

Abstract
Multiple system tauopathy with presenile dementia (MSTD) is an inherited disease caused by a (g) to (a) transition at position +3 in intron 10 of Tau. It belongs to the spectrum of frontotemporal dementia and parkinsonism linked to chromosome 17 with mutations in Tau (FTDP-17T). Here we present the longitudinal clinical, neuropsychological, neuroimaging, neuropathological, biochemical and genetic characterization of the MSTD family. Presenting signs were consistent with the behavioural variant of frontotemporal dementia in 17 of 21 patients. Two individuals presented with an atypical form of progressive supranuclear palsy and two others with either severe postural imbalance or an isolated short-term memory deficit. Memory impairment was present at the onset in 15 patients, with word finding difficulties and stereotyped speech also being common. Parkinsonism was first noted 3 years after the onset of symptoms. Neuroimaging showed the most extensive grey matter loss in the hippocampus, parahippocampal gyrus and frontal operculum/insular cortex of the right hemisphere and, to a lesser extent, in the anterior cingulate gyrus, head of the caudate nucleus and the posterolateral orbitofrontal cortex and insular cortex bilaterally. Neuropathologically, progressive nerve cell loss, gliosis and coexistent neuronal and/or glial deposits consisting mostly of 4-repeat tau were present in frontal, cingulate, temporal and insular cortices, white matter, hippocampus, parahippocampus, basal ganglia, selected brainstem nuclei and spinal cord. Tau haplotyping indicated that specific haplotypes of the wild-type allele may act as modifiers of disease presentation. Quantitative neuroimaging has been used to analyse the progression of atrophy in affected individuals and for predicting disease onset in an asymptomatic mutation carrier. This multidisciplinary study provides a comprehensive description of the natural history of disease in one of the largest known families with FTDP-17T.
Keywords: frontotemporal dementia, progressive supranuclear palsy, hippocampus, voxel-based morphometry, Tau haplotype
Introduction
Frontotemporal dementia (FTD) is a clinical syndrome characterized by behavioural and personality changes, as well as an impairment of executive function, language and movement (Brun et al., 1994; Neary et al., 1998; Cairns et al., 2007). Following the demonstration of linkage to chromosome 17q21 in a family with an autosomal-dominant form of FTD (Wilhelmsen et al., 1994), additional kindreds with inherited FTD linked to the same region were identified (Petersen et al., 1995; Wijker et al., 1996; Yamaoka et al., 1996; Bird et al., 1997; Heutink et al., 1997; Murrell et al., 1997), resulting in the denomination ‘frontotemporal dementia and parkinsonism linked to chromosome 17’ (FTDP-17) for this syndrome (Foster et al., 1997). In 1998, genetic analysis of the Tau (MAPT) gene on chromosome 17q21 revealed mutations in affected individuals from 9 of the 13 families originally linked to chromosome 17 (Poorkaj et al., 1998; Hutton et al., 1998; Spillantini et al., 1998b; Clark et al., 1998). More recently, mutations in Progranulin (PGRN), another gene located on chromosome 17q21, have been associated with frontotemporal lobar degeneration with tau-negative, ubiquitin-positive inclusions (FTLD-U) (Baker et al., 2006; Cruts et al., 2006). A mutation in PGRN has also been found in affected members from three of the four original FTDP-17 kindreds without Tau mutations (Mukherjee et al., 2006; Bronner et al., 2007; Leverenz et al., 2007). Since FTDP-17 can be caused by mutations in two different genes, FTDP-17T is now being used to refer to the disease in individuals with a mutation in Tau. Currently, more than 40 different Tau mutations have been shown to cause FTDP-17T in individuals from over 100 families (Goedert and Spillantini, 2006).
During the past 13 years, we have studied the family with ‘multiple system tauopathy with presenile dementia’ (MSTD), one of the original FTDP-17T kindreds (Ghetti and Farlow, 1994; Murrell et al., 1997; Spillantini et al., 1997, 1998a, 1998b). The disease in this family is caused by a (g) to (a) transition at position +3 in intron 10 of Tau, which results in the overproduction of tau isoforms with 4 microtubule-binding repeats (Spillantini et al., 1998b). In normal adult human brain, 6 tau proteins are expressed, with similar levels of 3-repeat and 4-repeat isoforms (Goedert et al., 1989; Goedert and Jakes, 1990). Here, we have used clinical, neuropsychological, neuroimaging, neuropathological, biochemical and genetic data to provide a longitudinal characterization of the disease in a large number of individuals from the MSTD family.
Materials and Methods
Subjects
The MSTD pedigree comprises more than 200 individuals spread over seven generations; 51 individuals are known to have had a clinical history of frontotemporal dementia (Fig. 1). Clinical information has been collected on 24 affected family members (Table 1, Fig. 1). Individuals 1–10 underwent a baseline clinical and neuropsychological assessment, as well as an annual follow-up evaluation at the Indiana Alzheimer Disease Center (IADC). Magnetic resonance imaging (MRI) of the brain was carried out in eight of these subjects (numbers 1–4, 6–9) and in one asymptomatic mutation carrier, whose personal data are not disclosed to preserve anonymity; this individual underwent baseline and annual follow-up clinical and neuropsychological assessment at the IADC. Clinical information on patients 11–24 was obtained by interviewing family members and through examination of clinical records. Data from the neurological examination, neuropsychological assessment, informant interviews and past medical records were used to establish a diagnosis of FTD using standard criteria (Neary et al., 1998). A sub-diagnosis of behavioural variant FTD (bv-FTD) was also established using standard criteria (McKhann et al., 2001). Pathological studies were carried out on 14 individuals whose brains were collected at autopsy (numbers 2, 5, 11, 13–17, 19–24). The spinal cord was dissected from five individuals (numbers 11, 14, 19, 20, 21). Informed consent was obtained, either from the patients themselves, or from their next of kin. This study was approved by the Indiana University–Purdue University–Indianapolis (IUPUI) Institutional Review Board.
Fig. 1.

Pedigree of the kindred with multiple system tauopathy with presenile dementia (MSTD). Affected individuals are distributed over six consecutive generations. For reasons of confidentiality, not all affected individuals in generations 5 and 6 are shown. Numbers 1–24 correspond to the patients with MSTD listed in Table 1.
Table 1.
Demography of the examined MSTD patients
| Subject | C | M | R | P | Gender | Age at onset (years) | Age at death (years) | Disease duration (years) | Brain weight (grams) | MAPT haplotype | Apo E genotype |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | + | + | F | 50 | NA | 6a | NA | H1H2 | 3/3 | ||
| 2 | + | + | + | M | 45 | 54 | 9 | NA | H2H2 | 3/3 | |
| 3 | + | + | M | 42 | NA | 8a | NA | H1H2 | 3/3 | ||
| 4 | + | + | F | 51 | NA | 2a | NA | H1H2 | 3/3 | ||
| 5 | + | + | M | 54 | 69 | 15 | 1160 | H1H2 | 3/3 | ||
| 6 | + | + | F | 46 | NA | 2a | NA | H1H2 | 2/3 | ||
| 7 | + | + | M | 45 | NA | 7a | NA | H2H2 | 3/4 | ||
| 8 | + | + | M | 39 | NA | 6a | NA | H1H2 | 3/3 | ||
| 9 | + | + | F | 53 | NA | 5a | NA | H1H2 | 3/3 | ||
| 10 | + | M | 50 | NA | 10a | NA | H1H2 | NA | |||
| 11 | + | + | M | 47 | 48 | 1 | NA | H1H2 | NA | ||
| 12 | + | M | 42 | 47 | 5 | NA | H1H2 | 3/3 | |||
| 13 | + | + | M | 48 | 54 | 6 | 1150 | H1H2 | NA | ||
| 14 | + | + | F | 52 | 58 | 6 | 1150 | H1H2 | 3/3 | ||
| 15 | + | + | M | 50 | 57 | 7 | NA | H1H2 | NA | ||
| 16 | + | + | M | 50 | 58 | 8 | 1000 | H2H2 | 3/3 | ||
| 17 | + | + | F | 58 | 66 | 8 | 933 | H2H2 | 3/3 | ||
| 18 | + | F | 58 | 69 | 11 | NA | H1H2 | 2/3 | |||
| 19 | + | + | F | 42 | 54 | 12 | 935b | H2H2 | NA | ||
| 20 | + | + | M | 52 | 64 | 12 | 1015 | H2H2 | 3/3 | ||
| 21 | + | + | F | 48 | 61 | 13 | 945 | H2H2 | 3/3 | ||
| 22 | + | + | M | 51 | 66 | 15 | NA | H1H2 | NA | ||
| 23 | + | + | F | 39 | 59 | 20 | 1120b | NA | NA | ||
| 24 | + | + | F | 43 | 64 | 21 | NA | H1H2 | NA |
C = subjects clinically examined at the IADC; M = subjects studied by MRI; R = subjects whose clinical information has been retrospectively collected; P = subjects pathologically examined; NA = not available or not applicable. aCurrent duration of illness. bWeighed after formalin fixation.
Clinical assessment
The neurological assessment was done by M.R.F. To obtain additional information, a structured interview was conducted by a research nurse with the patients’ caregivers. All patients underwent extensive neuropsychological testing. Tests were administered according to standard procedures and interpreted by comparison with control individuals of similar age and education. The clinicians were blind to the results of the genetic analysis.
Magnetic resonance imaging
A 1.5 Tesla GE Signa scanner was used to acquire a heavily T1-weighted, spoiled gradient echo sequence (SPGR; echo time = 8 ms, repetition time = 35 ms, flip angle = 30, bandwidth = 15, matrix = 256 × 128, field of view = 24 cm, slices = 124 ranging from 1.1 mm to 1.3 mm thicknesses to assure whole brain coverage). Images were registered at baseline and subsequent annual follow-up visits. SPM2 was used to conduct optimized and modulated voxel-based morphometry (VBM) (Ashburner and Friston, 2000; Good et al., 2001), aided by an automated script designed by Christian Gaser (http://dbm.neuro.uni-jena.de/vbm.html). For VBM studies, MRI was performed on seven MSTD patients [1 year (numbers 4, 6), 2 years (number 1) and 3 years (numbers 2, 3, 8, 9) after disease onset] and 19 age- and gender-matched controls (patients: four females, three males, mean age 49.0 ± 4.7 years; controls: 11 females, 8 males, mean age 49.4 ± 6.1 years). Details of the technique are described in supplementary information, Materials and Methods. Whole brain volume (WBV) and ventricular volume (VV) normalized for subject's head size, as well as two time-point percentage of brain volume changes (PBVC), were measured from serial MRI using the SIENAX and SIENA methods (Smith et al., 2002, 2004). They were assessed during the annual follow-up of the asymptomatic mutation carrier and symptomatic mutation carriers 1, 2, 3, 7, 9. Standard T2-weighted coronal MRI images were also acquired.
Neuropathology
The brains of 14 affected individuals were collected. Whole brain was harvested from five cases and fixed in formalin. For seven cases, one half of the brain was fixed in formalin and the other half was frozen. For the remaining two cases, only formalin-fixed, paraffin-embedded blocks of brain tissue were available. The spinal cord was collected from five cases. For two cases, the entire spinal cord was fixed in formalin; for the other three, representative segments were either fixed or frozen. Tissue samples for neuropathological studies were obtained from representative brain regions. The following methods were used: Weigert's hematoxylin-eosin, Woelcke–Heidenhain, Bodian, Gallyas and thioflavin S. For immunohistochemistry, antibodies against tau, Aβ, glial fibrillary acidic protein (GFAP), prion protein, ubiquitin, CD68 and TAR DNA-binding protein-43 (TDP-43) were used. Tau immunohistochemistry was carried out using the following phosphorylation-dependent (p), phosphorylation-independent and conformation-dependent anti-tau antibodies: AT8 (Innogenetics, Antwerp, Belgium; 1:400) pS202/pT205; PHF1 (1:10) pS396/pS404; AT180 (Innogenetics; 1:500) pT231; AT270 (Innogenetics, 1:500) pT181; AT100 (Innogenetics, 1:500) pT212/pS214/pT217; 12E8 (Elan Pharmaceuticals, South San Francisco, California; 1:500) pS262/pS356; CP3 (1:10) pS214; PG5 (1:10) pS409; BR134 (1:500) against the carboxy-terminus of tau; RD3 (Upstate, Charlottesville, Virginia; 1:3,000) against 3-repeat tau isoforms; RD4 (Upstate 1:100) against 4-repeat tau isoforms; Alz50 (1:10) conformational. Additional information is provided in supplementary information, Materials and Methods.
