

Abstract
The development of new methods to skeletally diverse sultams based on a central α-halo benzene sulfonamide building block is reported. Several salient features of this building block are utilized in multiple reaction pathways, including the Heck reaction, C- and O-arylation, Sonogashira-Pauson-Khand, Sonogashira-intramolecular hydroamination, lithiative cyclization and domino aza-Michael Heck for the generation of 5-, 6- and 7-membered benzofused bicyclic and tricyclic sultams.
1. Introduction
In recent years, there has been a rapidly growing demand for libraries of small molecules for high-throughput screening (HTS). This demand has presented challenging opportunities in molecular library development. In this regard, Diversity-Oriented Synthesis (DOS)i,ii has emerged as an enabling platform for the production of multiple scaffolds displaying skeletal diversity, where a lack there of has been cited as a bottleneck in the drug discovery process.i Among several features that define DOS, functional group pairingiii has surfaced as a significant component, which aims to selectively pair functional groups resulting in the generation of multiple scaffolds. Interest in the facile production of new sultams for biological screening has provided recent impetus for exploring new FG-pairing cyclization pathways of α-halo benzene sulfonamides. We herein report studies towards this goal revealing α-halo benzene sulfonamides as versatile starting materials whose features can be exploited in multiple reaction pathways to produce an array of 5-, 6- and 7-membered benzofused sultams. Ultimately, we aim to utilize these methods in a broader DOS strategy for library production of sultams.
Sultams (cyclic sulfonamides) have emerged as privileged structures in drug discovery due to their diverse biological properties.iv In particular, a number of benzofused sultams have recently been reported that exhibit broad inhibitory properties against a variety of enzymes including: COX-2,v HIV integrase,vi lipoxygenase,vii Calpain Iviii and MMP-2.ix Moreover, a number of additional sultams have shown promising bioactivity, such as antiviral,x anticancer,xi antimicrobial,xii antimalarial,xiii antileukemicxiv and AMPA receptor modulatory properties with potential for treating disorders of the brain.xv This impressive biological profile is augmented by a number of chemical properties inherent to sulfonamides including facile coupling/alkylation pathways for sulfonamide formation, stability to hydrolysis, polarity and their crystalline nature.xvi
α-Haloarylsulfonamides represent an attractive building block for the production of benzofused sultams,xvii due to a number of key features. The most prominent include: (i) click coupling between starting α-halobenzenesulfonyl chlorides and amines under mild conditions generating the corresponding sulfonamides in quantitative yield, (ii) the α-halo group enhances the acidity of the aryl sulfonamide N-H enabling Mitsunobu and conventional alkylation reactions to occur under mild conditions, (iii) the α-halo group can be utilized in metal-catalyzed cross coupling or Li-halogen exchange chemistry and (iv) a number of substituted α-halo benzene sulfonyl chlorides are commercially available. Taken collectively, these attributes guided our efforts in this study.
2. Results and discussion
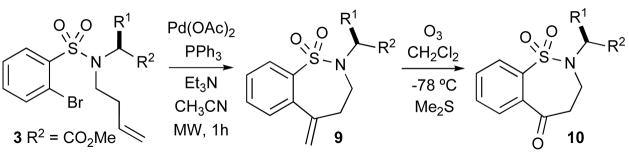
As outlined in Scheme 1, we began our initial investigation exploring Pd(0)-catalyzed Heck reactions with olefin containing sulfonamides 2, 3 and 4 (Scheme 1). Substrates 2 and 3 were designed to alleviate any potential issues with regioselective Heck reactions. Substrate 4 was a more promiscuous Heck substrate and deserves mention first. Sulfonamide 4 was prepared via simple coupling of sulfonyl chloride 1 with L-amino methyl ester•HCl under standard conditions (CH2Cl2, Et3N, DMAP) and allylation with allyl bromide. Subsequent Heck conditions were explored [Pd(OAc)2, Et3N, 0.25 M in CH3CN at 100 °C in a microwave] and produced a mixture of products via 6-exo-trig (5) and 7-endo-trig (6) pathways, along with product 7 resulting from deallylation.xviii
Scheme 1.

Intramolecular Heck
(a) Amino ester, Et3N, DMAP, CH2Cl2; (b) Ethyl 4-bromocrotonate, K2CO3, CH3CN, 60 °C; (c) Amino ester, Et3N, DMAP, CH2Cl2; (d) 4-Buten-1-ol, DIAD, PPh3, CH2Cl2, RT; (e) Allyl bromide, K2CO3, CH3CN, RT; (f) Pd(OAc)2, PPh3, Et3N, CH3CN. (g) O3, CH2Cl2, −78 °C then Me2S.
The regioselectivity concern of 6-exo versus 7-endo cyclization was not an issue when sulfonamide 2 was subjected to standard Heck reaction conditions. Thus, treatment of 1 with L-amino methyl ester•HCl under the aforementioned conditions, followed by alkylation with ethyl 4-bromocrotonate, yielded the alkylated sulfonamide 2, which upon subjection to Heck conditions in refluxing CH3CN, afforded the desired δ-sultam 8 as the sole product via a selective 6-exo cyclization pathway.
The straightforward syntheses of the 7-membered sultams 9 were also readily prepared as outlined in Scheme 1. Homo-allylation of the sulfonamide with 4-bromo-1-butene produced the alkylated product 3 in low yield. However, we found that performing the Mitsunobu reaction with 3-buten-1-ol in the presence of DIAD and PPh3 gave superior yields of 3.xix Subsequent intramolecular Heck reaction of 3, cleanly afforded the 7-exo-trig cyclization products 9 in excellent yields (Table 1). Ozonolysis of 9 produced the benzothiazepenones 10 in good to excellent yields. Similarly, the inseparable mixture of Heck products 5–6 was subjected to ozonolysis furnishing benzothiazenone 11 in pure form after isolation.
Table 1.
Pd(0)-catalyzed Heck cyclizations of substrate 3.a
 | ||||
|---|---|---|---|---|
| entry | SM | product | 9 yieldb % | 10 yieldc % |
| 1 | 3a | R1 = CH3 | 9a (72) | 10a (84) |
| 3 | 3b | R1 = CH2CHMe2 | 9b (91) | 10b (82) |
| 4 | 3c | R1 = H | 9c (70) | 10c (78) |
| 5 | 3d | R1 = H, R2 = C6H5 | 9d (62) | 10d (78) |
Isolated yields
Heck conditions: substrate (1.00 mmol), Pd(OAc)2 (0.1 mmol; 10.0 mol %), PPh3 (0.2 mmol) in CH3CN (0.25 M) at 100 °C for 1 h under microwave.
O3, −78 °C, CH2Cl2, then Me2S.
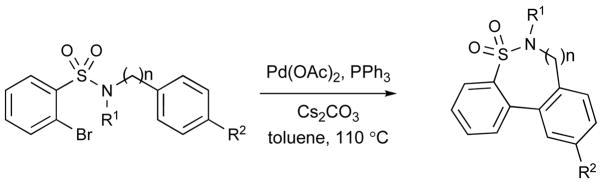
The chemistry of benzenesulfonamide was further extended to an intramolecular arylation reaction whereby the α-bromo aryl sulfonamide group was paired with an aromatic group.xx The literature contains a single example of a 6-membered sultam produced from an α-halobenzenesulfonamide using Pd(0)-catalysis.xxi However, in this intramolecular direct C-arylation, the yield was low. Treating 1 with aniline and subsequent alkylation with methyl iodide or ethyl iodide produced the corresponding sulfonamide 12, which was subjected to Pd(OAc)2, PPh3, Cs2CO3 in toluene at 110 °C affording cyclized product 13 via a C-arylation pathway. The present method offers a practical method for synthesis of dibenzothiazine dioxides in excellent yields (Table 2).
Table 2.
Pd(0)-catalyzed C-arylation of substrates 12 and 14.a
 | |||
|---|---|---|---|
| entry | SM | product | yieldb (%) |
| 1 | 12a | n = 0, R1 = Me, R2 = H | 13a (83) |
| 2 | 12b | n = 0, R1 = Et, R2 = H | 13b (77) |
| 3 | 14a | n = 1, R1 = Me, R2 = H | 15a (85) |
| 4 | 14b | n = 1, R1 = Me, R2 = OMe | 15b (56) |
| 5 | 14c | n = 1, R1 = Bn, R2 = H | 15c (95) |
Isolated yields
conditions: substrate (1.0 mmol), Pd(OAc)2 (0.1 mmol; 10.0 mol %), PPh3 (0.2 mmol), Cs2CO3 (2.0 mmol) in toluene (4.0 mL) at 110 °C in sealed tube for 12–24 h.
With this result in hand, we attempted the direct C-arylation to furnish a 7-membered sultam 15. Treatment of 1 with benzyl amine and subsequent alkylation with methyl iodide furnished 14, which was again subjected to Pd(OAc)2 conditions at 110 °C for 24 h to afford the 7-membered benzofused sultam 15 in 85% yield.
The method was next applied to an intramolecular O-arylation reactionxxii whereby the α-bromo aryl sulfonamide group was paired with an alcohol. Thus, 1 was coupled with phenylalanine methyl ester, followed by Mitsunobu reaction with 3-buten-1-ol and reduction with LiAlH4 to furnish sulfonamide 16 in 41% over 3 steps. Sulfonamide 16 was treated with CuI (20 mol%) in the presence of HOCH2CH2OH as a ligand in DMF at 120 °C to afford the O-arylation, cyclized product 17 in 49 % yield.
