

Abstract
In Alzheimer's disease (AD), the insidious impairment of declarative memory coincides with the accumulation of extracellular amyloid-β protein (Aβ) and intraneuronal tau aggregates. Dementia severity correlates strongly with decreased synapse density in hippocampus and cortex. Although numerous studies show that soluble Aβ oligomers inhibit hippocampal long-term potentiation, their role in long-term synaptic depression (LTD) remains unclear. Here, we report that soluble Aβ oligomers from several sources (synthetic, cell culture, human brain extracts) facilitated electrically-evoked LTD in the CA1 region. Aβ-enhanced LTD was mediated by mGluR or NMDAR activity, depending on the induction protocol. Both forms of LTD were prevented by an extracellular glutamate scavenger system. Aβ-facilitated LTD was closely mimicked by the action of the glutamate reuptake inhibitor TBOA, including a shared dependence on extracellular calcium levels and activation of PP2B and GSK-3 signaling. In accord, synaptic glutamate uptake was significantly decreased by soluble Aβ. We conclude that soluble Aβ oligomers perturb synaptic plasticity by altering glutamate recycling at the synapse and promoting synapse depression.
Alzheimer's disease (AD), the most common neurodegenerative disorder, is characterized by progressive memory and cognitive impairment and cerebral accumulation of extracellular amyloid plaques and intraneuronal neurofibrillary tangles. Although the specific molecular initiators of AD remain unknown in most patients, extensive research suggests that the amyloid β-protein (Aβ) plays an early and essential pathogenic role. Of note, dementia severity in AD correlates more strongly with cortical levels of soluble Aβ species than with insoluble amyloid plaque burden (Lue et al., 1999; McLean et al., 1999). Experimentally, soluble Aβ oligomers have been specifically shown to block hippocampal long-term potentiation (LTP), an electrophysiological correlate of learning and memory (e.g., Lambert et al, 1998; Walsh et al., 2002; Wang et al., 2002; Townsend et al., 2006; Shankar et al, 2008). In accord, impairment of synaptic plasticity can be detected in vivo before the formation of insoluble Aβ deposits in APP transgenic mouse models (e.g., Hsia et al., 1999; Mucke et al, 2000). Synthetic Aβ aggregates have been reported to inhibit N-methyl-D-aspartate receptor (NMDAR)-dependent LTP, but not NMDAR-independent LTP (Chen et al., 2002; Wang et al., 2004a; but see Raymond et al., 2003). This finding is consistent with evidence that Aβ can affect surface expression of NMDARs (Snyder et al., 2005; Dewachter et al., 2009) and may increase (Molnar et al., 2004; Wu et al., 1995) or decrease (Chen et al., 2002; Raymond et al., 2003) NMDAR conductance.
A principal neuropathological finding in AD subjects is cortical atrophy associated with degeneration of neurites, decreased dendritic spine density and frank neuronal loss (Terry et al, 1991; Knobloch and Mansuy, 2008). Anatomical studies in normal rodents suggest that the induction of LTP is associated with spine formation and increased spine volume, whereas the induction of long-term synaptic depression (LTD) results in decreased spine volume and spine elimination (Matsuzaki M, 2004; Nagerl et al., 2004; Zhou et al., 2004; Bastrikova et al., 2008). Similar to LTP, the induction of LTD in the CA1 region of hippocampus requires activation of NMDAR and/or metabotropic glutamate receptors (mGluR), depending on the stimulation protocol and recording conditions (Kemp and Bashir, 2001; Anwyl, 2007; Citri and Malenka, 2008). Mechanistically, synapse potentiation vs. depression may ultimately depend on alterations in cytosolic Ca2+ concentration and the differential activation of certain kinases and phosphatases, including p38 mitogen-activated protein kinase (MAPK) and calcineurin (protein phosphastase 2B (PP2B)) (reviewed in Kemp and Bashir, 2001; Citri and Malenka, 2008).
Although numerous reports describe the effects of soluble Aβ species on LTP, only few studies have examined LTD induction, which have yielded inconsistent results. For example, synthetic Aβ peptides were reported to facilitate LTD induction in an NMDAR-dependent manner in vivo (Kim at al., 2001), whereas other studies found no effect on LTD in slices (Raymond et al., 2003; Wang et al., 2002; 2004a). We recently extracted buffer-soluble Aβ directly from the brains of typical AD patients and showed that this extract, which contains soluble Aβ dimers and trimers, facilitated LTD induction in the CA1 region of mouse hippocampus by an mGluR-dependent mechanism (Shankar et al., 2008). Given that both NMDAR and mGluR activation have been implicated in the effects of Aβ on LTD, we asked whether glutamate clearance mechanisms are perturbed by Aβ. In addition to affecting synaptic plasticity, excitotoxic effects of glutamate are believed to contribute to progressive neuronal loss in AD (Pomara et al., 1992; Harkany et al., 2000). Furthermore, gene expression and protein levels of excitatory amino acid transporters (EAAT1 and EAAT 2) are altered in the hippocampus and frontal cortex of AD subjects (Jacob et al., 2007). Here, we provide evidence that small, soluble Aβ assemblies from several sources enhance synaptic depression through a novel mechanism involving altered glutamate uptake at hippocampal synapses. Our results have both mechanistic and therapeutic implications for the initiation of hippocampal synaptic failure in AD and in more subtle forms of age-related Aβ accumulation.
Methods
Aβ preparations
Secreted human Aβ peptides were collected and prepared from the conditioned medium (CM) of a CHO cell line (7PA2) that stably expresses human APP751 containing the V717F AD mutation (Podlisny et al, 1995). Cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum, 1% penicillin/streptomycin, 2 mM L-glutamine, and 200 mg/ml G418 for selection. Upon reaching ∼95% confluency, the cells were washed and cultured overnight (∼15 h) in serum-free medium. CM was collected, spun at 1500 × g to remove dead cells and debris, and stored at 4°C. The CM was concentrated 10-fold with a YM-3 Centricon filter (Walsh et al., 2005). Aliquots of concentrated 7PA2 CM were stored at −80°C. Synthetic Aβ1-42 (Biopolymer Lab., Dept. of Neurology, UCLA) was dissolved in water and incubated at 37°C for 10 days, then aliquoted (50 μl) and stored at −80 °C.
Hippocampal slice electrophysiology
The procedures for the hippocampal slice preparation and field excitatory postsynaptic potentials (fEPSPs) recordings in the CA1 region of mouse hippocampus were described previously (Li et al., 2006; Shankar et al., 2008). A bipolar stimulating electrode (FHC Inc., Bowdoin, ME) was placed in the Schaffer collaterals to deliver test and conditioning stimuli. A borosilicate glass recording electrode filled with artificial cerebrospinal fluid was positioned in stratum radiatum of CA1. Test responses were recorded for 30-60 min prior to beginning the experiment to ensure stable responses. The field potentials were amplified 100× using an Axon Instruments 200B amplifier and digitized with Digidata 1322A. Traces were obtained by pClamp 9.2 and analyzed using the Clampfit 9.2 program. To induce LTD, either 300 or 900 pulses at low frequency (1 Hz) stimulation (LFS) were applied to the slices. LTP was induced by two sequential high-frequency stimulations (HFS: 100 Hz for 1 s, then repeated after 20 s). All LTD/LTP values represent fEPSP slopes measured 50 min after the conditioning stimulus unless stated otherwise. Two-tailed Student's t-test and one-way analysis of variance (ANOVA) were used to determine statistical significance.
Whole-cell recordings were made from the soma of visually identified pyramidal neurons located in CA1 of the hippocampus. Patch pipettes (5∼7 MΩ) were filled with an internal solution containing (in mM): 110 Cs-gluconate, 20 CsCl, 10 HEPES, 4 NaCl, 0.5 EGTA, 2 MgCl2, 2 Na2ATP and 0.25 NaGTP, titrated with KOH to pH 7.4. The excitatory postsynaptic currents (EPSCs) were recorded following stimulation of Schaffer collaterals ∼150 μM from the CA1 cell body with a bipolar electrode. AMPA-mediated EPSCs were recorded at a holding potential of −70 mV in the presence of 10 μM bicuculline (BIC) and 50 μM AP5. NMDA-mediated EPSCs were recorded at +45 mV in ACSF containing 10 μM BIC and 10 μM NBQX, or else at −70 mV in 0.1 mM Mg2+ ACSF containing BIC and NBQX. Series resistance was kept 15∼30 MΩ and monitored throughout each recording. Cells were excluded from data analysis if the series resistance changed by >20% during the course of the experiment. All patch clamp experiments were performed at 24°C.
Glutamate uptake assay
Preparation of crude synaptosomal fractions and glutamate uptake assay were performed as described by Yang et al. (2005) with minor modifications. Briefly, hippocampal slices were treated for 2 hours with soluble Aβ, TBOA or DMSO, then homogenized in 0.32 M sucrose, 1 mM EDTA, 4 mM Tris and 10 mM glucose (pH 7.4) on ice. Homogenates were centrifuged at 1000 × g for 10 min and subsequent supernatants were spun at 9000 × g for 10 min. Final pellets were resuspended in HEPES buffer solution to yield a protein concentration of 0.25 mg/ml. To avoid treatment washout, crude synaptosomes were treated with the same respective agents for 45 min at 37 °C. Then, they were incubated with 10 nM [3H]glutamate (20-60 Ci/mmol; Perkin Elmer, Boston, MA) and 30 μM unlabeled glutamate for 5 min. The uptake was stopped by placing the samples on ice and spinning at 15,000 rpm for 10 min at 4 °C. Pellets were washed and resuspended in ice-cold PBS. Glutamate uptake was counted on an LS6500 multi-purpose scintillation counter. The absolute number of counts taken up varied from 500 to 40,000 counts per minute (cpm), with a background of 20 cpm.