Genetics
Genetic analysis was carried out on 59 members of the MSTD kindred. Genomic DNA was extracted from blood (53 individuals) or brain [fresh and/or formalin-fixed, paraffin-embedded (six individuals)] using standard protocols (Murrell et al., 1991). The Tau exon 10 sequence and adjoining intronic sequences were determined as described (Spillantini et al., 1998b). Standard amplification reactions were done with 50 ng genomic DNA. The amplified products were gel purified using the Qiaquick Gel Extraction Kit (Qiagen, Valencia, CA, USA) and asymmetric amplification performed using the DTCS Quick Start Kit (Beckman Coulter, Fullerton, CA, USA). Products were loaded onto a CEQ 2000XL DNA analysis system (Beckman Coulter) and sequences compared to the known Tau sequence. Tau haplotypes H1 and H2 were identified by the presence or absence of a 238 bp deletion in intron 9. When DNA was extracted from formalin-fixed, paraffin-embedded tissue, haplotypes were determined by assessing single nucleotide polymorphisms in exons 1, 9 and 11 using previously described primers (Baker et al., 1999). Digested products were separated on a 2% agarose gel. Amplification products were digested with AluI (SNPs 1 and 11) or MspI (SNP 9) and analysed on a 4% agarose gel. Tau haplotype was further analysed by genotyping SNPs rs1467967, rs242557, rs3785883, rs2471738 and rs7521 (Pittman et al., 2005) using restriction enzyme digestion of amplified products (DraI for rs1467967, ApaLI for rs242557, BsaHI for rs3785883, BstEII for rs2471738 and PstI for rs7521). ApoE genotyping was performed as described (Hixson and Vernier, 1990).
Biochemistry
Tissue samples from the following brain areas were used: caudate/putamen, cerebellum, frontal cortex, frontal white matter, hippocampus and parahippocampus, occipital cortex, occipital white matter, parietal cortex, parietal white matter, temporal cortex and thalamus. Sarkosyl-insoluble material was prepared as described (Goedert et al., 1992) from brain areas of cases number 2 (11 areas), number 5 (nine areas), number 14 (eight areas), number 17 (nine areas), number 20 (nine areas) and number 21 (eight areas). The spinal cords from numbers 20 and 21 were also used. In addition, sarkosyl-insoluble material was prepared from brain areas of two unaffected family members without the +3 mutation (10 areas from each case) and from the frontal cortex of a case with sporadic Alzheimer's disease. Sarkosyl-insoluble material was run on 10% Tris/glycine gels and blotted onto polyvinylidene fluoride (PVDF) membranes. The blots were incubated with anti-tau antibodies BR133 (against the amino-terminus of tau, 1:1000) or BR134 (1:5000) for 1 h at room temperature and developed using enhanced chemiluminescence (GE Healthcare).
Electron microscopy
Aliquots of the sarkosyl-insoluble dispersed filament preparations were processed for immunoelectron microscopy, as described (Crowther, 1991). Micrographs were recorded on a Philips EM208S microscope at a nominal magnification of ×40 000. Antibody BR134 was used at 1:100.
Results
Clinical findings in 10 longitudinally assessed patients with MSTD
The average age at disease onset in this group of 10 patients was 47.5 ± 4.9 years (range 39–54 years). Disease duration in the eight patients who are still alive ranges from 2 to 10 years. Two individuals have died 9 years (number 2) and 15 years (number 5) after developing the disease. Behavioural changes and/or deficits in executive function constituted the presenting signs in nine patients (numbers 1–6, 8–10). They were associated with memory loss in five patients (numbers 4–6, 9, 10) and with memory loss and dysnomia in one patient (number 1). Subject number 7 presented with dizziness, hearing loss, tinnitus, pressure-like headache, up-beating nystagmus and short-term memory loss. He was diagnosed with Ménière's disease. He underwent electronystagmography, electrocochleography and brainstem auditory evoked potentials studies, which indicated central and peripheral dysfunction.
Neurobehavioural changes
All 10 patients were diagnosed with frontotemporal dementia. Disinhibition, social conduct disorder, dysexecutive symptoms, memory impairment and progressive reduction of speech output were evident in patients affected for >3 years, with the exception of subject number 7, whose disease course was slower. For this individual, mild short-term memory problems and slight mental inflexibility were the only cognitive symptoms during the first 6 years of disease, with disequilibrium being the most pressing complaint. More pronounced personality changes and word-finding difficulties developed later.
Physical signs and symptoms
Findings were compiled from all 10 subjects seen at various stages of disease (for a more detailed description, see supplementary information, Table 1). Hypomimia and gait abnormalities were observed in the third year. Bradykinesia, symmetric rigidity and postural instability were observed later in the majority of subjects. Resting tremor was not present. One subject developed urinary incontinence. Reduced limb coordination was present in half of the subjects and was seen as early as the third year. Two individuals had speech dysfunction, with one individual developing slurred speech 6 years after the onset of disease, and the other showing a progressive reduction in speech volume after 3 years. Dysarthria developed progressively, culminating in anarthria by the sixth year. Eye movement abnormalities were noted in 4 patients; they consisted of square-wave jerks of intrusive saccades during pursuit and/or vertical gaze reduction. Both abnormalities were first noted after 5 years of disease. Four individuals suffered from a severe inversion of the normal sleep-wake cycle characterized by dramatically reduced nighttime sleep and increased daytime sleepiness. Two subjects complained of dizziness and light-headedness. These symptoms were occasional and mild in one case, but frequent and severely worsened by rapid movements of the head or after optokinetic stimulation in the other. One subject developed dysphagia. Affected individuals displayed mild disinhibition and reduced concentration during physical examination, but compliance to physician instructions was preserved until the later stages of disease.
Neuropsychological assessment
Of the 10 patients, nine received a clinical diagnosis of FTD at the time of their first assessment. One individual (number 7) was only diagnosed with FTD during follow-up. In order to identify the early features of cognitive dysfunction, a cross-sectional analysis was carried out on patients with a disease duration of <3 years (n = 7). Comparison with a matched control group is shown in Table 2. Despite a relatively short duration of disease (mean of 2 years) and mild dementia (mean MMSE of 25.6), the MSTD patients had statistically significantly lower scores than the controls across a broad range of cognitive domains. The greatest deficits were noted in verbal memory (Word List Memory, Word List Recall), visual memory (Visual Reproduction Recall), confrontation naming (Boston Naming Test) and verbal fluency [Controlled Oral Word Association (COWA)].
Table 2.
Neuropsychological performance of MSTD patients at early disease stage
| Affected n = 7 | Controls n = 23 | P value | |
|---|---|---|---|
| Mean age (years) | 48.5 ± 4.4 | 49.9 ± 6.1 | 0.58 |
| Gender (% female) | 57.1 | 69.6 | 0.54 |
| Elapsed disease duration (years) | 2.1 ± 1.0 | n.a. | n.a. |
| MMSE | 25.6 ± 2.3 | 29.0 ± 0.9 | <0.001 |
| Digit Span | 13.0 ± 5.4 | 16.0 ± 3.5 | 0.095 |
| Word List Memory | 12.9 ± 4.9 | 23.0 ± 3.5 | <0.001 |
| Word List Recall | 3.7 ± 1.4 | 8.4 ± 1.2 | <0.001 |
| Logical Memory | 19.4 ± 6.7 | 30.6 ± 7.4 | <0.005 |
| Logical Memory Recall | 12.9 ± 7.1 | 27.4 ± 8.2 | <0.001 |
| Visual Reproduction | 28.6 ± 4.4 | 34.0 ± 3.2 | <0.005 |
| Visual Reproduction Recall | 12.2 ± 13.0 | 29.0 ± 5.7 | <0.001 |
| Boston Naming Test | 38.6 ± 12.1 | 55.6 ± 3.0 (n = 22) | <0.001 |
| Animal Category | 11.3 ± 2.5 | 21.7 ± 4.3 | <0.001 |
| COWA | 21.6 ± 10.7 | 39.5 ± 12.0 (n = 22) | <0.005 |
| Block Design | 26.0 ± 12.1 | 35.0 ± 7.4 (n = 22) | <0.05 |
| Constructional Praxis | 10.3 ± 1.1 | 10.5 ± 0.7 | 0.53 |
| Constructional Praxis Delayed | 5.1 ± 3.9 | 10.9 ± 2.1 | <0.001 |
| Trail Making A | 38.9 ± 14.3 | 24.6 ± 6.5 | <0.001 |
| Trail Making B | 128.4 ± 60.7 | 54.9 ± 16.1 | <0.001 |
| WCST Category | 3.2 ± 2.6 (n = 6) | 5.4 ± 1.2 (n = 21) | <0.01 |
| WCST Pers. Resp. | 53.5 ± 56.5 (n = 6) | 10.4 ± 8.2 (n = 21) | <0.005 |
| Stroop Word | 68.0 ± 8.5 (n = 6) | 96.6 ± 21.6 (n = 19) | <0.005 |
| Stroop Colour | 41.5 ± 11.3 (n = 6) | 77.6 ± 12.1 (n = 19) | <0.001 |
| Stroop Colour-Word | 21.7 ± 5.5 (n = 6) | 43.1 ± 8.2 (n = 19) | <0.001 |
The longitudinal trajectory of cognitive changes was investigated in the unaffected mutation carrier (see Fig. 2C) and two patients (numbers 3, 5) (see supplementary information, Table 2) over a 7- and a 5-year period, respectively. Subject number 3, when assessed 3 years after disease onset, showed a subtle weakness in confrontation naming and a prominent defect in conceptual reasoning with severe perseveration. Subsequently, impairments in memory and psychomotor speed became notable. Subject number 5, when assessed 5 years after the onset of symptoms, had performances on confrontation naming and memory below the fifth percentile of healthy controls. Over time, scores on tests of verbal fluency and response inhibition fell below threshold of significant impairment.
Fig. 2.
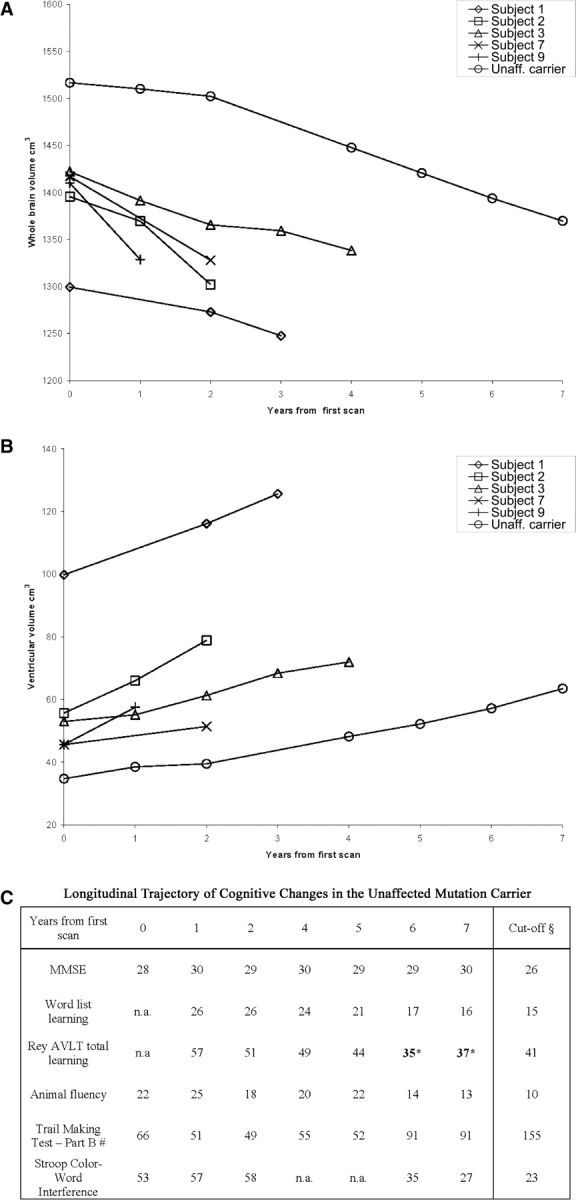
Longitudinal measurement of brain (A) and ventricular (B) volumes in five patients with MSTD and one asymptomatic mutation carrier. Subjects 1, 2, 3 and 9 presented with FTD, whereas subject 7 presented with disequilibrium and memory loss. Progression of cognitive decline in the unaffected mutation carrier is shown in table (C). § = 5th percentile of healthy controls. * = in bold, performance below the 5th percentile of controls. # = scores are expressed in seconds.