The chemistry of α-halobenzenesulfonamide was further extended to both Pauson-Khand (PK) and intramolecular hydroamination (IHA) reactions.xxiii In this method, the α-bromoaryl sulfonamide group was attached to an alkyne under Sonogashira conditions and subsequently paired with both an alkene (PK) as well as an N-H (IHA). The reaction sequence began with the sulfonylation of methylamine with tosyl chloride 18 followed by iodination to afford 2-iodosulfonamide 19 in 80% over 2 steps. xxiv Compound 19 was subjected to Sonogashira reaction with trimethylsilyl acetylene in presence of PdCl2(PPh3)2 and CuI to furnish sulfonamide 20.xxv Alkylation of 20 with allyl bromide in presence of K2CO3 in CH3CN at 50 °C afforded allylated enyne product 21 along with a small amount of the corresponding IHA product 22. However, when the reaction of 20 was carried out in the same conditions in the absence of allyl bromide, sultam 22 was produced as the sole product in 78% yield, representing a formal intramolecular hydroamination of an acetylene via a 5-exo-cyclization pathway. Finally, treatment of sulfonamide enyne 21 with [Co2(CO)8] under thermal conditions (90 °C) furnished the tricyclic Pauson-Khand product 23 in 67% yield.
The production of sultam 22 via an intramolecular hydroamination pathway prompted us to explore the development of a one-pot protocol that would take advantage of the inherent nucleophilicity of the sulfonamide N-H. In this regard, we have has previously reported a one-pot domino Heckaza-Michael protocol for the synthesis of 1,2-benzisothiazoline-3-acetic acid 1,1-dioxides 24.xxvi Such domino protocols are attractive strategies for the incorporation of multiple points of diversity in a one-pot multi-component transformation. Thus, we investigated the application of α-haloaryl sulfonamides toward a domino aza-Michael-Heck protocol, where by initial Michael addition of the sulfonamide into methyl propiolate, followed by an intramolecular Heck reaction yields the corresponding benzofused sultam. Thus, N-benzyl-2-iodobenzenesulfonamide was reacted with methyl propiolate under standard reaction conditions at 110 °C to cleanly afford the desired sultam 25 in 74% yield. We are currently investigating additional variants of this protocol.
As a final example, we explored additional chemistry of the 7-membered benzothiazepenones using a classical aldol reaction to produce enone 26. The underlying principle behind this was a reagent-based DOS approach towards skeletal diversity. Thus, treatment of 10e with benzaldehyde in the presence of KOH furnished 26 bearing an unsaturated ketone as a single isomer in 80% yield. This aldol condensation set the stage for further modifications as outlined in Scheme 5.iia Subsequent hetero-Diels-Alder (HDA) reaction of ethyl vinyl ether and 26 under neat conditions at 70 °C generated the tricyclic benzosultam 27 as a single regio- and diastereoisomer in 62% yield. Condensation reaction of 26 with phenyl hydrazine produced the pyrozoline-containing sultam 28 in 82% yield as a 1:1 mixture of diastereomers.xxvii Finally, reaction of malononitrile with 26 produced the tricyclic sultams 29 in 68% yield.
Scheme 5.

Reagent-Based DOS Approach
Conclusion
In conclusion, we have developed a variety of new reaction pathways toward the synthesis of skeletally diverse benzofused sultams. These methods employ commercially available α-haloaryl sulfonyl chlorides in achieving multiple reaction pathways such as Heck, lithiation, Sonogashira-NH addition, Sonogashira-Pauson-Khand, C- and O-arylations to produce 5, 6, 7-membered sultams in good to excellent yields. We also demonstrated that Heck reaction products undergo facile ozonolysis/aldol to produce an intermediate unsaturated ketone, which was utilized in a reagent-based DOS approach to furnish additional diverse sultams. All the sultam reported herein have been submitted to NIH biological outreach partners for biological screening through the NIH Molecular Library Screening Network (NIH-MLSCN). The utilization of the reported method is currently being employed in library production and the results will be reported in due course.
3. Experimental Section
General 3.1
All reactions were carried out in flame-dried glassware under argon. Toluene, THF, Et2O, and CH2Cl2 were purified by passage through a purification system (Solv-Tek) employing activated Al2O3.xxviii Et3N was distilled from CaH2. Flash column chromatography was performed with Merck silica gel (230–400 mesh). Thin layer chromatography was performed on silica gel 60F254 plates. 1H and 13C spectra were recorded in CDCl3 on either a Bruker DRX-400 or a Bruker AM-500 spectrometer operating at 400/100 MHz and 500/125 MHz, respectively. High-resolution mass spectrometry (HRMS) and FAB spectra were obtained on a VG Instrument ZAB double-focusing mass spectrometer. Infrared data was obtained on a Nicolet 320 Fourier Transform Infrared Spectrophotometers. Melting points were obtained on a Thomas Hoover capillary melting point apparatus. Optical rotations were carried out on a Rudolph Automatic Polarimeter (AUTOPOL IV).
3.2 Synthetic Studies
(S,E)-ethyl-4-(2-bromo-N-(1-methoxy-3-methyl-1-oxobutan-2-yl)phenylsulfonamido)but-2-enoate (2)
Into a flame dried flask was added, 1 (2.0 g, 7.82 mmol), H-Leu-OMe (1.42 g, 7.82 mmol) and CH2Cl2 (20 mL). After stirring for 5 mins at RT, Et3N (2.32 mL, 16.4 mmol) was added and the reaction flask stirred at RT for 2 h. The crude reaction mixture was filtered and concentrated under reduced pressure. A portion of the crude material (208 mg, 0.56 mmol) was added to a flame-dried flask, to which Cs2CO3 (294 mg, 1.165 mmol) and dry CH3CN (6 mL, 0.1 M) was added. After stirring for 5 mins, (E)-ethyl 4-bromobut-2-enoate (0.66 mmol) was added and the reaction mixture was stirred at 60 °C until completion of reaction (as monitored by TLC). The crude reaction mixture was filtered through a pad of Celite, washed with CH2Cl2, concentrated under reduced pressure and subjected to flash chromatography (1:1 EtOAc: Hexane) to provide 2 (201 mg, 0.44 mmol, 78%) as a clear oil. FTIR (neat): 2972, 1739, 1720, 1342,1163 cm−1; 1H NMR (500 MHz, CDCl3) δ ppm 8.11 (dd, J = 1.7, 7.9, 1H), 7.71 (dd, J = 7.8, 1.3 Hz, 1H), 7.43 (td, J = 7.7, 1.3 Hz, 1H), 7.37 (dt, J = 7.5, 1.8 Hz, 1H), 6.84 (ddd, J = 15.8, 7.8, 5.1 Hz, 1H), 5.87 (dt, J = 15.8, 1.5 Hz, 1H), 4.41 (dddd, J = 18.4, 9.0, 6.4, 1.5 Hz, 2H), 4.14 (q, J = 7.1 Hz, 2H), 4.09 (d, J = 10.3 Hz, 1H), 3.54 (s, 3H), 2.15 – 2.06 (m, 1H), 1.29 – 1.23 (m, 3H), 1.02 (d, J = 6.6 Hz, 3H), 0.91 (dd, J = 9.0, 3.8 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ ppm 171.0, 165.7, 144.4, 139.2, 135.4, 133.8, 133.1, 127.6, 123.1, 120.3, 65.5, 60.4, 51.7, 46.5, 28.9, 19.8, 19.3, 14.2; HRMS calculated for C18H24BrNNaO6S (M+Na)+ 484.0405; found 484.0379.
(S)-methyl-2-(2-bromo-N-(but-3enyl)phenylsulfonamido) propanoate (3a)
Into a flame-dried flask under argon was added 1 (7.82 mmol), amino ester/amine (7.82 mmol) and CH2Cl2 (20 mL). Et3N (16.4 mmol) was added and the reaction flask stirred at RT for 2 h. The crude reaction mixture was filtered and concentrated under reduced pressure. A portion of the crude (2.56 g 7.64 mmol) was added to a flame-dried flask under argon (2.56 g 7.64 mmol), to which was added PPh3 (2.21 g, 8.45 mmol) and dry CH2Cl2 (40 mL). After stirring for 5 mins, 3-buten-1-ol (0.72 mL, 8.45 mmol) was added followed by drop wise addition of diisopropyl azodicarboxylate (DIAD) (1.71 mL, 8.45 mmol) at RT. The reaction mixture was stirred for 3 h, after which time the crude mixture was concentrated under reduced pressure and purified by flash chromatography (1:1 EtOAc: Hexane) to provide 3a (2.34 g, 79% yield) as a white solid. FTIR (neat): 3064, 2950, 1795, 1448, 1434, 1342, 1163 cm− 1; 1H NMR (500 MHz, CDCl3) 1H NMR (500 MHz, CDCl3) δ 8.15 (dd, J = 7.9, 1.7 Hz, 1H), 7.71 (dd, J = 7.8, 1.3 Hz, 1H), 7.44 (td, J = 7.7, 1.3 Hz, 1H), 7.37 (td, J = 7.6, 1.7 Hz, 1H), 5.62 (ddt, J = 17.2, 10.5, 7.7 Hz, 1H), 4.96 (dd, J = 3.4, 1.8 Hz, 1H), 4.94 (dq, J = 11.8, 1.7 Hz, 1H), 4.82 (q, J = 7.4 Hz, 1H), 3.63 (s, 3H), 3.57 – 3.47 (m, 1H), 3.17 (ddd, J = 15.3, 9.3, 7.2, 1H), 2.28 – 2.21 (m, 2H), 1.50 (d, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 172.06, 135.44, 134.40, 133.47, 132.16, 127.50, 116.92, 55.43, 52.29, 45.14, 34.69, 16.48; HRMS calculated for C14H18BrNNaO4S (M+Na)+ 398.0038; found 398.0045.