Drug treatments
Paired control and experimental hippocampal slices from a single mouse were maintained together in a single chamber, except during individual drug treatments. The following antagonists were purchased from Tocris (Ellisville, MO): DL-2-amino-5-phosphonopentanoic acid (AP5), MK-801, MCPG, Cyclothiazide, NBQX, (-)-Bicuculline methiodide, SB203580, SB415286, Ryanodine, U73122, DL-threo-β-benzyloxyaspartic acid (TBOA) and Dihydrokainic acid (DHK). Glutamic-pyruvic transaminase, pyruvic acid, Ifenprodil and FK506 were purchased from Sigma (St Louis, MO).
Results
Soluble human Aβ from several sources facilitates hippocampal LTD
Small, soluble Aβ assemblies have been obtained from a variety of sources, including synthetic peptides, cell culture medium, and human brain extracts (Lambert et al., 1998; Podlisny et al., 1995; McLean et al., 1999; Shankar et al., 2008). A common feature among the soluble Aβ preparations used in the current study is the presence of low-n assembly forms, i.e., Aβ oligomers, that are slightly larger than the monomer but much smaller than amyloid fibrils. When examined on denaturing gels, these oligomers run principally as SDS-stable dimers and trimers (Fig. 1A). We recently reported that soluble oligomers extracted directly from human (AD) cortex facilitate LTD induction through activation of mGluR when a subthreshold (300 pulses, 1 Hz) stimulation is applied to hippocampal slices (Shankar et al., 2008). To investigate the mechanisms underlying the enhancement of LTD by soluble human Aβ, we first used cell-secreted Aβ species obtained from a CHO cell line that stably expresses hAPP with the V717F amyloidogenic AD mutation (7PA2 cells). These cells release readily detectable levels of monomeric and oligomeric Aβ species into the medium in the absence of insoluble aggregates (Podlisny et al., 1995; Walsh et al., 2002). In accord with our recent work using soluble Aβ extracted from human cortex, we found that 7PA2 CM [but not the CM of untransfected (CHO-) cells] facilitated the induction of hippocampal LTD following a subthreshold, low-frequency stimulus (LFS) (300 pulses at 1 Hz) that otherwise does not induce LTD (79.7 ± 2.4%, n=6, vs. 93.4 ± 4.2%, n=7, of baseline fEPSP slope; p<0.05) (Fig. 1B). We next sought to establish the effects of soluble Aβ on the expression of LTD using a standard LTD-inducing protocol known to be NMDAR dependent (Kemp and Bashir, 2001), namely 900 pulses at 1 Hz. This form of LTD was equally inducible in slices perfused with artificial cerebrospinal fluid (ACSF) supplemented with 7PA2 CM or CHO- CM (73.6 ± 2.6%, n=9, vs. 77.1 ± 2.1%, n=9; p>0.05) (Fig. 1C). Similar results were observed using synthetic Aβ1-42 (Fig. 1D), soluble extracts of human AD cortex containing Aβ oligomers (Fig. 1E), or size-exclusion chromotagraphy (SEC) fractions of 7PA2 CM enriched in either oligomers or monomers (Figs. 1A, right panel, and Fig. 1F). These data are consistent with previous reports that synthetic Aβ does not significantly change the magnitude of NMDAR-dependent (i.e., 900-pulse) LTD achieved in vitro (Raymond et al., 2003; Wang et al., 2002, 2004a).
Figure 1. Soluble Aβ facilitates long-term depression in the CA1 region of hippocampal slices.
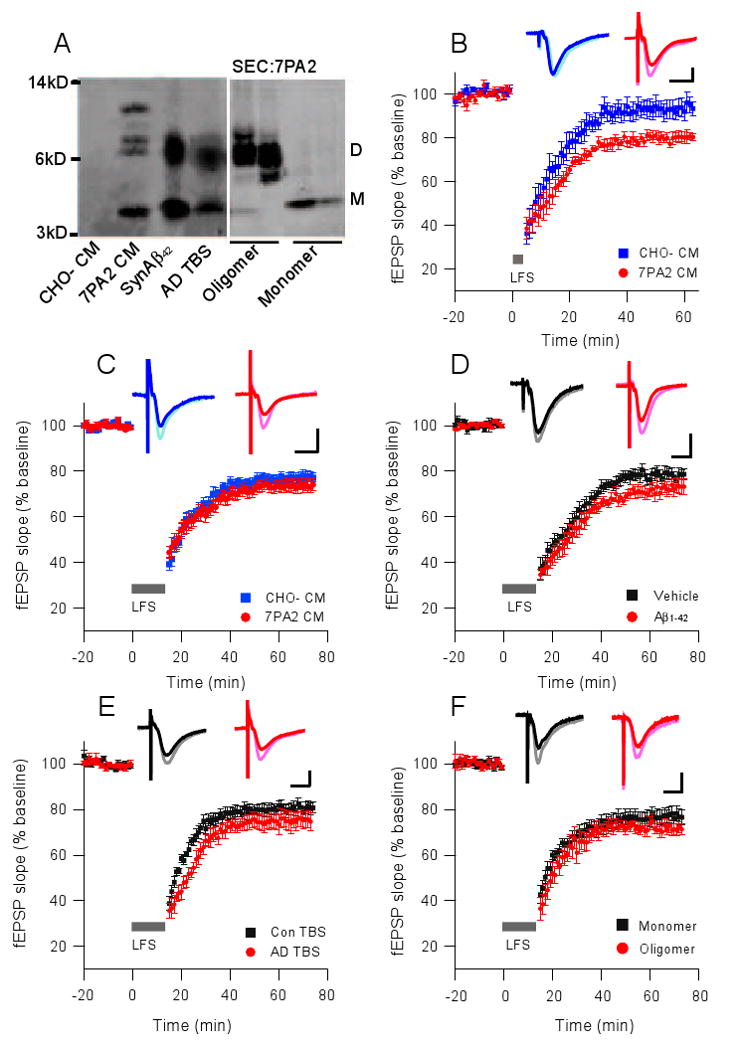
(A) Western blot of the 3 sources of soluble Aβ used for LTD experiments in this study. All 3 contain a SDS-stable band at ∼8 kDa, and this has been confirmed previously as an Aβ dimer by mass spectrometry (Shankar et al, 2008). 7PA2 CM also contains a higher, apparent trimer species. CHO- CM is devoid of these human Aβ species, as expected, and serves as a negative control throughout these studies. All samples were immunoprecipitated with polyclonal Aβ antibody AW8 and blotted with combined Aβ monoclonal antibodies 2G3 (Aβ40) and 21F12 (Aβ42). SEC: fractions rich in oligomers (left) or monomers (right) separated by SEC of 7PA2 CM (B) A train of 300 single pulses at 1 Hz (5 min; small grey bar) did not induce LTD in acute mouse hippocampal slices in the presence of CHO- CM (blue squares, n=6) but induced a significant LTD in the presence of 7PA2 CM (red circles, n=7). C-F A standard LTD protocol of 900 single pulses at 1 Hz (15 min; long grey bar) was applied to slices treated with 7PA2 CM (C), synthetic Aβ1-42 (D), AD brain TBS extract (E), or SEC fractions from 7PA2 CM (F). All are not significantly different from their respective controls. Insets in B-F represent typical field excitatory postsynaptic potentials (fEPSPs) recorded before (light) and 50 min after (dark) LFS. Horizontal calibration bar, 10 ms; vertical bar, 0.5 mV.
To probe the mechanism of Aβ-facilitated LTD, we used the non-selective group I/II mGluR antagonist MCPG (500 μM) and the NMDAR antagonist AP5 (50 μM). In agreement with our previous finding using soluble Aβ from AD cortex (Shankar et al, 2008), the facilitation of 300-pulse LTD by 7PA2 CM was dependent on activation of mGluR but not NMDAR, as the LTD was MCPG-sensitive and AP5-resistant (93.2 ± 6.1%, n=6, vs. 80.4 ± 4.8%, n=5; p<0.05) (Fig. 2A). Unlike the 300-pulse LTD, 900-pulse LTD induced in the presence of 7PA2 CM was not prevented by MCPG (78.5 ± 4.1%, n=6, vs. 73.6 ± 2.6%, n=9; p>0.05) (Fig. 2B). Interestingly, 50 μM AP5 fully blocked 900-pulse LTD in slices exposed to CHO- CM, but had no significant effect on the 900-pulse LTD in slices exposed to the Aβ-rich 7PA2 CM (100.6 ± 1.4%, n=8, vs. 75.4 ± 3.4%, n=6; p<0.01) (Fig. 2C). This AP5-resistance noted with 7PA2 CM was also observed with soluble synthetic Aβ1-42 (Supp. Fig. 1A) and soluble extracts of AD cortex containing Aβ oligomers (Supp. Fig.1B; data quantified in Supp. Fig. 1C). Furthermore, application of SEC fractions of 7PA2 CM revealed that the induction of AP5-resistant 900-pulse LTD was specific to Aβ oligomers, not monomers (Fig. 2D; Supp. Fig. 1C). These results using three distinct sources of soluble low-n oligomers suggest that the LTD facilitated by Aβ oligomers is mechanistically different from conventional NMDAR-dependent LTD that is sensitive to 50 μM AP5. Because the 900-LFS protocol for inducing LTD in the CA1 region is well known to be NMDAR-dependent (see Kemp and Bashir, 2001, and all control data in Figs. 2C and D and Supp Fig.1C), the resistance to AP5 of the LTD induced in the presence of the various soluble Aβ preparations was unexpected. It has been reported that synthetic Aβ can increase NMDAR activity by acting as an agonist or co-agonist of NMDARs (Cowburn et al., 1997) and increasing NMDA conductance (Wu et al., 1995). Consequently, 50 μM AP5 may not have been sufficient to completely block an enhanced NMDAR activation mediated by Aβ oligomers. When we used a more potent isoform of AP5, D-AP5, and increased the concentration to 100 μM, the soluble Aβ-facilitated LTD was now significantly prevented (96.5 ± 3.4%, n=5) (Fig. 2E). Indeed, the block of Aβ-facilitated LTD was dependent on AP5 dose (Fig. 2F). Rescuing LTD expression in the presence of 7PA2 CM required much higher AP5 concentrations than those needed to rescue LTD in the presence of CHO- CM (IC50: 17.4 vs. 72.5 μM; p<0.001), supporting the conclusion that active NMDARs are involved in LTD facilitation by soluble Aβ.