The unaffected mutation carrier displayed a trend for progressive impairment in memory, verbal fluency, conceptual shifting and response inhibition, as evidenced by the latest scores either approaching or below the 5th percentile of healthy controls (Fig. 2C).
Retrospective evaluation of clinical findings in 14 patients with MSTD
A summary of the information collected retrospectively on this group of 14 affected individuals (numbers 11–24) is presented in supplementary information, Table 3. The average age at disease onset was 48.6 ± 5.7 years (range 39–58 years) and the average age at death was 58.9 ± 6.6 years (range 47–69 years). The mean disease duration was 10.4 ± 5.7 years (range 1–21 years). Clinical data were available on 12 subjects (numbers 11–22). The presenting signs of disease are known for 11 individuals. Behavioural abnormalities were the initial symptom in 8 individuals (numbers 12, 13, 15–18, 20, 22). They were associated with short-term memory loss in two cases (numbers 18, 22) and with short-term memory loss and dysnomia in four cases (numbers 13, 15–17). Two siblings (brother and sister, numbers 11, 14) presented with an atypical progressive supranuclear palsy (PSP)-like syndrome characterized by dizziness, pressure-like headaches, neck stiffness and postural imbalance (Litvan et al., 1996). In addition, subject number 11 reported episodes of tachycardia, increased perspiration, swallowing difficulties, sleep disorder and urinary incontinence. Subject number 14 also presented with short-term memory impairment. Subject number 21 presented with memory loss only. Over time, 10 subjects (numbers 12, 13, 15–22) developed a full frontotemporal syndrome with behavioural alterations and language deficits, leading to memory loss and late mutism.
Dizziness and neck stiffness were the predominant early symptoms in subject numbers 11 and 14. Unsteadiness and light-headedness were initially experienced in association with rapid turning of the head; they became more frequent and severe later on and ended up being continuous. To reduce discomfort, both patients required proper adjustment when sitting and could not use a car because of severe motion sickness. They experienced neck muscle spasms, as well as numbness and tingling sensations in hands, arms and shoulders; grip strength and coordination were also reduced. On physical examination, they had increased muscle tone, particularly in the neck, as well as hyperactive deep tendon reflexes. They experienced weakness of hand muscles and, to a lesser extent, upper and lower extremities, in the absence of atrophy and visible fasciculations. The Hoffmann sign was present bilaterally, but no Babinski sign was noted. Electromyography revealed signs of denervation at multiple levels of the cervical spinal cord and scattered fasciculations of hand muscles. Mild intentional tremor and impaired alternating movements were noted during the finger-to-nose test. Both patients exhibited limitation of neck extension, reduced superior gaze, pursed lips and swallowing difficulties. Sensory findings were normal. Subject number 11 died of aspiration pneumonia almost 1 year after disease onset without displaying cognitive alterations. Subject number 14 became unable to walk and developed resting and intentional tremor, mild global cognitive decline, reduced verbal fluency and naming. She died suddenly 6 years after disease onset.
Data from a neurological evaluation were available for 9 of the 14 patients. Parkinsonism was the most commonly reported finding, followed by frontal lobe signs, incontinence, dysphagia, dizziness and signs of upper and/or lower motor neuron dysfunction. Eye movement abnormalities, sleep disturbance and paresthesias were noted less commonly.
Summary of clinical data
For this group of patients with MSTD, the average age of disease onset was 48.1 ± 5.3 years (n = 24, range 39–58 years) and the mean age at death 59.2 ± 6.8 years (n = 16, range 47–69 years). The mean disease duration was 10.6 ± 5.4 years (n = 16, range 1–21 years). Clinical presentation was that of bv-FTD in 17 individuals. Five patients had a pure frontal syndrome, seven patients presented with additional short-term memory impairment and five with short-term memory impairment and dysnomia. Three individuals presented with dizziness (two of these patients also had early memory deficits). One subject presented with an isolated short-term memory deficit. Over the course of the disease, a diagnosis of FTD was made in 20 patients. An atypical PSP-like presentation (numbers 11, 14) and the lack of sufficient information (numbers 23, 24) precluded a clinical diagnosis of FTD in four individuals. Data from a neurological evaluation were available for 19 patients. Parkinsonism was the most common feature (12 patients), followed by incoordination (eight patients), frontal lobe signs (eight patients), sleep disturbances (seven patients), eye movement abnormalities or a PSP-like syndrome (six patients), dysphagia (five patients), signs of motor neuron disease (four patients) and paresthesias (two patients).
Neuroimaging
As depicted in Fig. 3, the seven MSTD patients studied in the early stages of disease with MRI and VBM (numbers 1, 2, 3, 4, 6, 8, 9) had a statistically significant loss of total grey matter volume and increased CSF volume when compared to 19 controls (P < 0.05), with the white matter volume not being significantly different between the two groups (see supplementary information, Table 4, for the stereotactic locations of the most significant grey matter loss). As shown in Fig. 4, the most significant focal points of large clusters of grey matter loss occurred bilaterally in medial temporal lobe (hippocampus/uncus; panels 2, 4), opercular cortex and insular cortex (panel 3). With a false discovery rate-corrected threshold (P < 0.05), these clusters covered a large expanse of the anterior cingulate gyrus (panel 1), the head of the caudate nucleus (panel 3) and the posterolateral orbitofrontal cortex (panel 4). Grey matter loss was also in evidence in the thalamus (panels 1, 2).
Fig. 3.
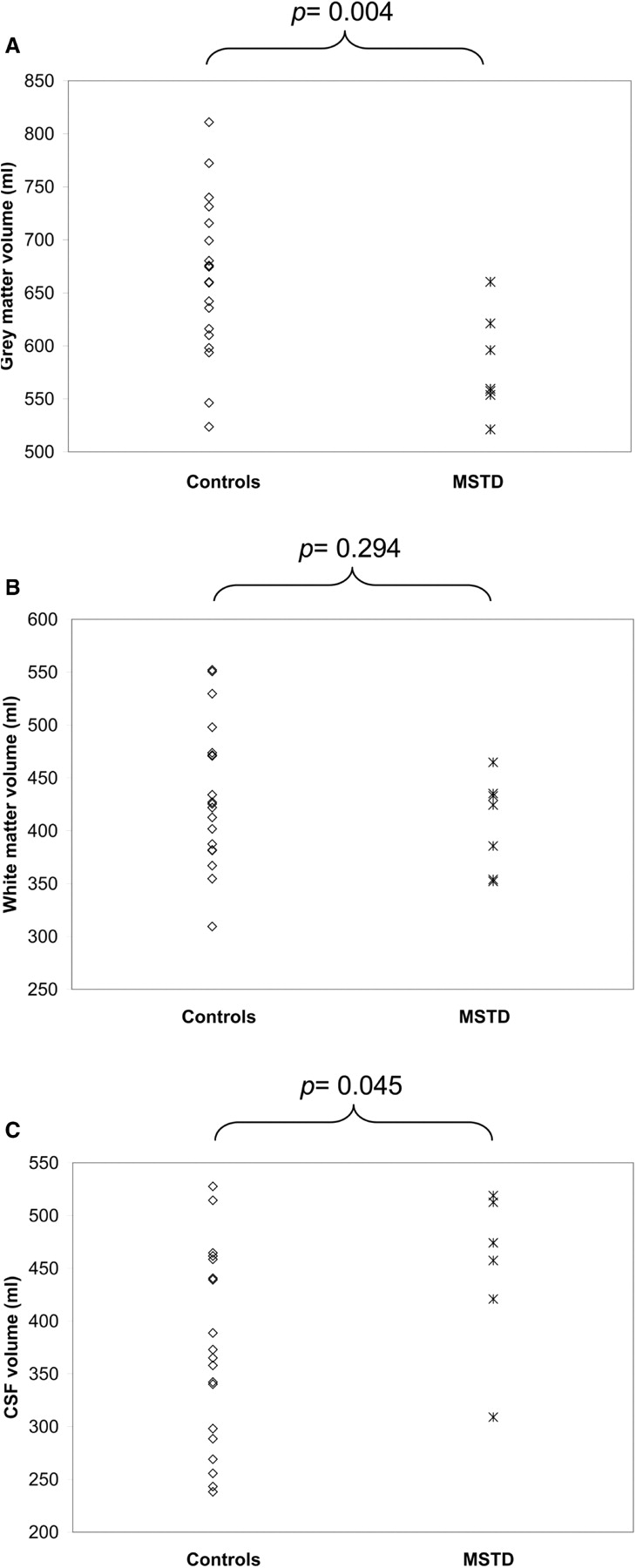
Grey matter (A), white matter (B) and CSF (C) volumes in MSTD patients in the early stages of disease (case numbers 1, 2, 3, 4, 6, 8, 9). The average disease duration was 2.1 ± 1.0 years (n = 7). Volumes were compared with those of age- and gender-matched controls (n = 19). Grey matter volume was significantly reduced and CSF volume increased (P < 0.05) in the MSTD patients, with no change in white matter volume.
Fig. 4.

Heat map showing the patterns of grey matter loss in MSTD patients in the early stages of disease (combined data from case numbers 1, 2, 3, 4, 6, 8, 9). The most significant clusters of grey matter atrophy were present in the medial temporal cortex (hippocampus/uncus, panels 2 and 4), the opercular cortex and the insular cortex (panel 3). Additional areas of significant loss of grey matter were in anterior cingulate gyrus (panel 1), thalamus (panels 1 and 2), the head of the caudate nucleus (panel 3) and posterolateral orbitofrontal cortex (panel 4). Displayed threshold: P < 0.05, false discovery rate-corrected.
Figure 5 illustrates the progression of whole brain atrophy and ventricular enlargement in patient number 3. Three years after disease onset, atrophy was particularly evident at the level of superior frontal and cingulate gyri, anterior temporal lobe, hippocampus and parahippocampal gyrus (Fig. 5A). Seven years after disease onset, atrophy had become more severe in frontal and temporal lobes and the lateral ventricles were now markedly enlarged especially in their anterior portion (Fig. 5B). In this subject with severe behavioural abnormalities, the right cerebral hemisphere was consistently more atrophic than the left.
Fig. 5.

MRI scans of MSTD case number 3 recorded at ages 45 [3 years after disease onset (A)] and 49 years [7 years after disease onset (B)]. The patient died aged 50. Severe cortical and subcortical atrophy is evident in (A), particularly at the level of superior frontal and cingulate gyri, anterior temporal lobes, hippocampus and parahippocampal gyrus. Marked progression of frontal and temporal lobar atrophy, as well as severe ventricular enlargement, are evident in (B). Note that the right cerebral hemisphere is more atrophic than the left. Images are in radiological orientation.