(S)-methyl 2-(2-bromo-N-(but-3-enyl)phenylsulfonamido)-4-methylpentanoate (3b)
Using a similar procedure as that used to produce sultam 3a, sultam 3b was produced in 69%. FTIR (neat): 3064, 2955, 1795, 1448, 1434, 1336, 1163 cm− 1; 1H NMR (500 MHz, CDCl3) δ ppm 8.14 (dd, J = 7.9, 1.7 Hz, 1H), 7.45 (td, J = 7.7, 1.3 Hz, 1H), 7.38 (td, J = 7.6, 1.7 Hz, 2H), 5.72 (ddt, J = 17.1, 10.2, 6.8 Hz, 1H), 5.04 (dddd, J = 16.0, 1.5, 1.5, 1.5 Hz, 1H), 5.03 (m, 1H), 4.56 (dd, J = 8.8, 5.6 Hz, 1H), 3.59 – 3.51 (m, 1H) 3.54 (s, 3H), 3.32 (ddd, J = 16.0, 11.0, 5.2 Hz, 1H), 2.57 – 2.48 (m, 1H), 2.34 – 2.25 (m, 1H), 1.78 – 1.69 (m, 2H), 1.65 – 1.56 (m, 1H), 0.93 (dd, J = 17.1, 6.2 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 171.7, 139.2, 135.5, 134.7, 133.4, 132.4, 127.5, 120.4, 117.0, 58.3, 52.1, 45.8, 39.4, 35.5, 24.5, 22.6, 21.7; HRMS calculated for C17H24BrNNaO4S (M+Na)+ 440.0507; found 440.0500.
Methyl-2-(2-bromo-N-(but-3-enyl)phenylsulfonamido) acetate (3c)
Using a similar procedure as that used to produce sultam 3a, sultam 3c was produced in 87%. FTIR (neat): 2951, 1753, 1340, 1161, 760 cm − 1; 1H NMR (400 MHz, CDCl3) δ ppm 8.19 (dd, J = 7.8, 1.8 Hz, 1H), 7.74 (dd, J = 7.8, 1.3 Hz, 1H), 7.45 (td, J = 7.5, 1.3 Hz, 1H), 7.40 (td, J = 7.5, 1.7 Hz, 1H), 5.59 (ddt, J = 17.0, 10.2, 6.8 Hz, 1H), 5.01 (ddd, J = 11.0, 6.6, 5.5 Hz, 2H), 4.28 (s, 2H), 3.71 (s, 3H), 3.57 – 3.38 (m, 2H), 2.34 – 2.11 (m, 2H); 13C NMR (100 MHz, CDCl3) δ ppm 169.5, 139.3, 135.5, 134.1, 133.6, 132.2, 127.5, 120.5, 117.5, 52.3, 48.5, 47.6, 32.0. HRMS calculated for C13H16BrNNaO4S (M+Na)+ 383.9881; found 383.9882.
N-Benzyl-2-bromo-N-(but-3-enyl)benzenesulfonamide (3d)
Using a similar procedure as that used to produce sultam 3a, sultam 3d was produced in 67%. FTIR (neat): 3064, 2927, 1641, 1448, 1434, 1332, 1124 cm − 1; 1H NMR (500 MHz, CDCl3) 1H NMR (500 MHz, CDCl3) δ 8.16 (dd, J = 7.8, 1.8 Hz, 1H), 7.76 (dd, J = 7.8, 1.4 Hz, 1H), 7.44 (td, J = 7.6, 1.4 Hz, 1H), 7.40 (td, J = 7.6, 1.9 Hz, 1H), 7.34 – 7.26 (m, 5H), 5.56 – 5.47 (m, 1H), 4.94 – 4.92 (m, 1H), 4.90 (t, J = 1.3 Hz, 1H), 4.57 (s, 2H), 3.26 (dd, J = 8.4, 6.8 Hz, 2H), 2.16 – 2.10 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 139.47, 135.90, 135.53, 134.30, 133.49, 132.34, 128.63, 128.26, 127.80, 127.51, 120.52, 117.13, 77.25, 77.00, 76.75, 51.26, 46.01, 31.90; HRMS calculated for C17H18BrNNaO2S (M+Na)+ 402.0140; found 402.0139
(S)-methyl 3-(N-allyl-2-bromophenylsulfonamido)-5-methyl-2-oxohexanoate (4)
Using a similar procedure as that used to produce sultam 2, sultam 4 was produced as a clear oil in 85% yield. [α]D20 - 42.8 (c 3.38, CH2Cl2), colorless oil FTIR (neat): 2966, 1740, 1340, 1163, 748 cm − 1; 1H-NMR (CDCl3, 400 MHz): δ (ppm) 8.01-7.28 (aromatic H, 4H), 5.80 (m, 1H), 5.06 (d, J = 17.2 Hz, 1H), 4.91 (d, J = 10.2 Hz, 1H), 4.49 (dd, J = 6.4, 3.9 Hz, 1H), 4.10 (dd, J = 16.4, 8.0 Hz, 1H), 3.93 (d, J = 10.4 Hz, 1H), 3.40 (s, 3H), 2.10 (m, 1H), 0.94 (d, J = 6.6 Hz, 3H), 0.80 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ (ppm) 170.7, 139.4, 135.5, 135.3, 133.6, 132.8, 127.5, 120.1, 117.3, 65.5, 51.3, 48.6, 28.5, 19.5, 18.8; HRMS (ESI) m/z calculated for C15H20BrNO4SNa 412.0194 (M+Na)+, found 412.0181.
Sultam (5)
Into a microwave vial was added sulfonamide 4 (1.77 g, 4.30 mmol), Pd(OAc)2 (0.43 mmol, 96 mg), PPh3 (0.86 mmol, 225 mg), CH3CN (17 mL) and Et3N (1.8 mL, 12.9 mmol). After stirring for 5 min., the reaction was run in the microwave at 100 °C for 1 h. After 1 h, the reaction mixture was cooled to RT and filtered through a small Celite pad and thoroughly washed with CH2Cl2. The reaction mixture was concentrated under reduced pressure and purified using flash chromatography (4:1 hexane:EtOAc) to provide a 1.24 g (89%) of a mixture of compounds 5/6/7 [1:0.14:0.74 based on 1H-NMR] as a yellow oil. FTIR (neat): 2964, 1740, 1340, 1170, 748 cm − 1; 1H-NMR (CDCl3, 400 MHz): δ (ppm) 8.06-7.41 (aromatic H, 4H), 5.78 (s, 1H), 5.25 (s, 1H), 4.67 (d, J = 6.7 Hz, 1H), 4.49 (d, J = 6.8 Hz, 1H), 4.11 (d, J = 10.6 Hz, 1H), 3.25 (s, 3H), 1.68 (m, 1H), 0.92 (d, J = 6.8 Hz, 3H), 0.06 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ (ppm) 170.2, 136.0, 134.3, 133.5, 131.1, 127.6, 125.3, 120.4, 113.8, 64.3, 51.4, 47.9, 27.9, 19.2, 18.8; HRMS (ESI) m/z calculated for C15H19NO4SNa 332.0932 (M+Na)+, found 332.0936.
Intramolecular Heck reaction to sultam 8
To the alkylated sulfonamide 2 was added Pd(OAc)2 (12.5 mg, 0.056 mmol), PPh3 (29 mg, 0.11 mmol) and Et3N (0.23 mL, 1.68 mmol) and the reaction mixture was heated at 70 °C for overnight. After 12 h the reaction mixture was cooled to room temperature and filtered through a small Celite pad and thoroughly washed with CH2Cl2. The reaction mixture was concentrated under reduced pressure and purified using flash chromatography (4:1 Hexane:EtOAc) to provide 8 (67% yield) as a yellow oil. [α]25 = − 12.1; (c 0.05, CHCl3); FTIR (neat): 2968, 1739, 1332, 1199, 1117, 1026 cm − 1. 1H NMR (500 MHz, CDCl3) δ ppm 7.94 (dd, J = 7.9, 0.9 Hz, 1H), 7.68 – 7.58 (m, 1H), 7.51 (ddd, J = 8.0, 6.4, 2.5 Hz, 2H), 6.96 (s, 1H), 4.56 (d, J = 10.4 Hz, 1H), 4.16 (q, J = 7.1 Hz, 2H), 3.78 (s, 3H), 3.59 (s, 2H), 2.24 (ddt, J = 13.3, 10.4, 6.7 Hz, 1H), 1.22 (t, J = 7.1 Hz, 3H), 0.97 (d, J = 6.6 Hz, 3H), 0.79 (d, J = 6.7 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ ppm 170.7, 170.6, 132.5, 132.1, 131.4, 128.1, 127.8, 124.0, 122.1, 113.5, 62.5, 61.2, 52.5, 36.6, 29.9, 19.1, 18.6, 14.1; HRMS calculated for C18H23NO6SNa (M+Na)+ 404.1144; found 404.1146.