Figure 2. Soluble Aβ enhances hippocampal LTD through mGluR or NMDAR, depending on the stimulation protocol.
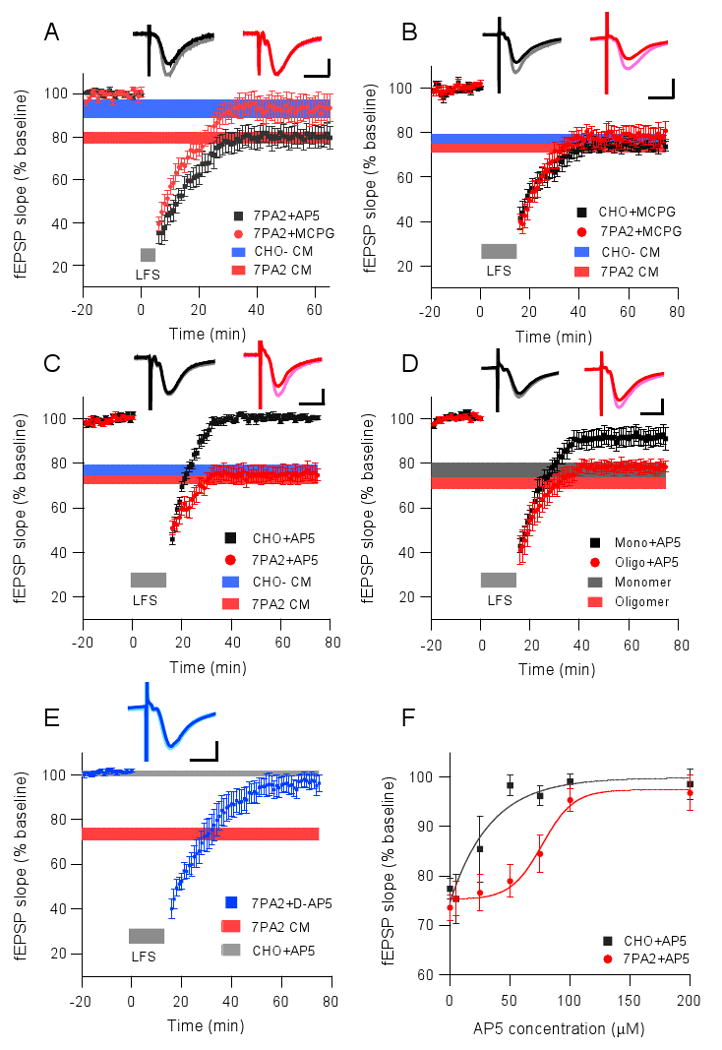
(A) LTD induced by the 300-pulse protocol (grey bar) in the presence of 7PA2 CM was blocked upon coadministration of the non-selective group I/II mGluR antagonist, MCPG (500 μM, red circles, n=6), but not the NMDAR antagonist, AP5 (50 μM, black squares, n=5). Horizontal colored bars represent the corresponding means ± SEMs from data shown in Fig.1B. (B) LTD induced by the 900-pulse protocol (grey bar) is independent of mGluR activation. Horizontal colored bars represent the corresponding means ± SEMs from data shown in Fig.1C. (C) The 900-pulse LTD induced in slices in CHO- CM was blocked by co-administering the NMDAR antagonist, AP5 (50 μM, black squares, n=8), whereas LTD in slices in 7PA2 CM was unaltered (red circles, n=6). Horizontal colored bars from data in Fig.1C. (D) 900-pulse LTD in Aβ monomer treated slices was blocked upon co-perfusing AP5 (50 μM, black squares, n=5), whereas the LTD in oligomer-treated slices was not (red circles, n=6). Horizontal colored bars represent the corresponding means ± SEMs from Fig.1F. (E) 7PA2 CM enhanced LTD was blocked by treatment with D-AP5 at 100 μM (n=5). Horizontal colored bars represent the corresponding means ± SEMs from Fig.2C. (F) Does-response curves of LTD blockade by AP5 in either CHO- CM (black squares) or 7PA2 CM (red circles). Insets in A-E represent typical fEPSPs recorded before (light) and 50 min after (dark) the LFS. Horizontal calibration bar, 10 ms; vertical bar, 0.5 mV.
An extracellular glutamate scavenger restores soluble Aβ-enhanced LTD to normal levels
Because Aβ oligomers enhanced LTD through activation of glutamate receptors, we asked whether this activity of soluble Aβ was dependent on extracellular glutamate concentration. To this end, we used an enzymatic glutamate scavenger system (glutamic-pyruvic transaminase [GPT] + pyruvate) to reduce extracellular glutamate levels (Overstreet et al., 1997; Min et al., 1998). Exposure of hippocampal slices to GPT alone (5 units/ml) had no effect on baseline synaptic activity (not shown). GPT treatment 15 minutes prior to the application of CHO- CM or 7PA2 CM affected neither baseline activity nor the magnitude of LTD induced by 900-LFS (CHO-: 82.6 ± 2.1%, n=5; 7PA2: 81.8 ± 4.5%, n=5) (Fig. 3A); however, the LTD induced in the presence of Aβ and GPT could now be fully blocked by 50 μM AP5 (95.1 ± 3.6%, n=5) (Fig. 3B). This result suggests that elevated extracellular glutamate concentrations contribute to the enhancement of LTD by soluble Aβ. Similarly, treatment with the GPT+pyruvate also prevented the 300-LFS Aβ-induced LTD (94.4 ± 2.8%, n=5) (Fig. 3C). We conclude that the enhancement of LTD by soluble Aβ in both mGluR-dependent and NMDAR-dependent conditions involves elevated extracellular glutamate levels.
Figure 3. Selective metabolism of extracellular glutamate prevents soluble Aβ facilitated-LTD.
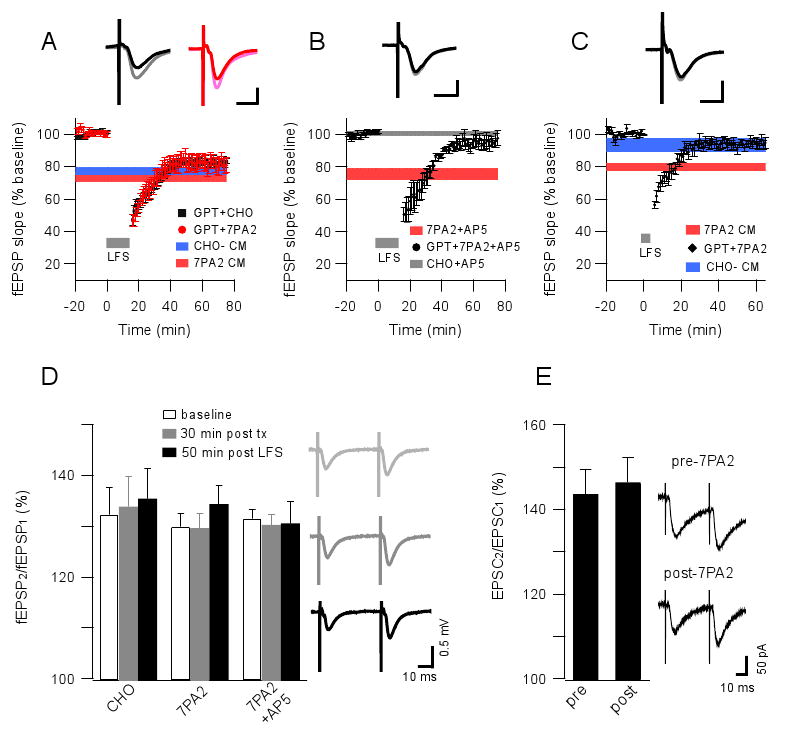
(A) A glutamate scavenger system (glutamic pyruvic transaminase [GPT, 5 unit/ml] + pyruvate [2 mM]) has no significant effect on the fEPSP baseline and 900-pulse LTD in CHO- or 7PA2 CM treated slices. Horizontal colored bars represent the corresponding means ± SEMs from Fig.1C. Insets in A-C represent typical fEPSPs recorded before (light) and 50 min after (dark) LFS; horizontal calibration bar, 10 ms; vertical bar, 0.5 mV. (B) 900-pulse LTD (grey bar) was blocked by AP5 (50 μM) in slices exposed to GPT + pyruvate for 15 min prior to 7PA2 CM (n=6). Red horizontal bar shows the LTD resistant to the same dose of AP5 in the absence of exposure to scavengers. (C) 300-pulse LTD (grey bar) was significantly prevented by co-administering glutamate scavengers with the 7PA2 CM. Horizontal colored bars represent the corresponding means ± SEMs from Fig.1B. (D) Paired-pulse facilitation in slices exposed to 7PA2 CM, CHO- CM or 7PA2 CM + AP5 (50 μM) measured before (white) or 30 min after (grey) adding these media or else measured 50 min after (black) a 900-pulse induction of LTD. At right are typical traces of these field recordings from the 7PA2 group. (E) Similar paired-pulse facilitation recorded by whole-cell voltage clamping at -70 mV in CA1 pyramid cells before and 30 min after 7PA2 CM exposure. At right are typical traces. Data are means ± SEMs.
Paired-pulse facilitation (PPF) reveals whether alterations of synaptic transmission are presynaptic. To assess whether soluble Aβ oligomers increase extracellular glutamate levels by affecting presynaptic release probability, we measured PPF in slices exposed to CHO- CM, 7PA2 CM or 7PA2 CM + AP5 (50 μM) at baseline, 30 min after these exposures, and then 50 min after a 900-pulse LTD induction. PPF was not significantly different between these three conditions (Fig. 3D). To establish further that soluble Aβ does not alter presynaptic release, we performed whole-cell recordings of pyramidal cells and found that PPF ratios before and 30 min after 7PA2 CM exposure were not significantly different (Fig. 3E). The experiments described so far suggest that the facilitation of LTD by soluble oligomeric Aβ involves elevated levels of extracellular glutamate, but not by altering presynaptic release probability.