Longitudinal changes in 5 symptomatic patients (numbers 1, 2, 3, 7, 9) and the asymptomatic mutation carrier are illustrated in Fig. 2. For affected mutation carriers, the average changes in WBV and VV were −34.1 ml/year (2.47% decrease per year) and +7.46 ml/year (11.87% increase per year), respectively. They were −19.9 ml/year (1.38% decrease per year) and +4.07 ml/year (8.57% increase per year) for the unaffected mutation carrier. Interestingly, in the first 2 years of this subject's examination, we observed an average change in WBV and VV of −7.2 ml/year (0.47% decrease per year) and +2.4 ml/year (6.77% increase per year), respectively. In the latest 5 years of this subject's follow-up, the average change in WBV and VV were −26.3 ml/year (1.83% decrease per year) and +4.9 ml/year (9.47% increase per year), respectively.
Neuropathology
A case with a short disease duration (1 year)
Formalin-fixed, paraffin-embedded blocks of cerebral cortex, hippocampus, parahippocampal gyrus, medulla and spinal cord were available for subject number 11 who had suffered from an atypical PSP-like syndrome rather than from FTD. The cerebral cortex showed mild loss of neurons, gliosis and microvacuolar changes in the second cortical layer. More severe neuronal loss and gliosis were noted in hippocampus and parahippocampal gyrus. Tau-immunoreactive neurons and neuropil threads were seen in dentate gyrus, CA, subiculum and entorhinal cortex. Occasional astrocytic plaques were observed in the entorhinal cortex. In contrast, numerous coiled bodies were present in subcortical white matter of the parahippocampal gyrus. Only a few tau-positive neurons and neurofibrillary tangles were observed in the neocortex. In the medulla, neuronal loss was moderate in the dorsal motor nucleus of the vagus nerve, the hypoglossal nucleus, the nucleus ambiguus and the inferior olivary nucleus, as well as, to a lesser extent, in the vestibular nuclei and the intermediate reticular zone. Nerve cells displayed tau-immunoreactive inclusions and occasional neurofibrillary tangles. Tau-immunoreactive neuropil threads were also noted. In the anterior horn of the cervical and thoracic spinal cord, mild to moderate nerve cell loss was observed. Neurons in lamina IX displayed tau-immunoreactive granular deposits. Abundant tau-immunoreactive neurites and grain-like deposits, as well as occasional neurons, were also present in lamina VII. The corticospinal tracts were severely atrophic, while Clarke's columns were unremarkable.
Cases with an intermediate disease duration (5–9 years)
Six cases (numbers 2, 13–17) belong to this group (Table 1), five of whom had suffered from FTD. Case number 14, who had presented with an atypical PSP-like syndrome, is discussed separately. Macroscopic examination revealed moderate, symmetric atrophy of the frontal and temporal lobes, which was most prominent at the level of the temporal pole and the parahippocampal gyrus. Caudate nucleus and amygdala were the most atrophic among the subcortical nuclei. The substantia nigra was depigmented, while the locus coeruleus was not affected in all cases. The bulk of the centrum semiovale was reduced, with frontal and temporal horns of the lateral ventricles being greatly enlarged. The histological changes are summarized in Table 3. Neuronal loss mostly affected cortical layers II, III, V and VI. Gliosis was particularly evident at the level of the junction between cortical grey and white matter. Spongiosis of the second cortical layer was a common finding. Ballooned neurons were detected in the cerebral cortex of three cases (numbers 13, 16, 17).
Table 3.
Distribution and severity of neuronal loss and gliosis in the MSTD patients
| Intermediate disease duration | Long disease duration | |||
|---|---|---|---|---|
| Neuronal loss | Gliosis | Neuronal loss | Gliosis | |
| Superior/middle frontal gyri | ++ | + | +++ | ++ |
| Superior/middle temporal gyri | ++ | + | +++ | ++ |
| Superior parietal lobe | – | – | ++ | + |
| Occipital cortex | – | – | + | + |
| Cingulate gyrus | ++ | + | +++ | +++ |
| Insular cortex | ++ | + | +++ | +++ |
| Hippocampus CA 1–4 | + | + | + | + |
| Dentate gyrus | + | + | + | + |
| Entorhinal cortex | ++ | + | +++ | ++ |
| Amygdala | ++ | ++ | +++ | +++ |
| Caudate nucleus | ++ | ++ | +++ | +++ |
| Putamen | ++ | ++ | +++ | +++ |
| Globus pallidus | + | + | ++ | ++ |
| Substantia innominata | + | + | ++ | ++ |
| Thalamus | – | – | + | + |
| Subthalamic nucleus | – | – | + | + |
| Hypothalamus | – | – | + | + |
| Cerebellar cortex | – | – | + | – |
| Dentate nucleus | – | – | + | – |
| Substantia nigra | +++ | + | +++ | ++ |
| Oculomotor nucleus | – | – | ++ | + |
| Locus coeruleus | ++ | – | +++ | ++ |
| Motor dorsal nucleus vagus | ++ | – | +++ | ++ |
| Hypoglossal nucleus | + | – | + | + |
| Vestibular nuclei | + | – | + | + |
| Inferior olivary nucleus | + | – | ++ | + |
+ = mild, ++ = moderate, +++ = severe.
The distribution and severity of tau pathology are summarized in Fig. 6A and exemplified in Fig. 7. In neocortex, tau-immunoreactive deposits were most abundant in neurons of layers II, III, V and VI (Fig. 8A and B). They were also rarely noted among large pyramidal neurons of the motor cortex. Most neuronal inclusions had a granular appearance, while neurofibrillary tangles (NFTs) were only occasionally seen in temporal cortex, parahippocampal gyrus, subiculum and amygdala (Fig. 8F). Abundant grain-like neuropil threads were observed in frontal and temporal cortices, hippocampus and subiculum. Tau-positive astrocytic inclusions were present in the cerebral cortex of all subjects with an intermediate disease duration, with tufted astrocytes and astrocytic plaques being present in subjects 2 and 17 (Figs. 8G and 9B). Tufted astrocytes were more commonly noted in frontal and insular cortices and in caudate/putamen. Astrocytic plaques were mostly detected in frontotemporal and entorhinal cortices. A consistent finding was the presence of numerous coiled bodies in frontal, temporal and parietal lower cortical layers and subcortical white matter, as well as in corpus callosum, centrum semiovale, internal and external capsules and descending fibers of the striatum (Fig. 9D). Tau deposits in brainstem were limited almost entirely to nerve cells, either in a granular cytoplasmic form, or as neuropil threads. NFTs were rare.
Fig. 6.

Distribution and severity of tau pathology in cerebrum and brainstem of MSTD patients who presented with FTD. (A) Cases of intermediate disease duration (numbers 2, 13, 15, 16, 17). Subject number 14, who presented with an atypical PSP-like syndrome, was excluded. (B) cases of long disease duration (numbers 5, 19, 20, 21, 22, 23, 24). Abbreviations: III = oculomotor nucleus; X = dorsal motor nucleus of the vagus nerve; XII = hypoglossal nucleus; A1/C1 = noradrenaline and/or adrenaline cells; AG = ambiens gyrus; AMB = nucleus ambiguus; BSC = superior colliculus; CG = cingulate gyrus; CP = cerebral peduncle; DMN = deep mesencephalic nucleus; DRN = dorsal raphe nucleus; DTN = dorsal tegmental nucleus; Ent = entorhinal cortex; FUG = fusiform gyrus; GRN = gigantocellular reticular nucleus; ICP = inferior cerebellar peduncle; IG = insular gyrus; ION = inferior olivary nucleus; IRN = intermediate reticular nucleus; ITH = inferior temporal gyrus; LC = locus coeruleus; LDTN = laterodorsal tegmental nucleus; LFP = longitudinal fibres of the pons; LPGN = lateral paragigantocellular nucleus; LRN = lateral reticular nucleus; ME5 = mesencephalic trigeminal nucleus; MFG = middle frontal gyrus; MGN = medial geniculate nucleus; ML = medial lemniscus; MRN = median raphe nucleus; MTG = middle temporal gyrus; MVe = medial vestibular nucleus; P = pyramidal tract; PAG = periaqueductal grey; PCF = pontocerebellar fibres; PCL = paracentral lobule; PCRN = parvocellular reticular nucleus; PMRN = paramedian raphe nucleus; PN = pontine nuclei; PoG = postcentral gyrus; POP = parietal operculum; PPTN = pedunculopontine tegmental nucleus; PrG = precentral gyrus; PRNo = pars oralis of pontine reticular nucleus; PUL = pulvinar; RN = red nucleus; ROb = raphe obscurus nucleus; SCP = superior cerebellar peduncle; SFG = superior frontal gyrus; SNc = substantia nigra, pars compacta; SNr = substantia nigra, pars reticulata; SP5 = spinal trigeminal nucleus; SpVe = spinal vestibular nucleus; STG = superior temporal gyrus.
Fig. 7.

Immunohistochemistry for phosphorylated tau in coronal sections from a case of MSTD of long disease duration. The sections were taken at the level of the anterior (A) and posterior (B) hippocampus of case number 19. Marked atrophy is present at the level of the superior frontal gyrus and the insular cortex (particularly in their most anterior portions), the cingulate gyrus, the middle and inferior temporal gyri, the hippocampus and the parahippocampal gyrus. Note the extensive dilatation of the lateral ventricle and the severe reduction of the corpus callosum (most prominent in the anterior portion). The body of the caudate nucleus is greatly reduced. Note the corresponding distribution of tau pathology in cortex and subcortical white matter, with partial sparing of the postcentral gyrus and the superior temporal gyrus (in B). Anti-tau antibody = AT8. Scale bars: 0.5 cm.
Fig. 8.
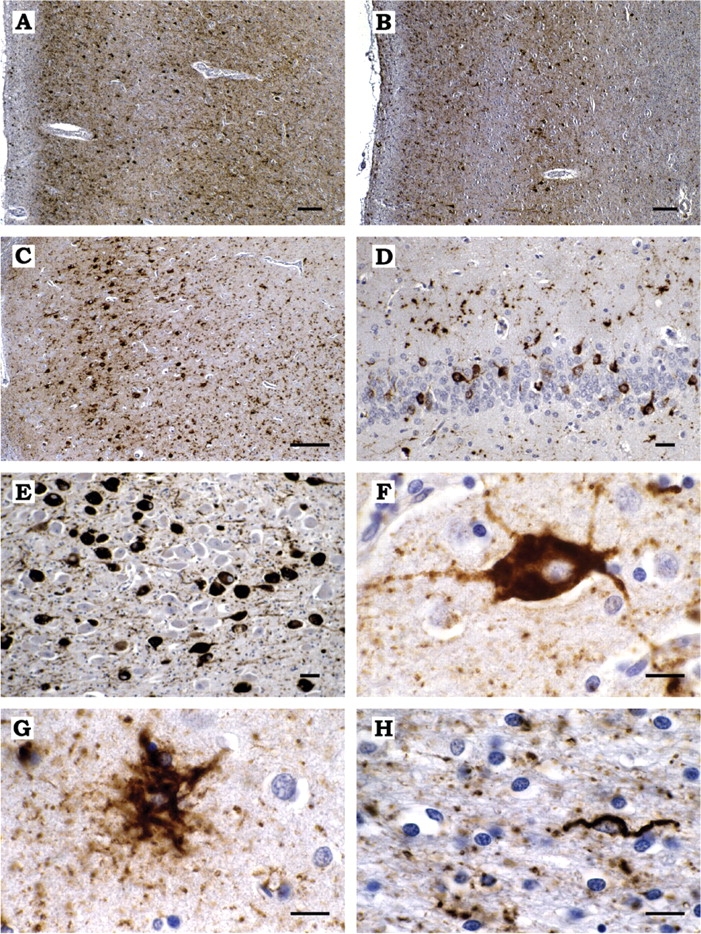
(A, B) Abundant tau-immunoreactive neurons and neuropil threads in layers II, IV and V of the cingulate gyrus (A) and layers II and V of the temporal cortex (B). (C) Numerous tau-positive neurons and grain-like threads, as well as occasional tau-immunoreactive astrocytes, in the pyramidal layer of hippocampal sector CA1. (D), Many perinuclear neuronal tau deposits in the granule cell layer of the dentate gyrus and a few tau-positive astrocytes in the molecular layer of the dentate gyrus. (E) Granular tau-immunoreactive neuronal deposits and occasional neurofibrillary tangles in the substantia innominata. (F) A nerve cell from frontal cortex with granular cytoplasmic tau immunoreactivity. (G) A tufted astrocyte in frontal cortex. (H) A coiled body in the striatal white matter. All the sections are from the brain of MSTD case number 5. Anti-tau antibody = PHF1. Scale bars: A, B, C = 50 μm; D, E = 10 μm; F, G, H = 5 μm.