(S)-1,2-Benzothiazepine-2(3H)-acetic acid, 4,5-dihydro-5-methylene-α-(2-methylpropyl)-, methyl ester, 1,1-dioxide (9b)
Into a microwave vial was added sulfonamide 3b (1.8 g, 4.30 mmol), Pd(OAc)2 (0.43 mmol, 96 mg), PPh3 (0.86 mmol, 225 mg), CH3CN (17 mL) and Et3N (1.8 mL, 12.9 mmol). After stirring for 5 min., the reaction was run in the microwave at 100 °C for 1 h. After 1 h, the reaction mixture was cooled to RT and filtered through a small Celite pad and thoroughly washed with CH2Cl2. The reaction mixture was concentrated under reduced pressure and purified using flash chromatography (4:1 hexane:EtOAc) to provide 1.29 g (89%) of the title compound 9b as a yellow oil. [α]25 = −35.4; (c 7.30, CHCl3); colorless liquid. FTIR (neat): 2954, 2869, 1741, 1467, 1153 cm − 1. 1H NMR (500 MHz, CDCl3) δ ppm 8.46 (dd, J = 7.8, 1.1 Hz, 1H), 8.03 (td, J = 7.5, 1.3 Hz, 1H), 7.97 – 7.89 (m, 2H), 5.86 (d, J = 0.7 Hz, 1H), 5.82 (d, J = 1.2 Hz, 1H), 5.23 (dd, J = 9.4, 6.0 Hz, 1H), 4.32 (ddd, J = 13.0, 8.2, 4.6 Hz, 1H), 4.19 (dt, J = 10.7, 5.1 Hz, 1H), 3.92 (s, 3H), 3.22 – 2.96 (m, 2H), 2.19 (dq, J = 17.1, 8.7 Hz, 2H), 2.04 (ddd, J = 13.6, 6.7, 6.5 Hz, 1H), 1.44 (d, J = 6.6 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ ppm 171.4, 147.0, 139.7, 139.7, 132.2, 129.9, 127.2, 126.1, 119.0, 57.5, 51.7, 45.0, 38.1, 35.0, 24.3, 22.8, 21.3; HRMS calculated for C17H23NNaO4S (M+Na)+ 360.1245; found 360.1224.
(S)-1,2-Benzothiazepine-2(3H)-acetic acid, 4,5-dihydro-α-methyl-5-methylene-, methyl ester, 1,1-dioxide (9a)
Using a similar procedure as that used to produce sultam 9b, sultam 9a was produced in 72%. [α]25 = −34.0; (c 3.45, CHCl3); colorless liquid. FTIR (neat): 2951, 1750, 1365, 1170, 1158 cm − 1. 1H NMR (500 MHz, CDCl3) δ ppm 7.88 (dd, J = 7.9, 1.1 Hz, 1H); 7.46 (td, J = 7.6, 1.3 Hz, 1H), 7.36 (td, J = 7.7, 1.5 Hz, 2H), 5.29 (s, 1H), 5.26 (s, 1H), 4.69 (q, J = 7.3 Hz, 1H), 3.70 (t, J = 4.3 Hz, 2H), 3.46 (s, 3H), 2.52 (ddd, J = 14.4, 6.2, 5.9 Hz, 2H), 1.33 (d, J = 7.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ ppm 171.7, 147.0, 139.8, 139.7, 132.2, 130.1, 127.3, 126.2, 119.3, 55.1, 52.0, 45.5, 35.5, 16.1; HRMS calculated for C14H17NNaO4S (M+Na)+ 318.0776; found 318.0780 (FAB).
(S)-1,2-Benzothiazepine-2(3H)-acetic acid, 4,5-dihydro-5-methylene-, methyl ester, 1,1-dioxide (9c)
Using a similar procedure as that used to produce sultam 9b, sultam 9c was produced in 70%. FTIR (neat) 2952, 1755, 1417, 1128, 860 cm − 1. 1H NMR (500 MHz, CDCl3) δ ppm 7.92 (dd, J = 7.8, 1.2 Hz, 1H), 7.52 (td, J = 7.5, 1.4 Hz, 1H), 7.44 – 7.35 (m, 2H), 5.36 (d, J = 0.9 Hz, 1H), 5.28 (d, J = 1.2 Hz, 1H), 3.83 (s, 4H), 3.70 (s, 3H), 2.58 (t, J = 4.0 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ ppm 168.9, 146.8, 140.2, 137.7, 132.8, 130.5, 127.5, 127.5, 119.7, 52.2, 50.5, 48.8, 32.6; HRMS calculated for C13H15NNaO4S (M+Na)+ 304.0619; found 304.0613.
1,2-Benzothiazepine, 2,3,4,5-tetrahydro-5-methylene -2-(phenylmethyl)-, 1,1-dioxide (9d)
Using a similar procedure as that used to produce sultam 9b, sultam 9d was produced in 62%. FTIR (neat): 2943, 1352, 1336, 1163, 727 cm − 1; 1H NMR (400 MHz, CDCl3) δ ppm 8.05 (dd, J = 7.7, 1.3 Hz, 1H), 7.56 (td, J = 7.5, 1.4 Hz, 1H), 7.45 (ddd, J = 16.2, 7.6, 1.3 Hz, 2H), 7.38 – 7.29 (m, 5H), 5.38 (d, J = 0.7 Hz, 1H), 5.31 (t, J = 2.0 Hz, 1H), 4.18 (s, 2H), 3.60 (s, 2H), 2.55 (t, J = 5.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ ppm 147.1, 140.6, 137.4, 135.7, 132.8, 130.6, 128.6, 128.3, 128.2, 128.2, 127.9, 127.6, 119.7, 50.0, 47.6, 31.5; HRMS calculated for C17H17NNaO2S (M+Na)+ 322.0878; found 322.0865.
(S)-1,2-Benzothiazepine-2(3H)-acetic acid, 4,5-dihydro-α-(2-methylpropyl))-5-oxo-, methyl ester, 1,1-dioxide (10b)
To a round bottom flask were added 9b (1.88 g 5.57 mmol), Sudan(III) (1 mg) and CH2Cl2 (50 mL). The reaction mixture was cooled down to − 78 °C and O3 was bubbled into the solution for 15 min. After such time, Me2S (5 mL) was added drop wise upon disappearance of the pink color of the crude reaction mixture. The reaction mixture was concentrated under reduced pressure and purified by column chromatography (4:1 hexanes:EtOAc) to provide 82% yield of the desired product (10c) as a colorless oil. [α]25 = 8.7; (c 1.39, CHCl3); FTIR (neat): 2956, 1741, 1693, 1342, 1203, 1172, 754 cm − 1; 1H NMR (500 MHz, CDCl3) δ ppm 8.00 (ddd, J = 7.0, 4.4, 2.5 Hz 1H), 7.90 (ddd, J = 8.8, 4.8, 2.2 Hz, 1 H), 7.65 – 7.62 (ddd, J = 5.0, 2.4, 2.1 Hz, 2 H), 4.78 (dd, J = 8.3, 7.3 Hz, 1H), 3.75 – 3.50 (m, 2H), 3.49 – 3.36 (m, 3H), 3.18 (dt, J = 13.9, 5.0 Hz, 1H), 1.81 – 1.57 (m, 4H), 1.02 (s, 3H), 1.00 (s, 3H); 13C NMR (125 MHz, CDCl3) δ ppm 198.9, 171.1, 140.8, 135.6, 132.4, 132.0, 129.4, 126.3, 58.9, 52.0, 42.3, 39.9, 38.0, 24.5, 23.1, 21.0.; HRMS calculated for C16H21NNaO5S (M+Na)+ 362.1038; found 362.1047.
(S)-1,2-Benzothiazepine-2(3H)-acetic acid, 4,5-dihydro-α-methyl-5-oxo-, methyl ester, 1,1-dioxide (10a)
Using a similar procedure as that used to produce sultam 10b, sultam 10a was produced in 84% yield as a colorless oil. [α]25 = 30.5; (c 0.67, CHCl3); FTIR (neat): 2935, 1693, 1589, 1315, 1280, 1153 cm − 1; 1H NMR (400 MHz, CDCl3) δ ppm 7.99 (ddd, J = 8.8, 4.8, 2.4 Hz, 1 H), 7.87 (ddd, J = 9.2, 4.8, 2.4 Hz, 1 H), 7.66 (ddd, J = 8.8, 4.4, 2.0 Hz, 2 H), 4.81 (q, J = 7.2 Hz, 1 H), 3.50 (s, 3 H), 3.45–3.64 (m, 3 H), 3.24 (ddd, J = 10.8, 6.4, 4.4 Hz, 1 H), 1.45 (d, J = 7.2 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ ppm 199.1, 171.1, 140.7, 135.7, 132.4, 132.0, 129.4, 126.2, 56.2, 52.2, 42.3, 40.0, 16.0; HRMS calculated for C13H15NNaO5S (M+Na)+ 320.0569; found 320.0570.
1,2-Benzothiazepine-2(3H)-acetic acid, 4,5-dihydro-5-oxo-, methyl ester, 1,1-dioxide (10c)
Using a similar procedure as that used to produce sultam 10b, sultam 10c was produced in 78 % yield as a colorless oil. FTIR (neat) 2954, 1751, 1693, 1342, 1163, 769 cm − 1; 1H NMR (500 MHz, CDCl3) δ ppm 7.95 (ddd, J = 9.0, 5.6, 3.2 Hz, 1H), 7.83 (ddd, J = 9.0, 6.4, 3.0 Hz, 1H), 7.68 (ddd, J = 10.0, 5.7, 3.3 Hz, 2H), 4.11 (s, 2H), 3.66 (s, 3H), 3.62 (t, J = 6.0 Hz, 2H), 3.34 (t, J = 6.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ ppm 199.8, 168.8, 138.8, 136.0, 132.7, 132.0, 129.5, 126.3, 52.3, 51.2, 45.3, 41.7; HRMS calculated for C12H13NNaO5S (M+Na)+ 306.0412; found 306.0425.