The Aβ-mediated increase in synaptic glutamate concentration alters properties of excitatory transmission
We next sought to determine more precisely the basis of soluble Aβ-mediated alterations in glutamatergic transmission through whole-cell recordings. AMPAR-mediated currents (AMPA-EPSC) and NMDAR-mediated currents (NMDA-EPSC) were recorded from pyramidal neurons in slices treated with CHO- CM or 7PA2 CM (Fig. 4A1). Both currents were significantly reduced by the soluble Aβ-rich 7PA2 CM compared to the CHO- CM (AMPA 67 ± 4%, n=18, p<0.001; NMDA 79 ± 3%, n=13, p<0.01) (Fig. 4A2). This result was unexpected, as the increased extracellular glutamate concentrations observed above should increase AMPA and/or NMDA currents. However, the result could be explained if the Aβ-mediated rise in glutamate levels leads to receptor desensitization. To address this possibility, we applied a well-characterized inhibitor of AMPAR desensitization, cyclothiazide (CTZ, 100 μM), to the slices before exposure to 7PA2 CM; the AMPA EPSC was no longer significantly decreased (CHO- CM: 115 ± 14 pA vs. 7PA2 CM + CTZ: 96 ± 3 pA, n=10; p>0.05) (Fig. 4A2). Interestingly, we observed spiking activity escaping voltage clamp in 6/10 cells recorded in the 7PA2 CM + CTZ condition (Supp. Fig. 2A, 2B), but not in the CTZ only condition (3 cells; not shown), further suggesting alterations in excitatory transmission due to supraphysiologic levels of extracellular glutamate in the presence of soluble Aβ. To assess whether AMPAR desensitization is involved in the induction of LTD by soluble Aβ, CTZ was applied to brain slices and 900-pulse LTD was induced; the LTDs were not significantly different in CHO- vs. 7PA2 CM (Supp Fig. 2C), suggesting that AMPAR desensitization is not required for the LTD induction.
Figure 4. Soluble Aβ oligomers alter NMDAR-mediated EPSCs in whole-cell voltage clamp recordings.
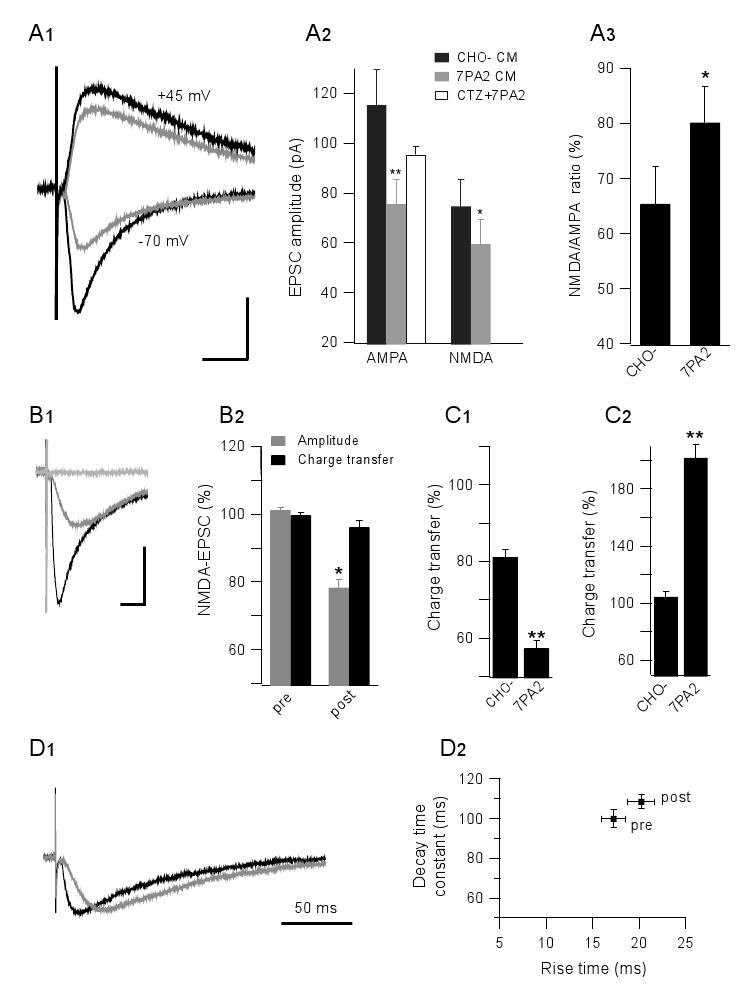
(A1) Isolated AMPA-EPSC and NMDA-EPSC were recorded from -70 mV and +45 mV holding potentials, respectively, plus pharmacological blockers. Black traces, CHO- CM treated cells; grey traces, 7PA2 CM treated cells. (A2) Summary data for CHO- CM (black) and 7PA2 CM (grey) groups, and for pre-treatment with cyclothiazide (CTZ, 100 μM) prior to 7PA2 CM (white). (A3) NMDA/AMPA EPSC ratios are different in CHO- CM and 7PA2 CM. (B1) Typical traces from CA1 pyramidal cells held at −70 mV (black), isolated NMDA-EPSC in low Mg2+ with NBQX (10 μM) and bicuculline (20 μM) at -70 mV (grey), and full blockage of the NMDA current by AP5 (50 μM) (light grey). (B2) Summary data for the isolated NMDA-EPSC before (pre) and 20-30 min after 7PA2 CM exposure. Peak amplitudes (grey) and total charge transfers (black) are expressed as means ± SEMs. (C1) The selective NR2B inhibitor, ifenprodil (3 μM), modestly reduces NMDA charge transfers in control but markedly reduces it in 7PA2 CM. (C2) 7PA2 CM significantly increases the extrasynaptic response when synaptic NMDAR are first blocked by MK-801. (D) NMDA-mediated EPSC kinetic analysis: left panel, representative scaled traces before (black) vs. after (grey) 7PA2 CM exposure; right panel, summary data of rise time plotted vs. decay time before (pre) and after (post)7PA2 CM exposure.
To preserve the neuronal glutamate uptake expected under normal physiological function, we repeated the single cell recordings at −70 mV rather than at positive potentials (Grewer and Rauen, 2005). NMDA-EPSC was isolated in low Mg2+ (0.1 mM) and with pharmacological blockade of AMPAR (with NBQX, 10 μM) and GABA-R (with BIC, 20 μM) (Fig. 4B1). Under these conditions, the NMDAR-EPSC was again found to be decreased by 7PA2 CM (78 ± 3%, n=10; p<0.01) (Fig. 4B2). The NMDA-EPSC reduction could be due to NMDAR desensitization in the presence of increased extracellular glutamate levels (Sarantis et al., 1993). To better understand the NMDA current changes, we quantified charge transfers, calculated as the area under the curve of the NMDA-EPSC. In contrast to the reduction in peak amplitude, the total charge transfer was not decreased by the Aβ-rich 7PA2 CM (96 ± 2% of pre-treatment values, n=11; p>0.05) (Fig. 4B2). Because NR2B receptors have a 2-3 fold higher sensitivity for glutamate than do NR2A receptors (Kutsuwada et al., 1992), we asked whether NR2B receptors played the principal role in the Aβ-mediated effect on NMDA currents. The selective NR2B receptor inhibitor, ifenprodil (3 μM), modestly inhibited the NMDA-EPSC total charge transfer under control (pre-treatment) conditions (81 ± 2%, n=8) (Fig. 4C1). However, ifenprodil strongly and significantly inhibited the NMDA-EPSC charge transfer in the presence of 7PA2 CM (55 ± 2%, n=9; p<0.05) (Fig. 4C1), suggesting enhanced activation of NR2B subunit-containing NMDA receptors. The NR2B subunit is considered to be predominantly present in extrasynaptic NMDA receptors (Tovar and Westbrook, 1999). We selectively blocked synaptic NMDAR with MK-801, an irreversible, use-dependent NMDA channel blocker, washed it out, and perfused with 7PA2 CM under the above NMDA-EPSC isolating conditions. The NMDA-EPSCs were markedly increased (202 ± 10% of the CHO- CM level, n=5; p<0.01) (Fig. 4C2). Taken together, the above results suggest that soluble Aβ oligomers increase NR2B-mediated NMDAR activity and enhance extrasynaptic responses.
Spillover of increased extracellular glutamate remained as the most parsimonious explanation for the relative increases in NMDA currents, activation of NR2B receptors, and extrasynaptic responses in the presence of Aβ oligomers. To further support this interpretation, we analyzed the NMDA EPSC kinetics, i.e., the 10-90% rise time and the decay time constant. While the peak amplitude decreased slightly after 7PA2 CM treatment, the rise time (16 ms vs 20 ms for pre vs. post-exposure; p<0.05) and decay time (100 ms vs 109 ms for pre vs. post-exposure; p<0.05) were significantly prolonged (Fig. 4D), suggesting increased diffusion distance and dwelling times, i.e., that neurotransmitter spillover is enhanced upon exposure to soluble Aβ oligomers.