Fig. 9.
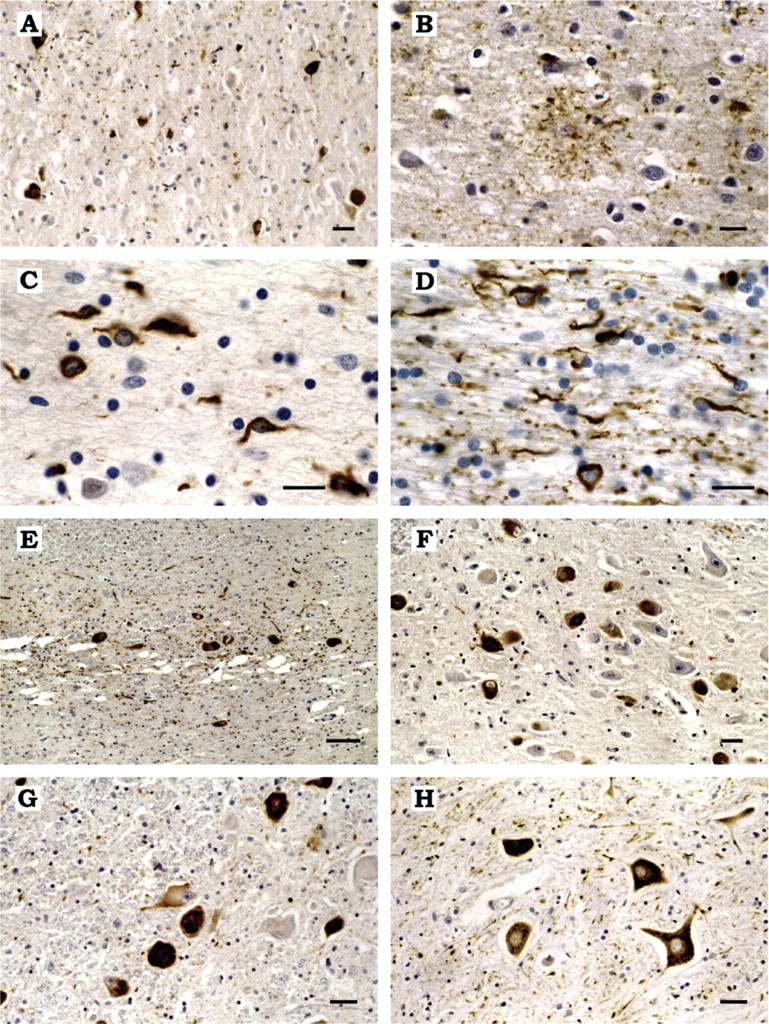
(A) Tau-immunoreactive neurons and grain-like threads in the cingulate gyrus. (B) Tau-positive astrocytic plaque in the temporal cortex. (C, D) Tau-immunoreactive coiled bodies in temporal subcortical white matter (C) and corpus callosum (D). (E) Severe neuronal loss, gliosis and abundant tau-positive neurons and neuropil threads in the substantia nigra. (F, G) Tau-immunoreactive nerve cells in the oculomotor nucleus (F) and the locus coeruleus (G). (H) Tau-immunoreactive neurons and grain-like threads in the anterior horn of the spinal cord. The sections in (A–G) are from the brain of MSTD case number 2. The spinal cord section in (H) is from MSTD case number 14. Anti-tau antibodies = RD4 specific for 4-repeat tau isoforms (A, C, D, E, F, G, H) and AT8 (B). No specific staining was obtained when antibody RD3, which recognizes tau isoforms with three repeats, was used. Scale bars: E = 25 μm; A, F, G, H = 10 μm; B,C,D = 5 μm.
Subject number 14 presented with an atypical PSP-like syndrome and developed mild cognitive decline in the last years of life. Pathologically, a small number of tau-immunoreactive neurons was present in frontal, temporal and parietal cortices and sparse coiled bodies in subcortical white matter. Large pyramidal neurons in the primary motor cortex were fewer than in controls, the remaining ones occasionally displaying granular tau-immunoreactive deposits. A few NFTs were seen in temporal cortex. Sparse tau-immunoreactive neurons and neuropil threads were also present in the CA and, more extensively, in subiculum and entorhinal cortex. A much greater burden of tau deposits was observed in subcortical nuclei and in brainstem. Abundant tau-immunoreactive nerve cells and tufted astrocytes were present in caudate/putamen. The subthalamic nucleus contained many tau-immunoreactive nerve cells and occasional NFTs. In brainstem, abundant tau-immunoreactive nerve cells and neuropil threads were seen in substantia nigra, periaqueductal grey, oculomotor nucleus, reticular formation, locus coeruleus and raphe nuclei. In the spinal cord, moderate nerve cell loss and gliosis were observed in the anterior horn. Residual neurons in lamina IX were strongly tau-immunoreactive. Abundant tau-positive nerve cells and neuropil threads were also present in lamina VII. Sections of the corticospinal tract, at the level of midbrain, pons, medulla and spinal cord, revealed mild loss of axons and myelin as well as presence of a limited number of CD68-immunoreactive macrophages and microglial cells.
Immunohistochemistry for TDP-43 was carried out on sections of frontal and temporal cortices, hippocampus and medulla from subject numbers 2 and 14, as well as spinal cord from subject 14: no TDP-43-immunoreactive deposits were observed.
Cases with a long disease duration (12–21 years)
Seven cases (numbers 5, 19–24) belong to this group (Table 1), all of whom had suffered from FTD. Severe knife-edge atrophy was observed in superior frontal, middle frontal, orbitofrontal, superior temporal and anterior cingulate gyri. Severe atrophy of the hippocampus and parahippocampal gyrus was also noted. The caudate nucleus was markedly atrophic, with a concave appearance of the medial border of its head in most cases. Among the remaining subcortical nuclei, only the thalamus was relatively spared on gross examination. Substantia nigra and locus coeruleus were severely depigmented. Centrum semiovale and corpus callosum were further reduced in bulk compared to cases with an intermediate disease duration, and the entire ventricular system was extensively dilated.
The histological findings are summarized in Table 3. Spongiosis of the upper cortical layers was commonly observed in frontal, temporal, entorhinal and, to a lesser extent, parietal cortices. Ballooned neurons were occasionally noted in the frontal and parietal cortices of patient numbers 19, 21 and 24. The distribution and severity of tau pathology are described in Fig. 6B. Tau-immunoreactive neurons and neuropil threads were frequently observed in neocortical layers II, III, V and VI. A few flame-shaped NFTs were also seen. In the primary motor cortex, occasional large pyramidal neurons displayed tau-immunoreactive deposits. Tufted astrocytes were detected in the frontal, temporal and cingulate cortices of patient numbers 5, 19, 20, 21 and 24. Astrocytic plaques were observed in the frontal, temporal and parietal cortices of cases 5, 19 and 21. The extent of pathology in the hippocampus varied among cases, with some displaying numerous tau-immunoreactive neurons and neuropil threads in the dentate gyrus and CA, and others presenting only a few tau-positive neurons in CA1 (Fig. 8C and D). Many coiled bodies were seen in lower cortical layers, subcortical white matter, internal capsule, corpus callosum and centrum semiovale. Abundant tau-immunoreactive neurons and neuropil threads were present in substantia nigra, reticular formation and anterior horn of the spinal cord. NFTs were detected in periaqueductal grey, reticular formation and raphe nuclei of most cases. Several patterns of tau pathology were observed in spinal cord. Subject number 19 displayed similar changes to those described above for patients 11 and 14. The spinal cord of subject number 20 was characterized by the involvement of lamina VII, with weaker tau labelling of motor neurons (Fig. 10C and D). Minimal loss of axons and myelin was observed in the lateral corticospinal tracts. In the spinal cord of subject number 21, laminae VII and IX were overall equally tau-immunoreactive. Only patient number 5 displayed Alzheimer-type pathology consisting of neuritic plaques in the neocortex (CERAD age-related plaque score B) and NFTs in transentorhinal and entorhinal cortex (Braak stage 2). Subject number 20 presented with rare clusters of Aβ-immunoreactive diffuse plaques in the cerebral cortex and mild amyloid angiopathy of leptomeningeal vessels. Immunohistochemistry for TDP-43 was carried out on sections of frontal and temporal cortices, hippocampus and spinal cord from subject 20: no TDP-43-immunoreactive deposits were observed.
Fig. 10.

(A, B) Antibodies RD3 (specific for tau isoforms with three repeats) and RD4 (specific for tau isoforms with four repeats) were used. Neurons and neurofibrillary tangles in the frontal cortex of subject number 20 were immunolabelled by RD3 (A) and RD4 (B): this subject constitutes an exception, since no other affected family member displayed such an extent of 3-repeat tau pathology in the cerebral cortex, in conjunction with the 4-repeat tau pathology typical of MSTD. Grain-like deposits in the neuropil were selectively labelled by RD4. (C, D) Spinal cord from subjects number 19 (C) and 20 (D) stained with RD4. Note the presence of extensive tau-immunoreactive deposits in neurons and threads in laminae VII and IX in (C) and the predominant involvement of lamina VII, with only weak immunoreactivity of motor neurons in (D). Scale bars: 20 μm.
Tau immunohistochemistry
Neuronal and glial tau deposits were intensely labelled by antibodies AT8, PHF1 and AT180. Antibody PG5 strongly stained cortical neurons and astrocytes, with weaker labelling of neuropil threads in dentate gyrus and molecular layer of the hippocampus. Antibody 12E8 labelled the majority of tau-immunoreactive neurons and oligodendrocytes in cortex, but failed to stain deposits in astrocytes. Weaker labelling was obtained with antibody CP3 and almost no labelling was observed with antibodies AT100, AT270 and Alz50. For the vast majority of cases, immunohistochemistry with antibodies RD3 and RD4 revealed that the neuronal and glial inclusions were composed solely of 4-repeat tau. The major exception was case number 20 with abundant RD3-immunoreactive nerve cell deposits in the substantia nigra and in layers II, V and VI of the cerebral cortex. RD3 labelled virtually all flame-shaped and perinuclear NFTs that were also RD4-immunoreactive (Fig. 10A and B). In addition, a few tau-immunoreactive neurons and flame-shaped NFTs in the neocortex of patient number 5, as well as a few neurons in the locus coeruleus of case number 2, were reactive with antibody RD3.
Genetic analysis and phenotypic correlation
A (g) to (a) transition at position +3 of the 5′ splice site of intron 10 of Tau was present in 30 of the 59 individuals investigated. At the time of writing, 24 of these had developed symptoms of MSTD (Table 1, Fig. 1). The Tau haplotype was determined in 29 mutation carriers; the H1H2 genotype was found in 20 individuals and the H2H2 genotype in nine individuals. To date, clinical symptoms and signs of MSTD have been observed in 16 mutation carriers with the H1H2 genotype and in seven mutation carriers with the H2H2 genotype. Nine mutation carriers with the H1H2 genotype and six mutation carriers with the H2H2 genotype have died. The mean age of onset was 48.5 years regardless of genotype. The mean disease duration was 9.7 ± 6.3 years for the H1H2 group and 10.3 ± 2.2 years for the H2H2 group. Tau sub-haplotypes were determined in 18 mutation carriers. Interestingly, subject numbers 11 and 14, who developed an atypical PSP-like syndrome instead of bv-FTD, inherited the same H1C Tau haplotype with the wild-type allele. The same was true of subject number 8, who suffered from bv-FTD (see supplementary information, Table 5). The ApoE genotype was determined in 23 mutation carriers. An ε3/ε3 genotype was observed in 18 individuals; two individuals had an ε2/ε3 genotype and three had an ε3/ε4 genotype.