1,2-Benzothiazepin-5-(2H)-one, 3,4-dihydro-2-(phenylmethyl)-, 1,1-dioxide (10d)
Using a similar procedure as that used to produce sultam 10b, sultam 10d was produced in 78% yield as a colorless oil. FTIR (neat): 2956, 1693, 1512, 1340, 1160 cm−1; 1H NMR (500 MHz, CDCl3) δ ppm 8.06 (ddd, J = 8.8, 5.6, 3.6 Hz, 1H), 7.83 – 7.60 (m, 3H), 7.49 – 7.24 (m, 5H), 4.30 (s, 2H), 3.49 (t, J = 6.0 Hz, 2H), 3.14 (t, J = 6.0 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ ppm 200.6, 137.5, 137.2, 135.0, 132.9, 131.7, 129.3, 128.8, 128.3, 128.2, 127.2, 52.5, 43.5, 41.2; HRMS calculated for C16H15NNaO3S (M+Na)+ 324.0670; found 324.0667 (FAB).
Sultam (11)
Using a similar procedure as that used to produce sultam 8, sultam 4 was produced and carried through crude using a similar procedure as that used to produce sultam 10, to yield 11 (39% over 2 steps) as a clear oil. FTIR (neat): 2964, 1739, 1469, 1340, 1170 cm−11H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 7.3 Hz, 1H), 7.82 (d, J = 7.5 Hz, 1H), 7.78 – 7.66 (m, 2H), 4.70 (d, J = 18.8 Hz, 1H), 4.39 (d, J = 18.9 Hz, 1H), 4.22 (d, J = 10.0 Hz, 1H), 3.20 (s, 3H), 2.28 – 2.03 (m, 1H), 1.05 (d, J = 6.7 Hz, 4H), 0.96 (d, J = 6.6 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 188.4, 170.3, 140.1, 134.2, 132.9, 129.9, 128.1, 123.4, 65.3, 54.0, 51.6, 28.4, 19.1, 19.1; HRMS calculated for C14H17NNaO5S (M+Na)+ 334.0725; found 334.0737.
N-Benzyl-2-bromo-N-methylbenzenesulfonamide 12a
Into a flame-dried flask under argon was added sulfonyl chloride 1 (1.0 g, 3.9 mmol), aniline (0.36 mL, 3.9 mmol), dry CH2Cl2 (39 mL, 0.1M) and pyridine (1.58 mL, 1.95 mmol). After stirring at RT for 2 h the crude reaction mixture was filtered, washed with water (2 × 20 mL), dried with MgSO4 and concentrated under reduced pressure. A portion of the crude material (1.0 g, 0.32 mmol) was added to a flame-dried flask followed by the addition of CH3I (1.0 mL, 1.61 mmol), K2CO3 (2.22 g, 1.61 mmol) and dry CH3CN (12 mL, 0.1 M) were added and reaction mixture was stirred at RT, until the starting material disappeared as monitored by TLC. The crude reaction mixture was filtered through a pad of celite and washed with CH2Cl2. The mixture was concentrated under reduced pressure and purified by flash chromatography (1:1 hexane:EtOAc) to provide 12a (88% yield) as a clear oil; FTIR (neat): 1493, 13339, 1157, 741, 565 cm − 1; 1H NMR (500 MHz, CDCl3) δ ppm 7.83 (dd, J = 7.1, 2.0 Hz, 1H), 7.66 (dd, J = 7.0, 2.0 Hz, 1H), 7.36 – 7.07 (m, 7H), 3.37 (s, 3H); 13C NMR (125 MHz, CDCl3) δ ppm 141.0, 138.2, 135.8, 133.9, 133.1, 129.4, 127.6, 127.6, 127.2, 120.7, 39.7; HRMS calculated for C13H12BrNNaO2S (M+Na)+ 347.9670; found 347.9636.
N-benzyl-2-bromo-N-ethylbenzenesulfonamide 12b
Using a similar procedure as that used to produce sultam 12a, sultam 12b was produced as a yellow oil in 85% yield. FTIR (neat): 2974, 1490, 1336, 1174, 741, 696 cm − 1; 1H NMR (500 MHz, CDCl3) δ ppm 7.77 (dd, J = 7.8, 1.8 Hz 1H), 7.65 (dd, J = 7.8, 1.3 Hz, 1H), 7.33 – 7.08 (m, 7H), 3.86 (q, J = 7.1 Hz, 2H), 1.09 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ ppm 138.8, 138.3, 135.6, 133.7, 133.1, 129.7, 129.4, 128.2, 127.5, 120.6, 47.7, 14.9; HRMS calculated for C14H14BrNNaO2S (M+Na)+ 361.9827; found 361.9830.
Intramolecular C-arylation to 6H-dibenzo[c,e][1,2] thiazine, 6-methyl-, 5,5-dioxide 13a
To 12a was added Pd(OAc)2 (3.02 mg, 0.0135 mmol,), PPh3 (7.08 mg, 0.027 mmol), Cs2CO3 (131 mg, 0.405 mmol) and toluene (4 mL). The crude reaction mixture was heated at 110 °C for 12–24 h, until the SM was consumed, as indicated by TLC. After 24 h the reaction mixture was cooled to RT and filtered through a small Celite pad and thoroughly washed with CH2Cl2. The reaction mixture was concentrated under reduced pressure and purified using flash chromatography (4:1 hexane:EtOAc) to afford the desired product 13a in 83% yield as a yellow oil. FTIR (neat): 1477, 1327, 1174, 754cm − 1; 1H NMR (500 MHz, CDCl3) δ ppm 8.03 (td, J = 6.5, 1.4 Hz, 2H), 7.98 (d, J = 8.0 Hz, 1H), 7.72 (td, J = 7.5, 1.4 Hz, 1H), 7.58 (td, J = 7.7, 1.1 Hz 1H), 7.52 (ddd, J = 8.1, 7.5, 1.5 Hz, 1H), 7.38 – 7.29 (m, 2H), 3.46 (s, 3H); 13C NMR (126 MHz, CDCl3) δ ppm 139.4, 134.1, 132.4, 132.3, 130.4, 128.2, 125.5, 125.4, 124.6, 123.9, 122.4, 119.3, 32.7; HRMS calculated for C13H11NNaO2S (M+Na)+ 268.0408; found 268.0430.
6H-dibenzo[c,e][1,2] thiazine, 6-ethyl-, 5,5-dioxide 13b
Using a similar procedure as that used to produce sultam 13a, sultam 13a was produced in 77% as a yellow oil. FTIR (neat): 1635, 1336, 1166, 1095, 977 cm − 1; 1H NMR (500 MHz, CDCl3) δ ppm 8.03-7.95 (m, 3H), 7.71 (td, J = 7.6, 1.3 Hz, 1H), 7.62 (dd, J = 7.7, 1.0 Hz, 1H), 7.57 (td, J = 7.7, 1.1 Hz, 1H), 7.50 (ddd, J = 8.3, 7.3, 1.5 Hz, 1H), 7.40 (dd, J = 8.0, 1.1 Hz, 1H), 7.37 (td, J = 7.6, 1.1 Hz, 1H), 3.99 (q, J = 7.1 Hz, 2H), 1.18 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ ppm 140.9, 139.4, 134.8, 133.5, 133.3, 130.2, 129.9, 129.5, 129.0, 128.5, 128.3, 127.5, 56.1, 39.0; HRMS calculated for C14H13NNaO2S (M+Na)+ 282.0565; found 282.0553.
2-Bromo-N-(4-methoxybenzyl)-N-methylbenzene-sulfonamide (14b)
Into a flame-dried flask under argon was added 2-bromobenzenesulfonyl chloride (150 mg, 0.58 mmol), methylamine (1.76 mmol), and dry CH2Cl2 (6 mL). Et3N (0.11 mL, 0.76 mmol) and DMAP (5 mg, 0.1 mmol) were added and the reaction flask stirred at RT for 2 hrs. The crude reaction mixture was filtered and washed with water. The mixture was dried (MgSO4), concentrated under reduced pressure and purified by flash chromatography (1:1 hexane:EtOAc) to provide the corresponding N-methyl sulfonamide in 98% yield as a yellow solid. To the 2-bromo-N-methylbenzene-sulfonamide (0.56 mmol) in dry CH3CN (6 mL, 0.1 M) was added Cs2CO3 (294 mg, 1.165 mmol). p-Methoxy benzyl bromide (0.66 mmol) was added and the reaction mixture was stirred at 60 °C, until the SM disappeared as monitored by TLC. The crude reaction mixture was filtered through a pad of Celite, washed with CH2Cl2, concentrated under reduced pressure and purified by flash chromatography (1:1 hexane:EtOAc) to provide 2-bromo-N-(4-methoxybenzyl)-N-methylbenzene-sulfonamide 14b in 88% yield. 1H NMR (500 MHz, CDCl3) δ 8.15 (dd, J = 7.8, 1.8 Hz, 1H), 7.78 (dd, J = 7.8, 1.3 Hz, 1H), 7.47 (td, J = 7.6, 1.3 Hz, 1H), 7.41 (td, J = 7.6, 1.8 Hz, 1H), 7.24 (d, J = 7.8 Hz, 1H), 6.89 (d, J = 7.5 Hz, 1H), 6.86 (d, J = 2.2 Hz, 1H), 6.83 (dd, J = 8.1, 2.2 Hz, 1H), 4.40 (s, 2H), 3.79 (s, 3H), 2.76 (s, 3H); 13C NMR (500 MHz, CDCl3) δ 159.8, 135.6, 133.6, 132.4, 129.6, 127.5, 120.5, 113.6, 113.2, 55.20, 54.04, 33.94; HRMS calculated for C15H16BrNNaO3S (M+Na)+ 391.9932; found 398.9906
N-benzyl-2-bromo-N-methylbenzenesulfonamide 14a
Using a similar procedure as that used to produce sultam 14b, sultam 14a was produced as a yellow oil in 71% yield. FTIR (neat): 3062, 2912, 1795, 1446, 1434, 1332, 1163 cm − 1; 1H NMR (500 MHz, CDCl3) δ 8.15 (dd, J = 7.8, 1.8 Hz, 1H), 7.78 (dd, J = 7.8, 1.3Hz, 1H), _7.47 (td, J = 7.6, 1.4 Hz, 1H), 7.41 (td, J = 7.6, 1.8 Hz, 1H), 7.36 – 7.34 (m, 1H), 7.33 – 7.32 (m, 2H), 7.31 (d, J = 1.0 Hz, 1H), 7.30 – 7.27 (m, 1H), 4.44 (s, 2H), 2.75 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 138.4, 135.8, 135.7, 133.6, 132.4, 128.7, 128.2, 127.8, 127.6, 120.3, 77.2, 77.0, 76.7, 54.1, 33.9; HRMS calculated for C14H14BrNNaO2S (M+Na)+ 361.9827; found 361.9843
N,N-dibenzyl-2-bromobenzenesulfonamide 14c
Using a similar procedure as that used to produce sultam 14b, sultam 14a was produced as a white solid in 96% yield. Mp: 110 °C; FTIR (neat): 1332, 1159, 698 cm − 1; 1H NMR (500 MHz, CDCl3) δ ppm 8.48 – 8.09 (m, 1H), 8.09 – 7.65 (m, 1H), 7.58 – 7.16 (m, 8H), 7.16 – 6.91 (m, 4H), 4.42 (s, 4H); 13C NMR (126 MHz, CDCl3) δ ppm 139.7, 135.6, 135.4, 133.6, 132.5, 128.6, 128.6, 127.8, 127.6, 120.8, 50.2; HRMS calculated for C20H18BrNNaO2S (M+Na)+ 452.0170; found 452.0238.