Soluble Aβ enhances LTD through inhibition of glutamate uptake
Our findings above that soluble Aβ-enhanced LTD may activate a larger population of NMDARs can be considered in terms of Aβ interrupting glutamate reuptake (Harkany et al., 2000; Gu et al., 2004). To investigate this possibility, we examined the effects of a well-characterized glutamate uptake inhibitor, TBOA. Soluble Aβ extracted from AD brain or present in 7PA2 CM (Fig. 1B) facilitates LTD in CA1 after a 300-LFS protocol, whereas this low stimulus induces weak or no LTD in control-treated slices. To test whether inhibiting glutamate uptake itself facilitates LTD, the 300-LFS protocol was applied after a 30 min TBOA treatment (15 μM). Closely similar to our results with soluble Aβ treatment in this 300-LFS mode (Fig. 1B), we found that TBOA alone induced LTD in an NMDAR-independent but mGluR-dependent manner (TBOA alone: 74 ± 4.5%, n=5; TBOA+AP5: 78.1 ± 3.5%, n=5; TBOA+MCPG: 91 ± 4.6%, n=6) (Fig. 5A). Similar to the 900-pulse LTD induced in the presence of soluble Aβ (Fig. 2), the 900-pulse LTD induced in the presence of TBOA was also resistant to 50 μM AP5 but was prevented by 100 μM D-AP5 (TBOA: 73.5 ± 3.4%, n=5; TBOA+AP5: 76.9 ± 5.6%, n=5; TBOA+D-AP5: 94.2 ± 2.8%, n=5; p<0.01 compared to TBOA+AP5) (Fig. 5B).
Figure 5. Soluble Aβ-enhanced LTD involves impaired glutamate reuptake.
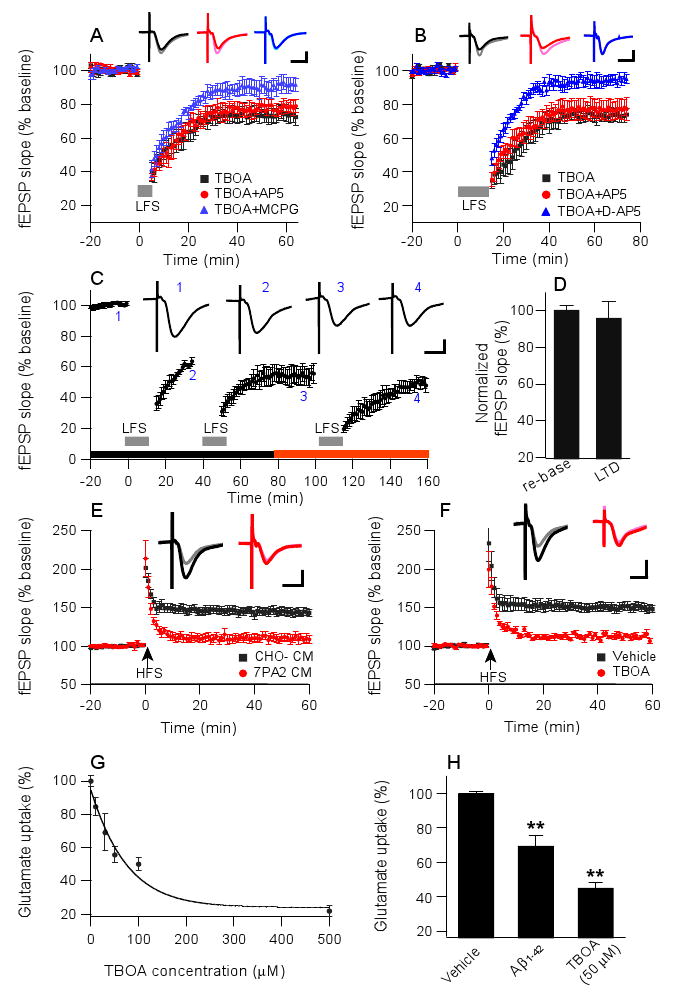
(A) LTD was induced by 300 pulses (grey bar) when just the glutamate uptake inhibitor, TBOA (15 μM), was perfused (black squares, n=5). This TBOA-mediated LTD was prevented by mGluR antagonist MCPG (500 μM, blue triangles, n=6), but not by NMDAR antagonist AP5 (50 μM, red circles, n=6). Insets in A-F represent fEPSPs recorded before (light) and 50 min after (dark) either LFS (in A-C) or HFS (in E and F). Horizontal calibration bars, 10 ms; vertical bars, 0.5 mV. (B) A 900-pulse LTD facilitated by TBOA was resistant to a standard dose of AP5 (50 μM, red circles, n=6), but it could be blocked by D-AP5 (100 μM, blue triangles, n=6). (C) Saturation of 900-pulse LTD occurring in the presence of TBOA (black horizontal bar) occludes further LTD in the presence of 7PA2 CM (red horizontal bar). (D) Summary data from occlusion experiments as in C: left bar, evoked fEPSP was re-normalized to baseline values (i.e., point 3 in C) before addition of 7PA2 CM to the perfusate and an additional LFS (right bar). (E) LTP induced by high-frequency stimulation (HFS) was prevented in slices in 7PA2 CM (red circles) but unaffected in slices in CHO- CM (black squares). (F) HFS-induced LTP was similarly prevented by TBOA. (G) Dose-dependent inhibition of glutamate uptake by TBOA in hippocampal synaptosomes. (H) Pretreatment with soluble synthetic Aβ1-42 impaired glutamate uptake in hippocampal synaptosomes in a fashion similar to TBOA treatment at 50 μM. Data are means ± SEMs as percentage of vehicle alone; **p<0.01.
To assess whether TBOA-enhanced LTD has mechanistic similarity with soluble Aβ, we recorded the TBOA-induced LTD in a repetitive 900-LFS paradigm to saturate the LTD response, then washed out the TBOA and replaced it with 7PA2 CM (Fig. 5C). When 900-LFS was then performed, there was no further increase in LTD (96.1 ± 9.1% of renormalized baseline measured 40 min after LFS, n=5; p>0.05) (Figs. 5C, D). This occlusion experiment suggests that these two forms of LTD share similar mechanisms. Interestingly, if we first applied 7PA2 CM and saturated the LTD, then the usual dose (15 μM) of TBOA significantly reduced baseline neurotransmission (to 78% of the renormalized baseline at 30 min) (Supp. Fig. 3A), and this effect was similar to that of a higher dose of TBOA alone (50 μM) on the baseline (81% at 30 min) (Supp. Fig. 3B). The latter results further substantiate a shared mechanism between TBOA and soluble Aβ on perturbation of neurotransmission. We also examined DHK (500 μM), an inhibitor of glial glutamate transporter (GLT-1), but the 900-pulse LTD observed in the presence of this agent was largely prevented by the standard 50 μM AP5 dose (89.2 ± 3.2%, n=6, vs. 77.5± 2.9%, n=6, p<0.05, Supp. Fig. 3C), suggesting specificity for neuronal, not glial, glutamate transporters in the enhancement of the 900-pulse LTD.
The effects of soluble Aβ in inhibiting HFS-induced LTP have been well established using several types of Aβ preparations, and we confirmed this here using soluble Aβ-rich 7PA2 CM (CHO- CM: 144.7 ± 5.4%, n=7, vs. 7PA2 CM: 108.1 ± 6.6%, n=7; p<0.001) (Fig. 5E). TBOA similarly inhibited HFS-induced LTP (vehicle: 151.3 ± 4.8%, n=6, vs. TBOA: 112.2 ± 3.1%, n=6; p<0.001) (Fig. 5F). These results support our conclusion that the effects of soluble Aβ on synaptic plasticity mimic those observed with the blockade of neuronal glutamate uptake.
To assess directly whether soluble Aβ inhibits glutamate uptake as all of the above data suggest, we measured the uptake of radiolabeled glutamate by isolated synaptosomes (see Methods). As a positive control, TBOA decreased glutamate uptake in the synaptosomal preparation in a dose-dependent manner (IC50=73.9 μM) (Fig. 5G). Synthetic Aβ1-42 significantly decreased glutamate uptake into synaptosomes (69 ± 6% of vehicle treatment, n=15; p<0.01), similar to the effects of TBOA (41 ± 4% at 50 μM, n=5; p<0.01) (Fig. 5H). This biochemical evidence further supports our hypothesis that soluble Aβ impairs neuronal glutamate uptake and can thereby increase extracellular glutamate concentration and subsequent NMDAR activation.
Extracellular calcium influx, not intracellular stores, contributes to soluble Aβ-facilitated LTD
To assess whether alterations in calcium influx through NMDARs mediate the facilitation of LTD by soluble Aβ, the ACSF calcium concentration was reduced to 0.5 mM prior to the application of soluble Aβ and 900-LFS to the brain slices. This reduction of extracellular calcium concentration prevented LTD induction in the presence of either 7PA2 or CHO- CM (Fig.6A). To test whether extracellular calcium is required for the NMDAR-mediated enhancement of LTD by soluble Aβ, we applied 50μM AP5 and varied the extracellular Ca2+ concentration between 0.5 mM and 6 mM (Fig. 6A). The LTD induced by 900-LFS in the presence of AP5 + CHO- CM was fully blocked when the Ca2+ concentration was 1 mM or 2.5 mM, and only a small AP5-resistant LTD (86.4 ± 4.5%, n=6) occurred at the highest Ca2+ concentration (6 mM). In contrast, LTD induced in presence of 7PA2 CM was significantly prevented by 50 μM AP5 at low (1 mM) Ca2+ concentration (7PA2 CM: 80 ± 4.2%, n=5, vs. 7PA2 CM + AP5: 93.2 ± 3.4%, n=6; p<0.05), but not at normal (2.5 mM: 75.4 ± 3.4%, n=6) or high (6 mM: 70.4 ± 3.4%, n=5) Ca2+ concentrations. These results indicate that Aβ-enhanced LTD is dependent on extracellular calcium concentration.
Figure 6. LTD facilitated by soluble Aβ or TBOA share similar signaling pathways.