Biochemistry and electron microscopy
Sarkosyl-insoluble material was prepared from a total of 54 brain areas of six individuals with MSTD and from a total of 20 brain areas of two unaffected family members lacking the +3 mutation in intron 10 of Tau. The spinal cords of cases 20 and 21 were also used. Three cases (numbers 2, 14, 17) were of intermediate disease duration and three cases (numbers 5, 20, 21) were of long disease duration. By immunoelectron microscopy, tau filaments from cases 2, 5, 14, 17 and 21 had a twisted ribbon or a half ribbon appearance (Fig. 11A–D and H). In brain and spinal cord of case 20, in addition to twisted ribbons and half ribbons, a substantial proportion of tau filaments consisted of Alzheimer-type paired helical and straight filaments (PHFs and SFs) (Figs 11E–G, I and 12). No tau filaments were detected in the sarkosyl-insoluble material prepared from brain regions of two unaffected members of the pedigree.
Fig. 11.
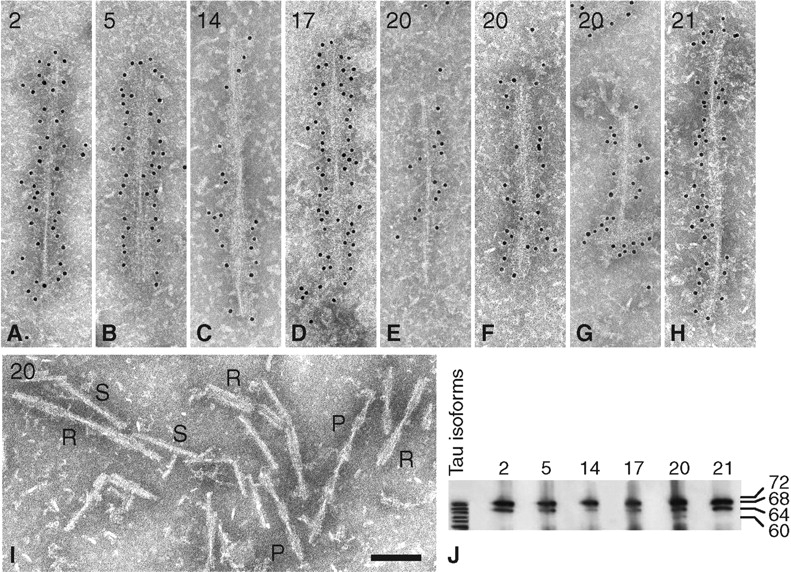
Immunoelectron microscopy and immunoblotting of sarkosyl-insoluble tau from brain of MSTD cases number 2,5,14,17,20 and 21. (A) Twisted ribbon from hippocampus of case number 2. (B) Twisted ribbon from temporal cortex of case number 5. (C) Twisted ribbon from parietal cortex of case number 14. (D) Twisted ribbon from frontal white matter of case number 17. (E) Half ribbon from thalamus of case number 20. (F) Paired helical filament from thalamus of case number 20. (G) Straight filament from frontal cortex of case number 20. (H) Twisted ribbon from putamen of case number 21. (I) Twisted ribbons [R], paired helical filaments [P] and straight filaments [S] in parietal white matter of case number 20. (J) Immunoblotting of sarkosyl-insoluble tau from frontal white matter (cases 2, 5, 17, 20, 21) and caudate/putamen (case 14). All six cases showed the presence of strong tau bands of 64 and 68 kDa molecular mass. For case number 20, an additional band of 60 kDa was also seen. Anti-tau antibody = BR134. Scale bar, 100 nm.
Fig. 12.
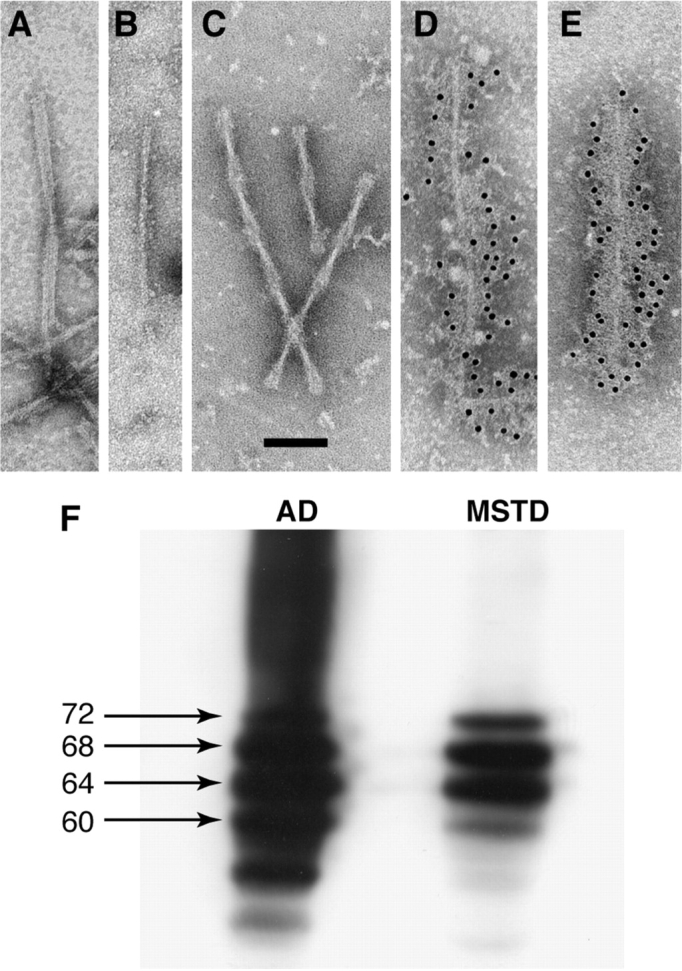
Immunoelectron microscopy and immunoblotting of sarkosyl-insoluble tau from the spinal cord of MSTD case number 20. (A) Twisted ribbon. (B) Straight filament. (C) Paired helical filaments. (D) Straight filament decorated with anti-tau antibody BR134. (E) Paired helical filament decorated with BR134. (F) Immunoblotting of sarkosyl-insoluble tau from the frontal cortex of a case of sporadic Alzheimer's disease (AD) and the spinal cord of MSTD case number 20 using antibody BR133. The abnormal tau bands of 60, 64, 68 and 72 kDa molecular mass are arrowed. Scale bar, 100 nm.
The distribution and relative abundance of tau filaments are shown in Table 4. Cases 2, 5, 17, 20 and 21 showed a substantial filament load in frontal and temporal lobes, with smaller numbers of filaments in parietal and occipital lobes. Filaments were particularly abundant in frontal and parietal white matter, probably reflecting the presence of abundant glial cell inclusions. Where examined, numerous filaments were present in caudate/putamen, hippocampus, parahippocampus, thalamus and spinal cord. The cerebellum was devoid of tau filaments. Overall, the distribution and relative abundance of tau filaments in 5 cases were consistent with a disease process prominently affecting the frontal and temporal lobes of the cerebral cortex. This was not true of case number 14, who lacked tau filaments in frontal and temporal lobes and had a filament load that was less severe than that of the other five cases. Clinically, individual number 14 did not have FTD, but presented with an atypical PSP-like syndrome.
Table 4.
Distribution and number of sarkosyl-insoluble tau filaments in brain and spinal cord of six MSTD patients
| Subject no. | 2 | 5 | 14 | 17 | 20 | 21 |
| Frontal cortex | ++ | ++ | 0 | ++ | +++ | + |
| Frontal white matter | +++ | +++ | +/− | ++ | +++ | ++ |
| Temporal cortex | +++ | ++ | 0 | ++ | +++ | ++ |
| Parietal cortex | + | + | + | n.d. | + | + |
| Parietal white matter | ++ | + | n.d. | ++ | +++ | n.d |
| Occipital cortex | +/− | + | 0 | + | n.d | +/− |
| Occipital white matter | + | n.d. | + | n.d. | + | n.d |
| Hippocampus/parahippocampus | ++ | n.d. | n.d | + | n.d. | n.d. |
| Caudate/putamen | ++ | ++ | + | ++ | + | ++ |
| Thalamus | + | + | +/− | + | + | + |
| Cerebellum | 0 | 0 | 0 | 0 | 0 | 0 |
| Spinal cord | n.d. | n.d. | n.d. | n.d. | + | n.d. |
+/− = <1 filament/grid square; + = 1–14 filaments/grid square; ++ = 15–40 filaments/grid square; +++ = >40 filaments/grid square; n.d. = not determined.
By Western blotting of sarkosyl-insoluble tau extracted from brain, five cases (numbers 2, 5, 14, 17, 21) showed two major pathological tau bands of 64 and 68 kDa, with a minor tau band of 72 kDa being visible in some cases (Fig. 11J). The same was true of sarkosyl-insoluble tau from the spinal cord of case number 21 (data not shown). In contrast, immunoblots of sarkosyl-insoluble tau from brain and spinal cord of case number 20 displayed three major tau bands of 60, 64 and 68 kDa, as well as the minor 72 kDa band, in keeping with the presence of PHFs and SFs by immunoelectron microscopy (Figs 11J and 12). The 60 kDa band was less intense than the 64 and 68 kDa bands. No pathological tau bands were detected in the equivalent brain areas from two unaffected members of the MSTD kindred.
Discussion
The MSTD family was the first known kindred with a +3 mutation in the intron following exon 10 of Tau (Spillantini et al., 1998b). Subsequently, two additional kindreds with this mutation were identified (Tolnay et al., 2000; Neumann et al., 2005). Here, we present a comprehensive study of 24 affected individuals of the MSTD kindred. Age at onset of disease ranged from 39 to 58 years and the disease duration was 1–21 years. The bv-FTD was the most common clinical presentation in the MSTD family. It was seen in 17 of the 21 affected individuals for whom sufficient clinical information was available. Five of these patients had a pure behavioural syndrome, with 12 patients also presenting with memory impairment. The second most common clinical presentation was an atypical PSP-like syndrome, which was present in two individuals, one of whom also had memory deficits. Another patient presented with severe postural imbalance; however, coexistent Ménière's disease may have contributed to the severity of symptoms. This patient also presented with memory loss and developed personality changes, as well as dysexecutive symptoms. One patient presented with an isolated short-term memory deficit. These findings indicate that memory loss is a common presenting sign in MSTD.
Early-stage MSTD patients with bv-FTD, studied by MRI-based VBM and compared to age-matched controls, had the most extensive grey matter loss in the hippocampus, parahippocampal gyrus and frontal operculum/insular cortex of the right hemisphere. This pattern strongly resembles that observed in a group of FTDP-17 subjects with the exon 10 + 16 Tau mutation (Whitwell et al., 2005). To a lesser extent, MSTD patients also displayed grey matter loss in the anterior cingulate gyrus, head of the caudate nucleus, posterolateral orbital cortex and insular cortex bilaterally. These data suggest a pattern of early grey matter loss in limbic, cortical and subcortical regions, which have extensive prefrontal-frontal connections. This was reflected in the patients’ early symptoms, which were marked by behavioural changes and memory loss, suggestive of prefrontal and hippocampal dysfunction. Bilateral atrophy of the orbitofrontal and insular cortices, as well as impaired metabolic activity of the same regions, have previously been reported in FTD in association with disinhibition (Rosen et al., 2002; Peters et al., 2006). Furthermore, behavioural changes have been associated with atrophy or impairment of the anterior cingulate gyrus (Salmon et al., 2003; Liu et al., 2004; Williams et al., 2005). In contrast, the early involvement of hippocampus and parahippocampus observed in MSTD has not been consistently seen in FTDP-17T or FTD generally.