Dibenzo[d,f][1,2]thiazepine, 6,7-dihydro-6-methyl-, 5,5-dioxide 15a
Using a similar procedure as that used to produce sultam 13a, sultam 15a was produced in 85% as a yellow oil. FTIR (neat): 1635, 1336, 1166, 1095, 977 cm − 1. 1H NMR (500 MHz, CDCl3) δ ppm 8.01 (dd, J = 7.8, 1.0 Hz, 1H), 7.73 (td, J = 7.6, 1.3 Hz, 1H), 7.62 (dd, J = 7.7, 1.0 Hz, 1H), 7.57 (td, J = 7.7, 1.3 Hz, 1H), 7.54 – 7.42 (m, 4H), 3.78 (s, 2H), 2.89 (s, 3H); 13C NMR (125 MHz, CDCl3) δ ppm 140.9, 139.4, 134.8, 133.5, 133.3, 130.2, 129.9, 129.5, 129.0, 128.5, 128.3, 127.5, 56.1, 39.0; HRMS calculated for C14H13NNaO2S (M+Na)+ 282.0565; found 282.0553.
Sultam 15b
Using a similar procedure as that used to produce sultam 13a, sultam 15b was produced in 56% yield as a yellow oil. FTIR (neat): 3335, 1636, 1340, 1120, 967 cm − 1; 1H NMR (500 MHz, CDCl3) δ ppm 8.00 (dd, J = 1.1, 7.8 Hz, 1H), 7.71 (td, J = 7.6, 1.3 Hz, 1H), 7.60 (dd, J = 7.7, 1.0 Hz, 1H), 7.56 (td, J = 7.7, 1.3 Hz, 1H), 7.34 (d, J = 8.3 Hz, 1H), 6.99 (d, J = 2.6 Hz, 1H), 6.93 (dd, J = 2.7, 8.3 Hz, 1H), 3.86 (s, 3H), 3.72 (s, 2H), 2.85 (s, 3H); 13C NMR (125 MHz, CDCl3) δ ppm 160.2, 142.1, 139.3, 134.7, 133.3, 131.4, 128.4, 127.5, 125.6, 114.6, 113.7, 55.5, 55.3, 38.9; HRMS calculated for C15H15NNaO3S (M+Na)+ 312.0670; found 312.0652.
Sultam 15c
Using a similar procedure as that used to produce sultam 13a, sultam 15c was produced in 95% yield as a yellow oil. FTIR (neat): 3058, 3028, 1446, 1334, 1168, 912, 746 cm − 1; 1H NMR (500 MHz, CDCl3) δ ppm 8.07 (dd, J = 7.8, 1.1 Hz, 1H), 7.71 (td, J = 7.6, 1.2 Hz, 1H), 7.58 (ddd, J = 10.5, 8.7, 4.4 Hz, 2H), 7.60 (d, J = 7.6 Hz, 1H), 7.57 (td, J = 7.6, 1.1 Hz, 1H), 7.42 (d, J = 7.1 Hz, 2H), 7.40 – 7.34 (m, 3H), 7.33 (d, J = 7.1 Hz, 1H), 7.24 (d, J = 7.5 Hz, 1H), 4.45 (s, 2H), 3.70 (s, 2H).; 13C NMR (125 MHz, CDCl3) δ ppm 135.8, 133.6, 133.2, 130.1, 129.9, 129.4, 129.0, 128.7, 128.6, 128.5, 128.3, 127.9, 127.0, 55.0, 52.7; HRMS calculated for C20H17NNaO2S (M+Na)+ 358.0878; found 358.0868.
(S)-2-Bromo-N-(but-3-enyl)-N-(1-hydroxy-3-phenylpropan-2 yl)benzenesulfonamide (16)
A flame-dried flask was charged with the homo allylated sulfonamide (0.917g, 2.027mmol) and Et2O (4 mL) and cooled to 0 °C. LiAlH4 (81 mg, 2.128 mmol) was added portion-wise to the reaction mixture over 10 min after which the reaction was allowed to warm to RT and stirred for 1 h. The reaction was quenched using the Fieser work up procedure. The organic layer was extracted with Et2O (5 mL × 2), dried (Na2SO4), concentrated under reduced pressure and purified by flash chromatography (1:1 hexane:EtOAc) to afford the alcohol in 95% yield. FTIR (neat): 3527, 3028, 1446, 1326, 1124, 919, 750 cm − 1; 1H NMR (500 MHz, CDCl3) δ 7.83 – 7.79 (m, 2H), 7.58 – 7.54 (m, 1H), 7.50 – 7.45 (m, 1H), 7.24 – 7.16 (m, 2H), 7.02 – 6.98 (m, 1H), 5.78 (ddt, J = 17.1, 10.2, 6.9 Hz, 1H), 5.12 (dd, J = 3.1, 1.5 Hz, 1H), 5.09 (td, J = 3.3, 1.9 Hz, 1H), 5.07 (s, 1H), 4.03 – 3.97 (m, 1H), 3.60 (t, J = 6.1 Hz, 1H), 3.41 (dt, J = 8.3, 7.3 Hz, 1H), 3.31 – 3.23 (m, 1H), 2.70 (dd, J = 13.5, 9.5 Hz, 1H), 2.61 (dd, J = 13.5, 5.3 Hz, 1H), 2.47 (dt, J = 14.5, 7.2 Hz, 1H), 1.97 (t, J = 5.9, 1H); 13C NMR (126 MHz, CDCl3) δ 140.5, 137.4, 134.9, 132.6, 129.1, 128.9, 128.6, 127.2, 126.7, 117.4, 77.3, 77.2, 77.0, 76.8, 62.2, 62.1, 44.3, 36.3, 35.4. HRMS calculated for C19H22NBrNaO3S (M+Na)+ 446.0401; found 446.0402.
Procedure for intramolecular O-arylation reaction 17
To sulfonamide 16 (0.301 g, 0.70 mmol) was added CuI (0.117 mmol, 22 mg), ethylene glycol (0.26 mL, 4.702 mmol), Cs2CO3 (1.53 g, 4.70 mmol) and DMF (3 mL) under Ar. The crude mixture was heated at 120 °C for 12–24 h, till the SM is consumed, as indicated on TLC. After 24 h the reaction mixture was cooled to RT, filtered through a small Celite pad, thoroughly washed with CH2Cl2, concentrated under reduced pressure and purified using flash chromatography (4:1 hexane:EtOAc) to provide 19 in 49% yield as a yellow oil. [α]25 = −56.7; (c 2.39, CHCl3); FTIR (neat): 3064, 1593, 1440, 1218, 1153, 1008, 918 cm − 1; 1H NMR (400 MHz, CDCl3) δ ppm 7.83 (dd, J = 7.9, 1.6 Hz, 1H), 7.36 (dddd, J = 19.4, 17.4, 12.1, 4.5 Hz, 6H), 7.17 – 7.11 (m, 1H), 7.03 – 6.97 (m, 1H), 5.56 (dddd, J = 16.3, 13.6, 9.5, 6.8 Hz, 1H), 4.94 (s, 1H), 4.91 (dd, J = 3.7, 2.1 Hz, 1H), 4.78 (t, J = 12.0 Hz, 1H), 4.34 (dd, J = 13.2, 4.6 Hz, 1H), 3.86 (s, 1H), 3.21 (dd, J = 13.7, 7.9 Hz, 1H), 3.06 (t, J = 7.7 Hz, 2H), 2.91 (dd, J = 13.6, 7.3 Hz, 1H), 2.14 – 2.03 (m, 1H), 1.96 (dt, J = 14.1, 7.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ ppm 155.5, 137.6, 134.4, 133.4, 132.1, 129.5, 129.4, 128.8, 128.7, 126.8, 123.2, 121.3, 117.0, 73.8, 64.6, 51.4, 37.3, 32.9; FT-IR (neat): 3064, 1593, 1440, 1218, 1153, 1008, 918 cm −1.; HRMS calculated for C19H21NNaO3S (M+Na)+ 366.1140; found 366.1136.
