ruan J, Coin L, Hériché J, et al. TreeFam: a curated database of phylogenetic trees of animal gene families. Nucleic Acids Res. 2006;34:D572–80. doi: 10.1093/nar/gkj118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dosztányi Z, Csizmók V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21(16):3433–4. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- 30.Letunic I, Doerks T, Bork P. SMART 6: recent updates and new developments. Nucleic Acids Res. 2009;37:D229–32. doi: 10.1093/nar/gkn808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perrodou E, Chica C, Poch O, Gibson T, Thompson J. A new protein linear motif benchmark for multiple sequence alignment software. BMC Bioinformatics. 2008;9:213. doi: 10.1186/1471-2105-9-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lockless S, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286(5438):295–9. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 33.Brown C, Takayama S, Campen A, Vise P, Marshall T, et al. Evolutionary rate heterogeneity in proteins with long disordered regions. J Mol Evol. 2002;55(1):104–10. doi: 10.1007/s00239-001-2309-6. [DOI] [PubMed] [Google Scholar]
- 34.Lockless S, Zhou M, MacKinnon R. Structural and thermodynamic properties of selective ion binding in a K+ channel. PLoS Biol. 2007;5(5):e121. doi: 10.1371/journal.pbio.0050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dyson H, Wright P. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6(3):197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 36.Tompa P, Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci. 2008;33(1):2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 37.Hegyi H, Schad E, Tompa P. Structural disorder promotes assembly of protein complexes. BMC Struct Biol. 2007;7:65. doi: 10.1186/1472-6807-7-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen J, Romero P, Uversky V, Dunker A. Conservation of intrinsic disorder in protein domains and families: II. functions of conserved disorder. J Proteome Res. 2006;5(4):888–98. doi: 10.1021/pr060049p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uversky V, Oldfield C, Dunker A. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit. 2005;18(5):343–84. doi: 10.1002/jmr.747. [DOI] [PubMed] [Google Scholar]
- 40.Daughdrill G, Narayanaswami P, Gilmore S, Belczyk A, Brown C. Dynamic behaviour of an intrinsically unstructured linker domain is conserved in the face of negligible amino acid sequence conservation. J Mol Evol. 2007;65(3):277–88. doi: 10.1007/s00239-007-9011-2. [DOI] [PubMed] [Google Scholar]
- 41.Takei K, Yoshida Y, Yamada H. Regulatory mechanisms of dynamin–dependent endocytosis. J Biochem. 2005;137(3):243–7. doi: 10.1093/jb/mvi052. [DOI] [PubMed] [Google Scholar]
- 42.Tomizawa K, Sunada S, Lu Y, Oda Y, Kinuta M, et al. Cophosphorylation of amphiphysin i and dynamin i by cdk5 regulates clathrin-mediated endocytosis of synaptic vesicles. J Cell Biol. 2003;163(4):813–24. doi: 10.1083/jcb.200308110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collins M, Yu L, Campuzano I, Grant S, Choudhary J. Phosphoproteomic analysis of the mouse brain cytosol reveals a predominance of protein phosphorylation in regions of intrinsic sequence disorder. Mol Cell Proteomics. 2008;7(7):1331–48. doi: 10.1074/mcp.M700564-MCP200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dataset of functional instances. List of the 116 instances, classified per structural class with phylogeny, sequence and motif identifiers.
(0.00 MB TXT)
Comparison of the IUPdiff distribution between the PLM and ALM sets. Kolmogorov-Smirnov test comparing the IUPdiff distribution of the PLM and ALM sets of each instance. The difference is the Kolmogorov-Smirnov statistic calculated from the cumulative distributions of the compared samples.
(0.03 MB PDF)
Mean and standard deviation of the correlation between PLM and ALM frequency profiles. Spearman correlation coefficient calculated between the PLM and ALM frequency profiles of each instance. Correlation of the frequency profiles of IUPdiff versus locCons and IUPdiff versus globCons are indicated as locCons corr and globCons corr respectively.
(0.02 MB PDF)
Effect of the stringency of the regular expression matching on the correlation between the PLM and ALM frequency profiles. Spearman correlation coefficient calculated between the PLM and ALM frequency profiles of each instance. Correlation of the frequency profiles of IUPdiff versus locCons and IUPdiff versus globCons are indicated as locCons corr and globCons corr respectively. Percentages indicate the stringency used to define a match to the ELM regular expression: 100% stringency supposes that a LM is present only if there is a perfect match to the ELM regular expression in the same position of the annotated instance; lower percentages consider that a LM is present also in case of partial match to the regular expression. Correlation values in bold show the biggest difference (more than 0.05) with the corresponding 100% stringency correlation value. Missing values can not be calculated due insufficient number of sequence pairs in the ALM set.
(0.05 MB PDF)