Little is known about the progression of anatomical changes in FTDP-17T along the disease course. In our study, the longitudinal evaluation of brain volume revealed a near linear increase in brain atrophy in affected mutation carriers, similar to what has been described for two siblings with the S305N mutation in Tau (Boeve et al., 2005). In an MSTD asymptomatic mutation carrier followed for 7 years, brain atrophy was slowly progressive for the first 2 years, but has been increasing in a near linear fashion since. Brain volume and rate of atrophy now approach those of affected mutation carriers. This trend parallels that of the progressive cognitive decline evidenced by neuropsychological assessment. Performance on a test of verbal learning is now below the fifth percentile of controls. Additional scores on tests for verbal memory, verbal fluency and response inhibition are now close to the cut-off of affected individuals. Several biomarkers have been proposed in order to identify FTDP-17 disease onset (Alberici et al. 2004; Arvanitakis et al., 2007). Our findings suggest the usefulness of quantitative brain imaging for determining and predicting disease onset in FTDP-17T.
A new finding emerging from this study is the identification of individuals affected with a phenotypic variant of MSTD, resembling an atypical PSP-like syndrome. The observed clinical heterogeneity extends knowledge derived from similar findings in other FTDP-17T families (Bugiani et al., 1999) and further supports an aetiological relationship between FTD and PSP (Kertesz, 2003). Previously, different mutations in Tau were also found to give rise to atypical PSP-like syndromes (Delisle et al., 1999; Stanford et al., 2000; Pastor et al., 2001; Poorkaj et al., 2002; Ros et al., 2005; Van Swieten and Spillantini, 2007). Genetic modifiers may have a strong influence on the initial clinical presentation of disease in the MSTD family. The mutation occurred on the background of at least one H2 Tau allele, with no statistically significant differences in age at onset and disease duration between mutation carriers with haplotypes H1H2 and H2H2. This finding is consistent with the results of a previous study (Baba et al., 2005). Since it has been shown that specific H1 Tau sub-haplotypes are differently associated with the risk to develop tauopathies (Pittman et al., 2005), we analyzed Tau sub-haplotypes in our group of MSTD individuals. Interestingly, the two subjects (brother and sister), who presented with an atypical PSP-like phenotype, were carriers of an H1C Tau haplotype wild-type allele, which has been associated with an increased risk to develop PSP (Pittman et al., 2005) and increased expression of 4-repeat tau isoforms (Myers et al., 2007). The synergistic effects of an H1C haplotype in the wild-type allele and the exon 10 + 3 mutation in the pathogenic allele may have played a role in giving rise to the clinical presentation of atypical PSP in these two siblings. However, a third subject with the H1C haplotype in the wild-type allele did not develop signs of PSP. This individual belongs to a different branch of the pedigree from that of the two siblings and has thus a different genetic background, suggesting that additional genetic modifiers may also play a role. The small sample size prevented us from investigating statistical correlations between specific Tau sub-haplotypes and clinicopathological findings. Since the large majority of affected subjects was homozygous for the ApoE ε3 allele, we were unable to study the role of ApoE genotype as a genetic modifier of MSTD.
Language dysfunction was found in 8 of the 21 patients at disease onset and in a total of 20 individuals along the disease course. It was mostly characterized by naming difficulties, stereotyped speech and reduced verbal fluency, resulting in late mutism. Reduced verbal fluency and, to a lesser extent, executive functioning have been reported as the most consistent early feature of cognitive dysfunction in FTDP-17T individuals (Van Swieten et al., 1999; Ferman et al., 2003). Parkinsonism was only noted in the third year of disease, with limb incoordination appearing in year 4, and frontal release signs, eye movement disorders and sleep abnormalities in year 5.
The neuropathological correlates of MSTD were investigated in 14 patients, 12 of whom had suffered from bv-FTD and two from atypical PSP. In cases with FTD of long duration, atrophy of the frontotemporal lobes and the cingulate gyrus was extensive, with less neurodegeneration in parietal and occipital lobes. Hippocampal formation and striatum were also severely affected, as were substantia nigra, locus coeruleus and dorsal motor nucleus of the vagus nerve.
The involvement of selected brainstem nuclei may explain some of the clinical features of MSTD. Thus, sleep-wake cycle disruption (7/19 subjects) may have resulted from neurodegeneration in locus coeruleus, raphe nuclei, pedunculopontine tegmental nucleus, laterodorsal tegmental nucleus and pontine reticular formation, as well as in the limbic system, all regions known to be important for sleep-wake cycle regulation (Sakurai, 2007).
Motor system pathology was characterized by mild cell loss and tau pathology in giant pyramidal cells of Brodmann area 4, as well as mild loss of axons and myelin in the corticospinal tract. The spinal cord consistently showed nerve cell loss in the anterior horn, similar to what has been described for a case with the N279K mutation in Tau (Slowinski et al., 2007). Vulnerability of motor neurons to tau-induced neurodegeneration is also supported by the phenotypes of some transgenic mouse models of human tauopathies (Lewis et al., 2000; Allen et al., 2002). Tau pathology was consistently detected in neurons of lamina VII. In most cases, motor neurons in lamina IX contained strongly tau-immunoreactive deposits. However, in one case, lamina IX was free of deposits. Additional studies are necessary to verify the existence of a heterogeneous pattern of tau pathology in the spinal cord of different MSTD patients. These findings may explain the presence of pyramidal/motor neuron disease signs in some patients and motor incoordination, possibly secondary to pathology of different descending pathways (e.g. reticulospinal and vestibulospinal), in others (Schoenen and Grant, 2004).
Overall, the severity of neurodegeneration in MSTD was proportional to disease duration. Cases with an atypical PSP-like syndrome were of either short (1 year) or intermediate (6 years) disease duration. They had much less pathology in the cerebral cortex than cases with FTD, with similar changes in subcortical areas. By immunohistochemistry, deposits made of hyperphosphorylated tau protein were present in neurons, astrocytes and oligodendrocytes in all affected brain regions. Oligodendroglial inclusions were particularly numerous in subcortical white matter in all cases, consistent with our previous findings (Spillantini et al., 1997; Spillantini et al., 1998a). In some cases, tufted astrocytes and astrocytic plaques were also present.
Tau inclusions were mostly made of 4-repeat tau, in keeping with the known effects of the +3 intronic mutation on the splicing of exon 10 of Tau (Spillantini et al., 1998b; Varani et al., 1999). Ultrastructurally, tau filaments had the appearance of twisted ribbons or half ribbons, in line with our earlier work (Spillantini et al., 1997). Relative filament numbers correlated well with the level of tau staining in a given brain region and the magnitude of neurodegeneration therein. One case showed a large number of Alzheimer-type PHFs and SFs, in addition to ribbon-like filaments. By immunohistochemistry, tau inclusions in this case were made of 3- and 4-repeat tau. By immunoblotting, pathological tau bands of 60, 64, 68 and 72 kDa were present, as is the case in Alzheimer's disease (Goedert et al., 1992). However, unlike what is observed in Alzheimer's disease, the 60 kDa band was weaker than the 64 and 68 kDa bands, consistent with the presence of more ribbon-like filaments than PHFs and SFs. This case constitutes an exception to the rule that the overexpression of human 4-repeat tau isoforms results in the formation of inclusions made solely of 4-repeat tau (Goedert and Spillantini, 2006; Van Swieten and Spillantini, 2007). The mechanisms leading to the coexistence of filaments made of 4-repeat tau only and of all 6 tau isoforms remain to be established.
In summary, this multidisciplinary and longitudinal characterization of the MSTD kindred has provided new insights into the neurodegenerative process: (i) mutation +3 in intron 10 of Tau can give rise to a clinical and neuropathological picture of either FTD or atypical PSP; (ii) Tau haplotype of the wild-type allele may act as a genetic modifier of disease presentation; (iii) early-onset memory loss and hippocampal atrophy are common findings in MSTD; (iv) quantitative neuroimaging may be useful in predicting disease onset in FTDP-17T; (v) neurodegeneration in MSTD involves various neurological systems, including prefrontal and limbic cortical/subcortical regions, hippocampus, parahippocampus, selected brainstem nuclei and spinal cord.
Supplementary material
Supplementary materials are available at Brain online.
Acknowledgements
We are indebted to the MSTD family members and caregivers for their enthusiastic commitment and participation in this study. We thank Brenda Dupree, Rose Richardson and Connie Alyea for histology and immunohistochemistry; Beatrice Terni for immunoblotting; Linda Bailey for secretarial work; Brad Glazier for editing manuscript and illustrations; Elizabeth Way for data retrieval and organization; Fredrik Skarstedt, Ryan Christy and Urs Kuderli for illustrations; Matthew Burger and Joseph Wade, for software assistance; Eileen H. Bigio, Thomas G. Beach and Charles W. White III for providing brain tissue. This paper is dedicated to Giancarlo Guazzi, professor of neurology and neuropathology. Supported by NIH PHS AG10133, UK Medical Research Council, the Alzheimer's Research Trust and the Parkinson's Disease Society. S.S. is a PhD student of and is in part supported by the Department of Neurological and Behavioral Sciences, University of Siena, Siena, Italy. Funding to pay the Open Access publication charges for this article was provided by the Indiana Alzheimer Disease Center (PHS P30AG10133).