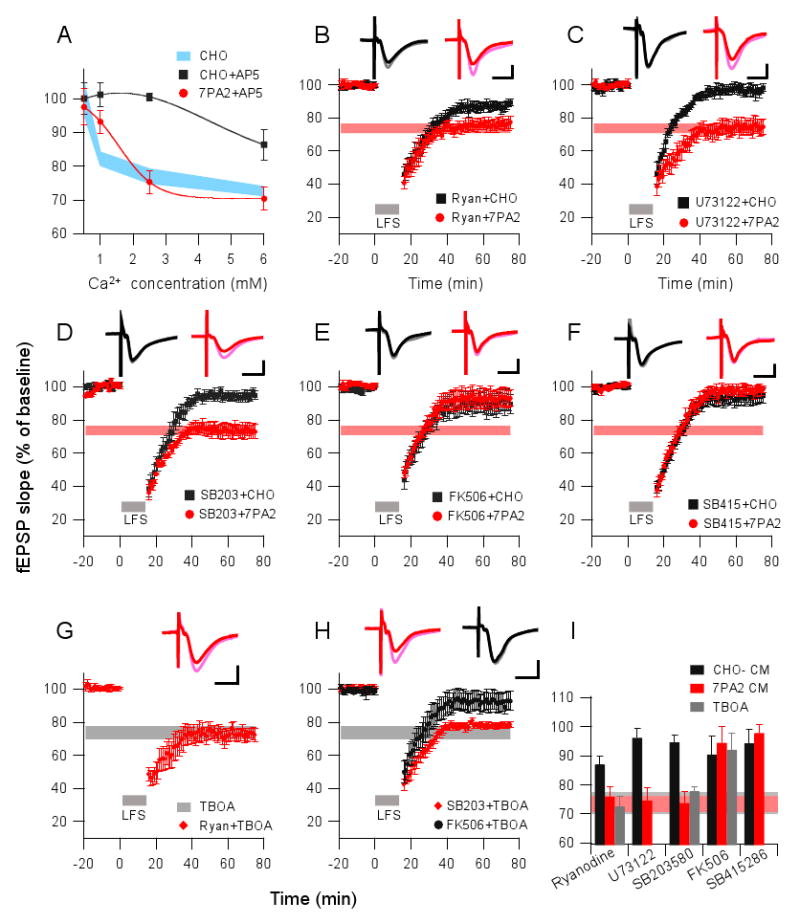
(A) Conventional LTD (in CHO- CM, black squares) and Aβ-enhanced LTD (in 7PA2 CM, red circles) induced by the 900-pulse LFS in the presence of AP5 (50 μM) were plotted as a function of increasing extracellular calcium concentrations in the perfusate. Light blue area represents the LTD obtained in the CHO- CM alone (without AP5). (B) Ryanodine (20 μM) given 30 min prior to 900 pulses partially blocked the LTD in CHO CM (black squares, n=5) but produced no block of the 7PA2-facilitated LTD (red circles, n=6). (C) U73122 (10 μM) (applied 45 min prior to 900 pulses) fully blocked LTD in the presence of CHO- CM (black squares, n=6) but had no effect in the presence of 7PA2 CM (red circles, n=6). (D) 900-pulse LTD was blocked by p38 MAPK inhibitor, SB203580 (5 μM), in CHO- CM (black squares, n=7) but not in 7PA2 CM (red circles, n=7). (E) Calcineurin inhibitor, FK-506 (20 μM) perfused 40 min prior to 900 pulses prevented LTD in both CHO- CM (black squares, n=5) and 7PA2 CM (red circles, n=6). (F) GSK-3β inhibitor, SB415286 (10 μM) perfused 60 min prior to 900 pulses prevented LTD in both CHO- CM (black squares, n=6) and 7PA2 CM (red circles, n=7). (G) LTD enhanced by TBOA (15 μM) was likewise resistant to ryanodine (n=5). (H) TBOA-enhanced LTD was prevented by the calcineurin inhibitor (FK 506, black circles, n=5) but not by the p38 MAPK inhibitor (SB203580, red diamonds, n=5). (I) Summary data for actions of the signaling pathway modulators on Aβ- and TBOA-enhanced LTD. The horizontal gray bar represents mean LTD in TBOA alone (Fig. 5B), and the horizontal light orange bar represents mean LTD in 7PA2 CM alone (Fig. 1C).
To assess whether intracellular calcium stores also regulate Aβ-mediated LTD, as they have been reported to do for Aβ neurotoxicity (Demuro et al., 2005), we tested two compounds that inhibit the release of intracellular Ca2+ stores, ryanodine and U73122. Although ryanodine (20 μM) and U73122 (10 μM) each prevented 900-LFS LTD induction in slices treated with CHO- CM, they did not alter the LTD facilitation by 7PA2 CM (87 ± 2.7%, n=5, vs. 76 ± 3.2%, n=5, for ryanodine, p<0.05) (Fig. 6B); 96.2 ± 3.4%, n=4, vs. 74.6 ± 4.6%, n=6, for U73122, p<0.01) (Fig. 6C). This suggests that soluble Aβ-facilitated LTD requires extracellular calcium influx but not intracellular calcium release.
Activation of calcineurin and GSK-3, but not p38 MAPK, is required for Aβ-mediated LTD
When cytosolic calcium reaches critical concentrations, certain LTD-related signaling pathways are activated (Kemp and Bashir, 2001). For example, NMDAR-dependent LTD in the CA1 region recruits calcineurin (PP2B) and p38 MAPK cascades (Mulkey et al., 1994; Bolshakov et al., 2000; Li et al., 2006). We examined these two signaling pathways using their respective inhibitors, FK506 (20 μM) and SB203580 (5 μM). Whereas blocking p38 MAPK activation with SB203580 prevented conventional 900-pulse LTD induction (i.e., in CHO- CM), it did not significantly affect the LTD induced in the presence of soluble Aβ-rich 7PA2 CM (94.6 ± 2.5%, n=7, vs. 73.9 ± 4%, n=6; P<0.01) (Fig. 6D). In contrast, PP2B function was required for LTD induced both in the absence and presence of soluble Aβ (Fig. 6E). Glycogen synthase kinase-3 (GSK3)-mediated signaling was recently reported to play an important role in LFS-induced LTD (Peineau et al., 2007), and it has also been implicated in Aβ-mediated neurotoxicity in cultured hippocampal slices (Nassif et al., 2007). Accordingly, we treated slices with the GSK-3β inhibitor, SB415286 (10 μM), for 30 min prior to applying either CHO- CM or 7PA2 CM, and then performed 900-LFS. LTD could not be induced in either condition (94.3 ± 4.7%, n=6, vs. 98.4 ± 2.6%, n=6) (Fig. 6F), suggesting that GSK-3β activity is required for both conventional and soluble Aβ-mediated LTD. To further verify that glutamate uptake inhibition shares similar mechanisms with soluble Aβ as regards LTD induction, ryanodine, SB203580, and FK506 were each applied to the slices prior to treatment with TBOA. Entirely consistent with the results for Aβ-mediated LTD, both intracellular Ca2+ release and p38 MAPK activation were not required for the TBOA-mediated LTD, whereas PP2B activity was (Fig. 6G-I).
To provide further information about the receptor mechanisms of LTD facilitation by soluble Aβ, we took advantage of Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) knockout mice, which are deficient in the ability to mount an LTD response (Li et al., 2006). Ras-GRF proteins are best known for their ability to activate Ras/Erk through their CDC25-GEF domains and activate the Rac/p38 cascade through their DH-GEF domains, which act as calcium sensors for different classes of NMDARs and drive the activation of distinct MAP kinase family members. As expected, hippocampal slices from these Ras-GRF1 -/- mice did not express LTD when 900-LFS was administered in the presence of CHO- CM. In contrast, LTD was elicited in the presence of the oligomer-rich 7PA2 CM (99.4 ± 2.9%, n=6, vs. 75.1 ± 2.9%, n=7; p<0.01) (Supp. Fig. 4A). This soluble Aβ-facilitated LTD in the Ras-GRF1 -/- mice was not blocked by 500 μM MCPG (Supp. Fig. 4B) nor by 50 μM AP5 (Supp. Fig. 4C), but it was significantly decreased by 100 μM AP5 (91.6 ± 3.1%, n=5, p<0.05) (Supp. Fig. 4C). Furthermore, the p38 MAPK inhibitor (SB203508) had no effect on the Aβ-facilitated LTD (80.2 ± 4.8%, n=5) (data not shown), whereas pretreatment with either the calcineurin inhibitor (FK506) or the GSK-3β inhibitor (SB415286) significantly prevented the LTD associated with 7PA2 CM (95.6 ± 4.8%, n=6, and 88.4 ± 4.7%, n=6, respectively) (Supp. Fig. 4D). Therefore, soluble Aβ bypassed the non-functional LTD induction pathway in Ras-GRF1 -/- mice, a pathway that is normally sensitive to 50 μM AP5 in wild-type mice.
Discussion
Given the mounting evidence that soluble Aβ oligomers mediate synaptic impairment in AD, elucidating the precise molecular pathways by which this occurs has important implications for treating and preventing the disease. Here, we demonstrate that soluble Aβ oligomers facilitate LTD in the hippocampus through a mechanism that appears to involve the inhibition of glutamate uptake. With a weak (300 pulse) LFS, Aβ-mediated LTD was prevented by MCPG but not AP5, and with a conventional (900 pulse) LFS, the LTD was prevented only by a high (100 μM), not a standard (50 μM), dose of AP5. Extracellular glutamate scavengers effectively prevented Aβ-enhanced LTD. Mechanistically, soluble Aβ oligomers caused glutamate spillover, and their ability to facilitate LTD required an influx of extracellular calcium but did not detectably require Ca2+ release from intracellular stores. Once Ca2+ enters the neuron, it is known to trigger PP2B and GSK3 signaling pathways, and we implicated these in the Aβ-facilitated LTD induction.
Importantly, Aβ facilitation of LTD was closely mimicked by pharmacologically inhibiting neuronal (but not glial) glutamate uptake, consistent with the rescue of Aβ-LTD by the glutamate scavenger. In accord, we found that Aβ significantly impaired glutamate uptake by synaptosomes, suggesting that Aβ facilitates LTD in part by altering synaptic glutamate recycling. These results support the hypothesis that excess synaptic glutamate caused by Aβ mechanistically alters LTD induction in hippocampus. It should be emphasized that the effects of soluble Aβ we describe in this study are attributable to soluble oligomers (Fig. 1A), as no larger assemblies (protofibrils, fibrils) were present, and SEC-isolated oligomers but not monomers conferred the effects on LTD (Fig. 2D). A summary mechanism incorporating all of our findings is proposed in Figure 7.