N,4-dimethyl-2 ((trimethylsilyl)ethynyl)benzene-sulfonamide 20
To a stirring solution of tosyl chloride (1.0 g, 5.24 mmol) in CH2Cl2 (10 mL) at 0 °C was added Et3N (2.2 mL) and methylamine (0.23 ml, 5.24 mmol). After stirring for 2 h, the reaction was quenched with H2O (10 mL), the organic layer removed and the aqueous layer extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were washed with sat. brine (10 mL), dried (MgSO4), filtered and concentrated under reduced pressure to give the desired sulfonamide as a pale yellow solid. A portion of this crude (0.8 g, 4.31 mmol) in Et2O (43 mL) was cooled to −78 °C and stirred for 15 mins. After such time n-BuLi (1.0 M, 4.31 mmol) was added drop wise to the crude mixture and the reaction was stirred for an additional 15 mins. Elemental I2 (0.65g, 5.17 mmol) was added to the reaction mixture and after 15 mins was warmed to RT and stirred for 2 h. The reaction was quenched with aqueous sat. NH4Cl (10 mL), extracted with EtOAc (2 × 20 mL) and the combined organic layers were washed with aqueous sat. Na2SO3 (10 mL), dried (MgSO4), filtered and concentrated under reduced pressure to afford sulfonamide 19 as pale yellow solid. To a portion of crude material (500 mg, 1.6 mmol) in DMF (8.2 mL) was added PdCl2(PPh3)2 (33.8 mg, 0.048 mmol), PPh3 (51 mg, 0.192 mmol), CuI (8.8 mg, 0.08 mmol) and Et3N (0.67 mL, 4.818 mmol). After the addition of TMS acetylene (0.68 mL, 4.818 mmol), the reaction was heated to 80 °C and stirred for 14 h after which time the crude mixture was cooled to RT and concentrated under reduced pressure. The crude material was purified using flash chromatography (1:1 hexane:EtOAc) to provided 20 in 46% yield over 3 steps as a yellow oil. FTIR (neat): 2359, 1731, 1330, 1315, 1174, 1141, 846 cm − 1; 1H NMR (500 MHz, CDCl3) δ ppm 7.72 (d, J = 8.1 Hz, 1H), 7.27 (dd, J = 1.1, 0.5 Hz, 1H), 7.09 – 7.05 (m, 1H), 2.40 (d, J = 5.5 Hz, 3H), 2.21 (s, 3H), 0.11 (s, 9H); 13C NMR (125 MHz, CDCl3) δ ppm 142.9, 137.2, 135.0, 129.6, 129.6, 120.0, 103.1, 101.5, 29.4, 21.1; HRMS calculated for C13H19NNaO2SSi (M+Na)+ 304.0803; found 304.0806.
Sultam 22
To a flame-dried Rb- flask charged with 20 (0.5 g, 0.177 mmol) was added CH3CN (2.0 mL) and K2CO3 (0.7 g, 0.523 mmol) and the reaction mixture was stirred at 60 °C for 2 h. After such time, the crude reaction mixture was filtered through a silica pad, concentrated under pressure and purified by flash chromatography (4:1 hexane:EtOAc) to provide 22 in 67% yield as a yellow oil. FTIR: (neat) 2358, 2341, 1298, 1140, 790 cm − 1; 1H NMR (500 MHz, CDCl3) δ ppm 7.62 (d, J = 8.0 Hz, 1H), 7.55 – 7.39 (m, 1H), 7.31 (dd, J = 8.0, 0.7 Hz, 1H), 4.89 (d, J = 2.8 Hz, 1H), 4.35 (d, J = 2.8 Hz, 1H), 3.05 (s, 3H), 2.41 (s, 3H); 13C NMR (125 MHz, CDCl3) δ ppm 144.0, 138.9, 131.3, 130.6, 129.4, 121.5, 120.7, 84.2, 25.8, 21.8; HRMS calculated for C10H11NNaO2S (M+Na)+ 232.0408; found 232.0392.
Sultam 23
To a flame-dried Rb-flask charged with 21 (45 mg, 0.14 mmol) and toluene (5 mL) was added Co2(CO)8 (52 mg, 0.15 mmol). The reaction mixture was heated at 90 °C for 2h, concentrated and the crude product was purified by column chromatography (1:1 hexanes:EtOAc) furnishing the tricyclic product in 67% yield as a yellow oil. FTIR (neat): 2948, 1780, 1323, 1147, 1122, 946, 906 cm−1; 1H NMR (500 MHz, CDCl3) δ ppm 8.03 (s, 1H), 7.81 (d, J = 8.2 Hz, 1H), 7.30 – 7.23 (m, 1H), 7.20 (s, 1H), 4.57 (d, J = 14.4 Hz, 1H), 3.46 – 3.33 (m, 1H), 3.29 (dd, J = 14.4, 1.3 Hz, 1H), 2.83 (s, 3H), 2.77 – 2.62 (m, 1H), 2.44 (s, 3H), 2.33 (dd, J = 17.6, 1.9 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ ppm 206.0, 164.4, 143.7, 143.1, 142.0, 130.4, 130.0, 129.9, 129.2, 60.4, 43.6, 40.3, 39.0, 21.4; HRMS calculated for C14H15NNaO3S (M+Na)+ 300.0670; found 300.0677.
(Z)-Methyl 2-(2-benzyl-3-benzisothiazoline-1-ylidene)-acetate 25
Into a 1 dram vial was added N-benzyl-2-idobenzenesulfonamide (50 mg, 0.13 mmol), Et3N (37μL, 0.26 mmol), Bu4NCl (50 mg, 0.13 mmol), Pd2(dba)3•CHCl3 (2 mol%, 2.8 mg, 0.0027 mmol) and dry DMF (0.98 mL). After stirring for 5 min at RT, methyl propiolate (33 μL, 0.40 mmol) was added and the reaction vial was placed immediately into a preheated reaction block. The reaction was stirred at 110 °C for 14 hours after which time the reaction was cooled and concentrated under reduced pressure. The crude product was purified using flash chromatography (8:1 hexane:EtOAc) to yield 25 as a yellow oil (31.6 mg, 76%, 0.096 mmol). FTIR (neat) 2362, 1730, 1291, 1172 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.26 (dd, J = 17.2, 1.3 Hz, 1H), 7.92-7.83 (m, 1H), 7.73-7.63 (m, 2H), 7.38-7.21 (m, 3H), 5.25 (s, 1H), 4.79 (s, 2H), 3.62 (s, 3H); 13C NMR (126 MHz, CDCl3) 166.1, 143.1, 133.9, 133.7, 132.8, 132.1, 129.6, 129.0, 128.1, 127.3, 126.9, 121.1, 96.1, 53.6, 44.4; HRMS calculated for C17H15NO4SNa (M+Na)+ 352.0619 found 352.0593 (TOF MS EI+).
Sultam 26
Into a flame-dried round bottom flask was added benzothiazepenone 10e (0.1 g, 0.33 mmol), benzaldehyde (0.5 mL, 0.49 mmol) and EtOH (2 mL). To this mixture was added KOH (0.48 mmol) and the reaction mixture was heated at 50 °C for 4 h. After such time, the crude mixture was filtered and recrystallized from EtOH to produce 26 (80% yield) as a white solid. Mp 171 °C; FTIR (neat): 3060, 1676, 1607 1172, 958 cm−1; 1H NMR (500 MHz, CDCl3) δ ppm 7.94 (s, 1H), 7.91 (m, 2H), 7.73 – 7.65 (m, 2H), 7.36 – 7.24 (m, 7H), 7.19 (m, 2H), 7.09 (t, J = 7.5 Hz, 1H), 4.24 (s, 2H), 3.52 (s, 2H); 13C NMR (125 MHz, CDCl3) δ ppm 192.1, 143.3, 136.9, 134.8, 134.6, 134.0, 133.4, 132.7, 130.8, 130.7, 129.9, 129.7, 129.5, 128.8, 128.6, 128.4, 126.3, 55.9, 47.8; HRMS calculated for C23H19NNaO3S (M+Na)+ 412.0984; found 412.0983.
Sultam 27
To a flame dried round bottom flask was added benzothiazepenone (0.1 g, 0.33 mmol) and benzaldehyde (0.5 mL, 0.49 mmol) was dissolved in EtOH (2 mL). The reaction mixture was treated with KOH (0.48 mmol) and heated at 50 °C for 4 h. The crude product was filtered and recrystallized from EtOH producing the desired aldol product in 80% yield. To a flame-dried screw cap vial, aldol product (0.1 mmol) and ethyl vinyl ether (0.30 mmol) were heated at 70 °C in neat condition for overnight. The TLC analysis indicated that the reaction was complete and the crude product was purified by column chromatography (2:1 hexanes:EtOAc) to afford desired hetero-Diels-Alder product in 62% yield as a yellow oil. FTIR (neat): 2974, 1336, 1296, 1168, 1081 cm−1; 1H NMR (500 MHz, CDCl3) δ ppm 7.95 (dd, J = 7.8. 1.1 Hz, 1H), 7.72 (d, J = 7.8 Hz, 1H), 7.56 (td, J = 7.7, 1.3 Hz, 1H), 7.44 (td, J = 7.7, 1.2 Hz, 1H), 7.35 – 7.29 (m, 2H), 7.29 – 7.17 (m, 3H), 7.06 – 7.01 (m, 3H), 6.77 (d, J = 7.1 Hz, 2H), 5.20 (t, J = 3.0 Hz, 1H), 4.13 (d, J = 14.6 Hz, 1H), 3.95 (dq, J = 9.5, 7.1 Hz, 1H), 3.82 (d, J = 14.6 Hz, 1H), 3.66 (dq, J = 9.5, 7.1 Hz, 1H), 3.51 (t, J = 8.2 Hz, 1H), 3.08 (d, J = 13.8 Hz, 1H), 2.93 (d, J = 13.7 Hz, 1H), 2.20 – 2.04 (m, 2H), 1.21 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ ppm 147.3, 142.3, 136.9, 135.7, 134.4, 132.4, 129.3, 128.8, 128.6, 128.6, 128.4, 127.8, 127.3, 127.0, 126.9, 111.3, 97.0, 64.3, 54.0, 48.9, 39.4, 36.0, 15.3; HRMS calculated for C27H27NNaO4S (M+Na)+ 484.1558; found 484.1534.