Glossary
Abbreviations:
- FTD
frontotemporal dementia
- MSTD
multiple system tauopathy with presenile dementia
- NFT
neurofibrillary tangle
- CA
cornu ammonis
References
- Alberici A, Gobbo C, Panzacchi A, Nicosia F, Ghidoni R, Benussi L, et al. Frontotemporal dementia: impact of P301L tau mutation on a healthy carrier. J Neurol Neurosurg Psychiatry. 2004;75:1607–10. doi: 10.1136/jnnp.2003.021295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, et al. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340–51. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanitakis Z, Witte RJ, Dickson DW, Tsuboi Y, Uitti RJ, Slowinski J, et al. Clinical-pathological study of biomarkers in FTDP-17 (PPND family with N279K tau mutation) Parkinsonism Relat Disord. 2007;13:230–9. doi: 10.1016/j.parkreldis.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Ashburner J, Friston JR. Voxel-based morphometry–the methods. Neuroimage. 2000;11:805–21. doi: 10.1006/nimg.2000.0582. [DOI] [PubMed] [Google Scholar]
- Baba Y, Tsuboi Y, Baker MC, Uitti RJ, Hutton ML, Dickson DW, et al. The effect of tau genotype on clinical features in FTDP-17. Parkinsonism Relat Disord. 2005;11:205–8. doi: 10.1016/j.parkreldis.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, et al. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999;8:711–5. doi: 10.1093/hmg/8.4.711. [DOI] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–9. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Bird TD, Wijsman EM, Nochlin D, Leehey M, Sumi SM, Payami H, et al. Chromosome 17 and hereditary dementia: linkage studies in three non-Alzheimer families and kindreds with late-onset FAD. Neurology. 1997;48:949–54. doi: 10.1212/wnl.48.4.949. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Tremont-Lukats IW, Waclawik AJ, Murrell JR, Hermann B, Jack CR, et al. Longitudinal characterization of two siblings with frontotemporal dementia and parkinsonism linked to chromosome 17 associated with the S305N tau mutation. Brain. 2005;128:752–72. doi: 10.1093/brain/awh356. [DOI] [PubMed] [Google Scholar]
- Bronner IF, Rizzu P, Seelaar H, van Mil SE, Anar B, Azmani A, et al. Progranulin mutations in Dutch familial frontotemporal lobar degeneration. Eur J Hum Genet. 2007;15:369–74. doi: 10.1038/sj.ejhg.5201772. [DOI] [PubMed] [Google Scholar]
- Brun A, Englund E, Gustafson L, Passant U, Mann DMA, Neary D, et al. Clinical, neuropsychological and neuropathological criteria of frontotemporal dementia. J Neurol Neurosurg Psychiat. 1994;57:416–8. doi: 10.1136/jnnp.57.4.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugiani O, Murrell JR, Giaccone G, Hasegawa M, Ghigo G, Tabaton M, et al. Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in Tau. J Neuropathol Exp Neurol. 1999;58:667–77. doi: 10.1097/00005072-199906000-00011. [DOI] [PubMed] [Google Scholar]
- Cairns NK, Bigio EH, Mackenzie IRA, Neumann M, Lee VMY, Hatanpaa KJ, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZZ, Miller BL, et al. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci USA. 1998;95:13103–7. doi: 10.1073/pnas.95.22.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther RA. Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc Natl Acad Sci USA. 1991;88:2288–92. doi: 10.1073/pnas.88.6.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–4. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- Delisle MB, Murrell JR, Richardson R, Trofatter JA, Rascol O, Soulages X, et al. A mutation at codon 279 (N279K) in exon 10 of the Tau gene causes a tauopathy with dementia and supranuclear palsy. Acta Neuropathol. 1999;98:62–77. doi: 10.1007/s004010051052. [DOI] [PubMed] [Google Scholar]
- Ferman TJ, McRae CA, Arvanitakis Z, Tsuboi Y, Vo A, Wszolek ZK. Early and pre-symptomatic neuropsychological dysfunction in the PPND family with the N279K tau mutation. Parkinsonism Relat Disord. 2003;9:265–70. doi: 10.1016/s1353-8020(02)00098-6. [DOI] [PubMed] [Google Scholar]
- Foster NL, Wilhelmsen K, Sima AAF, Jones MZ, D’Amato CJ, Gilman S, et al. Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Ann Neurol. 1997;41:706–15. doi: 10.1002/ana.410410606. [DOI] [PubMed] [Google Scholar]
- Ghetti B, Farlow MR. Hereditary presenile dementia and multiple system degeneration with neurofibrillary tangles. Brain Pathol. 1994;4:517. [Google Scholar]
- Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3:519–26. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9:4225–30. doi: 10.1002/j.1460-2075.1990.tb07870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159–68. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. 2006;314:777–81. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Good CD, Johnsrude IS, Ashburner J, Henson RN, Friston KJ, Frackowiak RS. A voxel-based morphometric study of ageing in 465 normal adult human brains. Neuroimage. 2001;14:21–36. doi: 10.1006/nimg.2001.0786. [DOI] [PubMed] [Google Scholar]
- Heutink P, Stevens M, Rizzu P, Bakker E, Kros JM, Tibben A, et al. Hereditary frontotemporal dementia is linked to chromosome 17q21–q22: a genetic and clinicopathological study of three Dutch families. Ann Neurol. 1997;41:150–9. doi: 10.1002/ana.410410205. [DOI] [PubMed] [Google Scholar]
- Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31:545–8. [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Kertesz A. Pick complex: an integrative approach to frontotemporal dementia, primary progressive aphasia, corticobasal degeneration, and progressive supranuclear palsy. Neurologist. 2003;9:311–7. doi: 10.1097/01.nrl.0000094943.84390.cf. [DOI] [PubMed] [Google Scholar]
- Leverenz JB, Yu CE, Montine TJ, Steinbart E, Bekris LM, Zabetian C, et al. A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain. 2007;130:1360–74. doi: 10.1093/brain/awm069. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, van Slegtenhorst M, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–5. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47:1–9. doi: 10.1212/wnl.47.1.1. [DOI] [PubMed] [Google Scholar]
- Liu W, Miller BL, Kramer JH, Rankin K, Wyss-Coray C, Gearhart R, et al. Behavioral disorders in the frontal and temporal variants of frontotemporal dementia. Neurology. 2004;62:742–8. doi: 10.1212/01.wnl.0000113729.77161.c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ, et al. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick's Disease. Arch Neurol. 2001;58:1803–9. doi: 10.1001/archneur.58.11.1803. [DOI] [PubMed] [Google Scholar]
- Mukherjee O, Pastor P, Cairns NJ, Chakraverty S, Kauwe JSK, Shears S, et al. HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol. 2006;60:314–22. doi: 10.1002/ana.20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell JR, Farlow MR, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science. 1991;254:97–9. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- Murrell JR, Koller D, Foroud T, Goedert M, Spillantini MG, Edenberg HJ, et al. Familial multiple system tauopathy with presenile dementia is localized to chromosome 17. Am J Hum Genet. 1997;61:1131–8. doi: 10.1086/301594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers AJ, Pittman AM, Zhao AS, Rohrer K, Kaleem M, Marlow L, et al. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis. 2007;25:561–70. doi: 10.1016/j.nbd.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–54. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- Neumann M, Mittelbronn M, Simon P, Vanmassenhove B, de Silva R, Lees A, et al. A new family with frontotemporal dementia with intronic 10 + 3 splice site mutation in the tau gene: neuropathology and molecular effects. Neuropathol Appl Neurobiol. 2005;31:362–73. doi: 10.1111/j.1365-2990.2005.00629.x. [DOI] [PubMed] [Google Scholar]
- Pastor P, Pastor E, Carnero C, Vela R, García T, Amer G, et al. Familial atypical progressive supranuclear palsy associated with homozigosity for the delN296 mutation in the tau gene. Ann Neurol. 2001;49:263–67. doi: 10.1002/1531-8249(20010201)49:2<263::aid-ana50>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Peters F, Perani D, Herholz K, Holthoff V, Beuthien-Baumann B, Sorbi S, et al. Orbitofrontal dysfunction related to both apathy and disinhibition in frontotemporal dementia. Dement Geriatr Cogn Disord. 2006;21:373–9. doi: 10.1159/000091898. [DOI] [PubMed] [Google Scholar]
- Petersen RB, Tabaton M, Chen SG, Monari L, Richardson SL, Lynches T, et al. Familial progressive subcortical gliosis. Presence of prions and linkage to chromosome 17. Neurology. 1995;45:1062–7. doi: 10.1212/wnl.45.6.1062. [DOI] [PubMed] [Google Scholar]
- Pittman AM, Myers AJ, Abou-Sleiman P, Fung HC, Kaleem M, Marlowe L, et al. Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J Med Genet. 2005;42:837–46. doi: 10.1136/jmg.2005.031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–25. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H, et al. An R5L Tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol. 2002;52:511–6. doi: 10.1002/ana.10340. [DOI] [PubMed] [Google Scholar]
- Ros R, Thobois S, Streichenberger N, Kopp N, Sánchez MP, Pérez M, et al. A new mutation of the tau gene, G303V, in early-onset familial progressive supranuclear palsy. Arch Neurol. 2005;62:1444–50. doi: 10.1001/archneur.62.9.1444. [DOI] [PubMed] [Google Scholar]
- Rosen HJ, Gorno-Tempini ML, Goldman WP, Perry RJ, Schuff N, Weiner M, et al. Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology. 2002;58:198–208. doi: 10.1212/wnl.58.2.198. [DOI] [PubMed] [Google Scholar]
- Sakurai T. The neural circuit of orexin (hypocretin): mantaining sleep and wakefulness. Nat Rev Neurosci. 2007;8:171–81. doi: 10.1038/nrn2092. [DOI] [PubMed] [Google Scholar]
- Salmon E, Garraux G, Delbeuck X, Collette F, Kalbe E, Zuendorf G, et al. Predominant ventromedial frontopolar metabolic impairment in frontotemporal dementia. Neuroimage. 2003;20:435–40. doi: 10.1016/s1053-8119(03)00346-x. [DOI] [PubMed] [Google Scholar]
- Schoenen J, Grant G. Spinal cord: connections. In: Paxinos G, Mai JK, editors. The human nervous system. London: Elsevier Academic Press; 2004. pp. 233–49. [Google Scholar]
- Slowinski J, Dominik J, Uitti RJ, Ahmed Z, Dickson DW, Wszolek ZW. Frontotemporal dementia and parkinsonism linked to chromosome 17 with the N279K tau mutation. Neuropathology. 2007;27:73–80. doi: 10.1111/j.1440-1789.2006.00742.x. [DOI] [PubMed] [Google Scholar]
- Smith SM, Zhang Y, Jenkinson M, Chen J, Matthews PM, Federico A, et al. Accurate, robust, and automated longitudinal and cross-sectional brain change analysis. Neuroimage. 2002;17:479–89. doi: 10.1006/nimg.2002.1040. [DOI] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TE, Johansen-Berg H, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage. 2004;23(Suppl 1):S208–19. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MJ, Ghetti B. Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci USA. 1997;94:4113–8. doi: 10.1073/pnas.94.8.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol. 1998a;8:387–402. doi: 10.1111/j.1750-3639.1998.tb00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998b;95:7737–41. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford PM, Halliday G, Brooks W, Kwok J, Storey C, Creasey H, et al. Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: expansion of the disease phenotype caused by tau gene mutations. Brain. 2000;123:880–93. doi: 10.1093/brain/123.5.880. [DOI] [PubMed] [Google Scholar]
- Tolnay M, Spillantini MG, Rizzini C, Eccles D, Lowe J, Ellison D. A new case of frontotemporal dementia and parkinsonism resulting from an intron 10 + 3-splice site mutation in the tau gene: clinical and pathological features. Neuropathol Appl Neurobiol. 2000;26:368–78. doi: 10.1046/j.1365-2990.2000.00109.x. [DOI] [PubMed] [Google Scholar]
- Van Swieten J, Spillantini MG. Hereditary frontotemporal dementia caused by Tau gene mutations. Brain Pathol. 2007;17:63–73. doi: 10.1111/j.1750-3639.2007.00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Swieten JC, Stevens M, Rosso SM, Rizzu P, Joosse M, deKoning I, et al. Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Ann Neurol. 1999;46:617–26. doi: 10.1002/1531-8249(199910)46:4<617::aid-ana10>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Varani L, Hasegawa M, Spillantini MG, Smith MJ, Murrell JR, Ghetti B, et al. Structure of tau exon 10 splicing regulatory element RNA and destabilization by mutations of frontotemporal dementia and parkinsonism linked to chromosome 17. Proc Natl Acad Sci USA. 1999;96:8229–34. doi: 10.1073/pnas.96.14.8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Josephs KA, Rossor MN, Stevens JM, Revesz T, Holton JL, et al. Magnetic resonance imaging signatures of tissue pathology in frontotemporal dementia. Arch Neurol. 2005;62:1402–8. doi: 10.1001/archneur.62.9.1402. [DOI] [PubMed] [Google Scholar]
- Wijker M, Wszolek ZK, Wolters ECH, Rooimans MA, Pals G, Pfeiffer RF, et al. Localization of the gene for rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration to chromosome 17q21. Hum Mol Genet. 1996;5:151–4. doi: 10.1093/hmg/5.1.151. [DOI] [PubMed] [Google Scholar]
- Wilhelmsen KC, Lynch T, Pavlou E, Higgins M, Nygaard TG. Localization of disinhibition-dementia-parkinsonism-amyotrophy complex to 17q21–22. Am J Hum Genet. 1994;55:1159–65. [PMC free article] [PubMed] [Google Scholar]
- Williams GB, Nestor PJ, Hodges JR. Neural correlates of semantic and behavioural deficits in frontotemporal dementia. Neuroimage. 2005;24:1042–51. doi: 10.1016/j.neuroimage.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Yamaoka LH, Welsh-Bohmer KA, Hulette CM, Gaskell PG, Murray M, Rimmler JL, et al. Linkage of frontotemporal dementia to chromosome 17: clinical and neuropathological characterization of phenotype. Am J Hum Genet. 1996;59:1306–12. [PMC free article] [PubMed] [Google Scholar]