Figure 7. Schematic of the principal pathways implicated by this study in conventional LTD (left) and in LTD facilitated by soluble Aβ oligomers (right).

Conventional LTD requires NMDAR-mediated influx of extracellular calcium and liberation of intracellular calcium stores. This ultimately activates PP2B, GSK-3β or p38 MAPK signaling pathways that induce LTD. Soluble Aβ oligomers lead to activation of more NMDAR, leading to extracellular calcium influx and activation of PP2B and GSK-3β pathways to facilitate LTD. Our data suggest that Aβ oligomers decrease glutamate uptake by neuronal transporters (red x's), resulting in the enhanced activation of NMDARs and thus facilitation of LTD-inducing pathways.
We focused on the NMDAR-dependent induction of LTD, a widely used protocol for LTD studies. There have been contradictory findings among the few studies of the effects of Aβ on LTD (Kim et al., 2001; Wang et al., 2002; Raymond et al., 2003; Wang et al., 2004a). Hsieh et al. (2006) showed that Aβ secreted by neurons in hippocampal slices transfected with mutant human APP could induce LTD via an mGluR-dependent mechanism involving AMPAR internalization. Moreover, Chang et al. (2006) reported that aged APP/Presenilin-1 double knock-in mice having reduced AMPAR mEPSCs were resistant to LTD induction, suggesting a floor effect for synapse depression following prolonged exposure to Aβ. Our new data indicate that soluble Aβ oligomers can induce hippocampal LTD, although the mechanisms do not follow canonical p38 MAPK pathways.
Synthetic Aβ has been shown to increase NMDAR-mediated currents in rat dentate gyrus (Wu et al., 1995) or enhance NMDA response in the hippocampus after a 15 min exposure in vivo (Molnar et al., 2004). Our current results show that soluble Aβ oligomers depressed AMPAR currents (Fig.4A, B), but this was likely due to desensitization. Earlier work has suggested that NR2B-containing NMDAR play a major role in normal LTD induction (Liu et al., 2004), although some recent evidence is not in agreement (Morishita et al., 2007). Our study suggests that soluble Aβ oligomers can significantly increase NR2B receptor activation, as demonstrated by an increased ifenprodil-sensitive component of the NMDAR-EPSC following exposure to soluble Aβ. NR2B receptors are specifically coupled to SynGAP (Kim et al., 2005), and their activation would thus inhibit the Ras-Erk signaling pathway known to be required for LTP, instead favoring LTD induction. Although some studies have colocalized Aβ with NMDAR at synapses (Dewachter et al., 2009; Lacor et al., 2007), it remains unclear whether these are a direct target of the peptide, and other studies have suggested Aβ interactions with voltage-dependent calcium channels (VDCCs) (Ueda et al., 1997), mGluRs (Wang et al., 2004b), nicotinic receptors (Dineley et al., 2001; Snyder et al., 2005) or insulin receptors (Townsend et al, 2007), all of which could modulate NMDAR activity. It remains plausible that the hydrophobic Aβ oligomers actually interact with lipid and/or protein targets upstream of these various receptors perturbing their function secondarily.
Extrasynaptic glutamate spillover is a phenomenon that has been implicated in cross-talk among hippocampal synapses (Asztely et al., 1997; Min et al., 1998; Scimemi et al., 2004). Inhibition of glutamate uptake at the synapse (e.g., by soluble Aβ) can significantly increase the extracellular glutamate concentration and thereby enhance glutamate spillover to neighboring synapses. Electrophysiologically, NMDA currents will be increased and their rise and decay time courses significantly prolonged, due to the increased distance between the glutamate release site and the target receptors. In addition, extrasynaptic NMDAR activity will be increased by synaptic glutamate accumulation. Our present results do not fully meet these criteria, as we found that soluble Aβ depressed peak NMDAR currents. We hypothesize that Aβ oligomers depress NMDA currents as a result of desensitization of the receptors. It is of related interest that several glutamate uptake inhibitors have been shown to depress NMDA currents (Sarantis et al., 1993; Maki et al., 1994; Asztely et al., 1997), as we show for soluble Aβ oligomers here. Thus, Aβ-mediated decreases in glutamate uptake and consequent accumulation of extracellular glutamate could induce NMDAR desensitization and/or activation of presynaptic mGluRs, thereby reducing the peak amplitudes of the NMDA currents.
Glutamate excitotoxicity has been hypothesized to have a role in AD pathogenesis. Dysfunction of glutamate transporters has been implicated in this pathway (Masliah et al., 1996; Jacob et al., 2007). Synthetic Aβ has been shown to inhibit glutamate uptake in cultured astrocytes (Harris et al., 1996; Harkany et al., 2000; but see Ikegaya et al., 2002) and oocytes (Gu et al., 2004). Extracellular glutamate concentration is tightly controlled in the brain by a family of membrane transporters, predominantly expressed by perisynaptic astrocytes (Danbolt, 2001). Their role is to regulate the glutamate released at synapses and to prevent spillover of the transmitter to extrasynaptic receptors. Our results showing that soluble Aβ facilitates LTD through both mGluR (300-LFS protocol) and NMDAR (900-LFS protocol) pathways is supported by our concomitant finding that a common upstream element, the glutamate transporter, is also regulated by soluble Aβ. In vivo microdialysis recently revealed that microinjection of the soluble Aβ-rich 7PA2 CM can significantly increase the interstitial fluid levels of glutamate -- but not GABA or aspartate -- in hippocampus (O'Shea et al., 2008). Inhibition of glutamate uptake by TBOA has been shown to increase spontaneous epileptiform discharges, and this increased excitability can be blocked by AP5 (Campbell and Hablitz, 2004). Interestingly, Aβ has also been found to significantly increase spontaneous non-convulsive seizure activity in cortical and hippocampal networks in APP transgenic mice (Palop et al., 2007). The precise mechanism by which soluble Aβ oligomers interfere with glutamate transporters at the synapse remains to be determined. Given the numerous distinct receptors and channel proteins reported to be altered by Aβ to date, we speculate that the hydrophobic Aβ oligomers bind principally to membrane lipids and thereby secondarily interrupt the structure and function of synaptic transmembrane transporters and channels, leading to changes in the transduction of intracellular signalling cascades. Such a mechanism would be consistent with the direct effect of soluble Aβ on glutamate uptake by isolated synaptosomes in vitro (Fig. 5H).
Calcium is a second messenger that controls many cellular processes, including neuronal excitability, synaptic plasticity and neuronal death. The increase in postsynaptic calcium that results in LTD can arise from a number of sources. For example, extracellular Ca2+ influx can occur through NMDARs or VDCCs (Kemp and Bashir, 2001), and intracellular Ca2+ release can stem from activation of the ryanodine receptor (RyR) and the inositol 1,4,5-trisphosphate receptor (IP3R). Our data (Fig. 6A-C) show that conventional hippocampal LTD requires extracellular Ca2+ influx through NMDARs and recruits intracellular stores from IP3R and possibly RyR, in line with previous studies on hippocampal LTD (Kemp and Bashir, 2001). However, among these possible calcium sources, Aβ-facilitated LTD only required NMDAR activation and enhanced extracellular calcium entry. An increase in cytosolic calcium can trigger signaling cascades involved in LTD induction, including PP2B, p38 MAPK and GSK3 (Mulkey et al., 1994; Bolshakov et al., 2000; Li et al., 2006; Peineau et al., 2007). The differences between our study and previous reports implicating Aβ in the activation of p38 MAPK (Hsieh et al. 2006; Origlia et al., 2008) could lie in the experimental conditions, for example, acute slices vs. dissociated neuronal cultures or field vs. whole-cell recordings.
In conclusion, the findings described here suggest that soluble Aβ oligomers facilitate LTD through both NMDARs and mGluRs by interfering with glutamate recycling at the synapse (Fig. 7). Our study also suggests that additional mechanisms may underlie LTD in AD pathological states beyond those described previously through the NMDAR-p38/MAPK pathway. The receptors and signaling molecules activated by Aβ as described in this study could represent additional therapeutic targets in Alzheimer's disease.
Supplementary Material
Acknowledgments
Supported by NIH grant AG027443 (DJS). We thank Dr. L. Feig (Tufts University) for providing Ras-GRF1 knockout mice, and Drs. M. J. Rowan (Trinity College) and B. L. Sabatini (Harvard Medical School) for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anwyl R. Induction and expression mechanisms of postsynaptic NMDA receptor-independent homosynaptic long-term depression. Prog Neurobiol. 2006;78:17–37. doi: 10.1016/j.pneurobio.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Asztely F, Erdemli G, Kullmann DM. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- Bastrikova N, Gardner GA, Reece JM, Jeromin A, Dudek SM. Synapse elimination accompanies functional plasticity in hippocampal neurons. Proc Natl Acad Sci U S A. 2008;105:3123–3127. doi: 10.1073/pnas.0800027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolshakov VY, Carboni L, Cobb MH, Siegelbaum SA, Belardetti F. Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3-CA1 synapses. Nat Neurosci. 2000;3:1107–1112. doi: 10.1038/80624. [DOI] [PubMed] [Google Scholar]
- Campbell SL, Hablitz JJ. Glutamate transporters regulate excitability in local networks in rat neocortex. Neuroscience. 2004;127:625–635. doi: 10.1016/j.neuroscience.2004.05.030. [DOI] [PubMed] [Google Scholar]
- Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, Shirao T, Aoki C, Huerta PT. AMPA receptor downscaling at the onset of Alzheimer's disease pathology in double knockin mice. Proc Natl Acad Sci U S A. 2006;103:3410–3415. doi: 10.1073/pnas.0507313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QS, Wei WZ, Shimahara T, Xie CW. Alzheimer amyloid beta-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiol Learn Mem. 2002;77:354–371. doi: 10.1006/nlme.2001.4034. [DOI] [PubMed] [Google Scholar]
- Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33:18–41. doi: 10.1038/sj.npp.1301559. [DOI] [PubMed] [Google Scholar]
- Cowburn RF, Wiehager B, Trief E, Li-Li M, Sundström E. Effects of beta-amyloid-(25-35) peptides on radioligand binding to excitatory amino acid receptors and voltage-dependent calcium channels: evidence for a selective affinity for the glutamate and glycine recognition sites of the NMDA receptor. Neurochem Res. 1997;22:1437–42. doi: 10.1023/a:1021942109490. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- Dewachter I, Filipkowski RK, Priller C, Ris L, Neyton J, Croes S, Terwel D, Gysemans M, Devijver H, Borghgraef P, Godaux E, Kaczmarek Deregulation of NMDA-receptor function and down-stream signaling in APP[V717I] transgenic mice. Neurobiol Aging. 2009;30:241–256. doi: 10.1016/j.neurobiolaging.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewer C, Rauen T. Electrogenic glutamate transporters in the CNS: molecular mechanism, pre-steady-state kinetics, and their impact on synaptic signaling. J Membr Biol. 2005;203:1–20. doi: 10.1007/s00232-004-0731-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu QB, Zhao JX, Fei J, Schwarz W. Modulation of Na(+), K(+) pumping and neurotransmitter uptake by beta-amyloid. Neuroscience. 2004;126:61–67. doi: 10.1016/j.neuroscience.2004.03.022. [DOI] [PubMed] [Google Scholar]
- Harkany T, Abrahám I, Timmerman W, Laskay G, Tóth B, Sasvári M, Kónya C, Sebens JB, Korf J, Nyakas C, Zarándi M, Soós K, Penke B, Luiten PG. beta-amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. Eur J Neurosci. 2000;12:2735–2745. doi: 10.1046/j.1460-9568.2000.00164.x. [DOI] [PubMed] [Google Scholar]
- Harris ME, Wang Y, Pedigo NW, Jr, Hensley K, Butterfield DA, Carney JM. Amyloid beta peptide (25-35) inhibits Na+-dependent glutamate uptake in rat hippocampal astrocyte cultures. J Neurochem. 1996;67:277–286. doi: 10.1046/j.1471-4159.1996.67010277.x. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegaya Y, Matsuura S, Ueno S, Baba A, Yamada MK, Nishiyama N, Matsuki N. Beta-amyloid enhances glial glutamate uptake activity and attenuates synaptic efficacy. J Biol Chem. 2002;277:32180–32186. doi: 10.1074/jbc.M203764200. [DOI] [PubMed] [Google Scholar]
- Jacob CP, Koutsilieri E, Bartl J, Neuen-Jacob E, Arzberger T, Zander N, Ravid R, Roggendorf W, Riederer P, Grünblatt E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer's disease. J Alzheimers Dis. 2007;11:97–116. doi: 10.3233/jad-2007-11113. [DOI] [PubMed] [Google Scholar]
- Kemp N, Bashir ZI. Long-term depression: a cascade of induction and expression mechanisms. Prog Neurobiol. 2001;65:339–365. doi: 10.1016/s0301-0082(01)00013-2. [DOI] [PubMed] [Google Scholar]
- Kim JH, Anwyl R, Suh YH, Djamgoz MB, Rowan MJ. Use-dependent effects of amyloidogenic fragments of (beta)-amyloid precursor protein on synaptic plasticity in rat hippocampus in vivo. J Neurosci. 2001;21:1327–1333. doi: 10.1523/JNEUROSCI.21-04-01327.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Knobloch M, Mansuy IM. Dendritic spine loss and synaptic alterations in Alzheimer's disease. Mol Neurobiol. 2008;37:73–82. doi: 10.1007/s12035-008-8018-z. [DOI] [PubMed] [Google Scholar]
- Kutsuwada T, Kashiwabuchi N, Mori H, Sakimura K, Kushiya E, Araki K, Meguro H, et al. Molecular diversity of the NMDA receptor channel. Nature. 1992;358:36–41. doi: 10.1038/358036a0. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Tian X, Hartley DM, Feig LA. Distinct roles for Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and Ras-GRF2 in the induction of long-term potentiation and long-term depression. J Neurosci. 2006;26:1721–1729. doi: 10.1523/JNEUROSCI.3990-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki R, Robinson MB, Dichter MA. The glutamate uptake inhibitor L-trans-pyrrolidine-2,4-dicarboxylate depresses excitatory synaptic transmission via a presynaptic mechanism in cultured hippocampal neurons. J Neurosci. 1994;14:6754–6762. doi: 10.1523/JNEUROSCI.14-11-06754.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer's disease. Ann Neurol. 1996;40:759–766. doi: 10.1002/ana.410400512. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Min MY, Rusakov DA, Kullmann DM. Activation of AMPA, kainate, and metabotropic receptors at hippocampal mossy fiber synapses: role of glutamate diffusion. Neuron. 1998;21:561–570. doi: 10.1016/s0896-6273(00)80566-8. [DOI] [PubMed] [Google Scholar]
- Molnár Z, Soós K, Lengyel I, Penke B, Szegedi V, Budai D. Enhancement of NMDA responses by beta-amyloid peptides in the hippocampus in vivo. Neuroreport. 2004;15:1649–1652. doi: 10.1097/01.wnr.0000134471.06244.d2. [DOI] [PubMed] [Google Scholar]
- Morishita W, Lu W, Smith GB, Nicoll RA, Bear MF, Malenka RC. Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharmacology. 2007;52:71–76. doi: 10.1016/j.neuropharm.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Nägerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Nassif M, Hoppe J, Santin K, Frozza R, Zamin LL, Simão F, Horn AP, Salbego C. Beta-amyloid peptide toxicity in organotypic hippocampal slice culture involves Akt/PKB, GSK-3beta and PTEN. Neurochem Int. 2007;50:229–235. doi: 10.1016/j.neuint.2006.08.008. [DOI] [PubMed] [Google Scholar]
- O'Shea SD, Smith IM, McCabe OM, Cronin MM, Walsh DM, O'Connor WT. Intracerebroventricular administration of amyloid b-protein oligomers selectively increases dorsal hippocampal dialysate glutamate levels in the awake rat. Sensors. 2008;8:7428–7437. doi: 10.3390/s8117428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Origlia N, Righi M, Capsoni S, Cattaneo A, Fang F, Stern DM, Chen JX, Schmidt AM, Arancio O, Yan SD, Domenici L. Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. J Neurosci. 2008;28:3521–3530. doi: 10.1523/JNEUROSCI.0204-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet LS, Pasternak JF, Colley PA, Slater NT, Trommer BL. Metabotropic glutamate receptor mediated long-term depression in developing hippocampus. Neuropharmacology. 1997;36:831–844. doi: 10.1016/s0028-3908(97)00031-2. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ. Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem. 1995;270:9564–9570. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- Pomara N, Singh R, Deptula D, Chou JC, Schwartz MB, LeWitt PA. Glutamate and other CSF amino acids in Alzheimer's disease. Am J Psychiatry. 1992;149:251–254. doi: 10.1176/ajp.149.2.251. [DOI] [PubMed] [Google Scholar]
- Raymond CR, Ireland DR, Abraham WC. NMDA receptor regulation by amyloid-beta does not account for its inhibition of LTP in rat hippocampus. Brain Res. 2003;968:263–272. doi: 10.1016/s0006-8993(03)02269-8. [DOI] [PubMed] [Google Scholar]
- Sarantis M, Ballerini L, Miller B, Silver RA, Edwards M, Attwell D. Glutamate uptake from the synaptic cleft does not shape the decay of the non-NMDA component of the synaptic current. Neuron. 1993 Sep;11(3):541–549. doi: 10.1016/0896-6273(93)90158-n. [DOI] [PubMed] [Google Scholar]
- Scimemi A, Fine A, Kullmann DM, Rusakov DA. NR2B-containing receptors mediate cross talk among hippocampal synapses. J Neurosci. 2004;24:4767–77. doi: 10.1523/JNEUROSCI.0364-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson N, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Soluble Amyloid β-Protein Dimers Isolated Directly from Alzheimer Disease Patients Potently Impair Synaptic Plasticity and Memory. Nat Medicine. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Mehta T, Selkoe DJ. Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J Biol Chem. 2007;282:33305–33312. doi: 10.1074/jbc.M610390200. [DOI] [PubMed] [Google Scholar]
- Ueda K, Shinohara S, Yagami T, Asakura K, Kawasaki K. Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J Neurochem. 1997;68:265–271. doi: 10.1046/j.1471-4159.1997.68010265.x. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, Hartley DM, Selkoe DJ. Certain inhibitors of synthetic amyloid beta-peptide (Abeta) fibrillogenesis block oligomerization of natural Abeta and thereby rescue long-term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL. Soluble oligomers of beta amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924:133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- Wang Q, Rowan MJ, Anwyl R. Beta-amyloid-mediated inhibition of NMDA receptor-dependent long-term potentiation induction involves activation of microglia and stimulation of inducible nitric oxide synthase and superoxide. J Neurosci. 2004a;24:6049–6056. doi: 10.1523/JNEUROSCI.0233-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004b;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Anwyl R, Rowan MJ. beta-Amyloid selectively augments NMDA receptor-mediated synaptic transmission in rat hippocampus. Neuroreport. 1995;6:2409–2413. doi: 10.1097/00001756-199511270-00031. [DOI] [PubMed] [Google Scholar]
- Yang CH, Huang CC, Hsu KS. Behavioral stress enhances hippocampal CA1 long-term depression through the blockade of the glutamate uptake. J Neurosci. 2005;25:4288–4293. doi: 10.1523/JNEUROSCI.0406-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.