Sultam 28
To a solution of aldol 26 (0.1 g, 0.25 mmol) in EtOH (3 mL) was added phenyl hydrazine (0.1 mL, 1.0 mmol) and a drop of sulfuric acid. The reaction mixture was stirred at RT overnight. The product was obtained after concentration of the solution, filtration and recrystallization from hexanes to afford 28 in 74% yield as a yellow oil. FTIR (neat): 1593, 1493, 1336, 1163, 1139, 746 cm−1; 1H NMR (500 MHz, CDCl3) δ ppm 8.08 – 7.91 (m, 2H), 7.54 (td, J = 7.6, 1.3 Hz, 1H), 7.43 (td, J = 7.6, 1.3 Hz, 1H), 7.32 – 7.17 (m, 6H), 7.16 – 7.00 (m, 6H), 6.95 (d, J = 7.8 Hz, 2H), 6.77 (t, I = 7.3 Hz, 1H), 4.69 (d, J = 7.0 Hz, 1H), 4.37 (d, J = 13.6 Hz, 1H), 3.97 – 3.74 (m, 2H), 3.61 (s, 1H), 3.36 (d, J = 8.7 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ ppm 145.8, 143.7, 140.6, 138.3, 135.0, 132.6, 130.8, 130.5, 129.5, 129.0, 128.8, 128.8, 128.5, 128.3, 128.2, 125.3, 120.4, 113.7, 76.8, 69.6 54.4, 48.7. HRMS calculated for C29H25N3NaO2S (M+Na)+ 502.1565; found 502.1560.
Sultam 29
To a solution of aldol product 26 (50 mg, 0.128 mmol) in EtOH (3 mL), was added malononitrile (9.3 mg, 0.14 mmol) and a drop of piperidine. The reaction mixture was stirred at RT for overnight, after which time the crude reaction mixture was concentrated and the crude product was purified by column chromatography (4:1 hexanes:EtOAc) furnishing the desired product 29 in 68% yield as a clear oil. FTIR (neat): 2974, 1650, 1512, 1336, 1168, 1081, 767 cm−1; 1H NMR (500 MHz, CDCl3) δ ppm 8.10 (dd, J = 7.8, 1.3 Hz, 1H), 8.02 (dd, J = 8.1, 1.0 Hz, 1H), 7.68 (td, J = 1.4, 7.8 Hz, 1H), 7.54 (td, J = 7.7, 1.1 Hz, 1H), 7.36 – 7.27 (m, 3H), 7.22 – 7.12 (m, 5H), 6.92 – 6.84 (m, 2H), 4.65 (s, 2H), 4.02 (d, J = 14.1 Hz, 1H), 3.86 (s, 1H), 3.80 (d, J = 17.8 Hz, 1H), 3.53 (d, J = 14.1 Hz, 1H), 3.45 (d, J = 17.7 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ ppm 158.7, 141.9, 140.3, 137.4, 134.5, 132.9, 129.2, 129.1, 128.8, 128.7, 128.6, 128.4, 128.4, 128.1, 128.0, 127.9, 127.7, 118.9, 116.40, 76.8, 60.4, 52.4, 48.2, 42.8.; HRMS calculated for C26H21N3NaO3S (M+Na)+ 478.1201; found 478.1211.
Figure 1.

Multiple cyclization pathways towards skeletally diverse Sultams.
Scheme 2.

C- and O-arylation reactions.
(a) Aniline, pyridine, CH2Cl2; (b) Methyl iodide or ethyl iodide, K2CO3, CH3CN, 50 °C; (c) benzyl amine, Et3N, DMAP, CH2Cl2; (d) phenyl alanine methyl ester, Et3N, DMAP, CH2Cl2; (e) DIAD, PPh3, 4-buten-1-ol (f) LiAlH4, THF, 0 °C to rt.
Scheme 3.

Sonogashira-PK and -IHA Strategies.
Scheme 4.

Domino aza-Michael-Heck Strategy
Acknowledgments
This work was generously supported by funds provided by the NIH Center for Chemical Methodologies and Library Development at the University of Kansas (KU-CMLD) (P50 GM069663), Pilot Scale Libraries Program (P41 GM076302), NIH COBRE award P20 RR015563 and the Umaer Basha Research fellowship for student support (H.S.). The authors thank Dr. David Vander Velde and Sarah Neuenswander for assistance with NMR measurements, Dr. Todd Williams for HRMS analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- (i).(a) Burke MD, Schreiber SL. Angew Chem Int Ed. 2004;43:46–58. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]; (b) Spiegel DA, Schroeder FC, Duvall JR, Schreiber SL. J Am Chem Soc. 2006;128:14766–14767. doi: 10.1021/ja065724a. [DOI] [PubMed] [Google Scholar]
- (ii).(a) Spandal RJ, Mónica DG, O’Connell KMG, Thomas GL, Spring DR. Chem Record. 2008;8:129–142. doi: 10.1002/tcr.20144. [DOI] [PubMed] [Google Scholar]; (b) Tan DS. Nat Chem Bio. 2005;1:74–84. doi: 10.1038/nchembio0705-74. [DOI] [PubMed] [Google Scholar]; (c) Spring DR. Org Biomol Chem. 2003;1:3867–3870. doi: 10.1039/b310752n. [DOI] [PubMed] [Google Scholar]
- (iii).Functional Group (FG) pairing: Nielsen TE, Schreiber SL. Angew Chem Int Ed. 2008;47:48–56. doi: 10.1002/anie.200703073.Comer E, Rohan E, Deng L, Porco JA., Jr Org Lett. 2007;9:2123– 2126. doi: 10.1021/ol070606t.
- (iv).Levy L. Drugs Future. 1992;17:451–454. [Google Scholar]
- (v).(a) Rabasseda X, Hopkins SJ. Drugs of Today. 1994;30:557–563. [Google Scholar]; (b) Inagaki M, Tsuri T, Jyoyama H, Ono T, Yamada K, Kobayashi M, Hori Y, Arimura A, Yasui K, Ohno K, Kakudo S, Koizumi K, Suzuki R, Kawai S, Kato M, Matsumoto S. J Med Chem. 2000;43:2040–2048. doi: 10.1021/jm9906015. [DOI] [PubMed] [Google Scholar]
- (vi).Brzozowski F, Saczewski F, Neamati N. Bioorg Med Chem Lett. 2006;16:5298–5302. doi: 10.1016/j.bmcl.2006.07.089. [DOI] [PubMed] [Google Scholar]
- (vii).Misu Y, Togo H. Org Biomol Chem. 2003;1:1342–1346. doi: 10.1039/b301330h. [DOI] [PubMed] [Google Scholar]
- (viii).Wells GJ, Tao M, Josef KA, Bihovsky R. J Med Chem. 2001;44:3488–3503. doi: 10.1021/jm010178b. [DOI] [PubMed] [Google Scholar]
- (ix).Cherney RJ, Mo R, Meyer DT, Hardman KD, Liu RQ, Covington MB, Qian M, Wasserman ZR, Christ DD, Trzaskos JM, Newton RC, Decicco CP. J Med Chem. 2004;47:2981. doi: 10.1021/jm049833g. [DOI] [PubMed] [Google Scholar]
- (x).Di Santo R, Costi R, Artico M, Massa S, Marongiu ME, Loi AG, De Montis A, La Colla P. Antivir Chem Chemoth. 1998;9:127–137. doi: 10.1177/095632029800900204. [DOI] [PubMed] [Google Scholar]
- (xi).Scozzafava A, Owa T, Mastrolorenzo A, Supuran CT. Curr Med Chem. 2003;10:925–953. doi: 10.2174/0929867033457647. [DOI] [PubMed] [Google Scholar]
- (xii).Zia-ur-Rehman M, Choudary JA, Ahmad S, Siddiqui HL. Chem Pharm Bull. 2006;54:1175–1178. doi: 10.1248/cpb.54.1175. [DOI] [PubMed] [Google Scholar]
- (xiii).Valente C, Guedes RC, Moreira R, Iley J, Gut J, Rosenthal PJ. Bioorg Med Chem Lett. 2006;16:4115–4119. doi: 10.1016/j.bmcl.2006.04.079. [DOI] [PubMed] [Google Scholar]
- (xiv).Silvestri R, Marfe G, Artico M, La Regina G, Lavecchia A, Novellino E, Morgante M, Di Stefano C, Catalano G, Filomeni G, Abruzzese E, Ciriolo MR, Russo MA, Amadori S, Cirilli R, La Torre F, Sinibaldi Salimei P. J Med Chem. 2006;49:5840. doi: 10.1021/jm0602716. [DOI] [PubMed] [Google Scholar]; (b) Lebegue N, Gallet S, Flouquet N, Carato P, Pfeiffer B, Renard P, Leonce S, Pierre A, Chavatte P, Berthelot P. J Med Chem. 2005;48:7363–7373. doi: 10.1021/jm0503897. [DOI] [PubMed] [Google Scholar]
- (xv).Francotte P, De Tullio P, Goffin E, Dintilhac G, Graindorge E, Fraikin P, Lestage P, Danober L, Thomas J-Y, Caignard D-H, Pirotte B. J Med Chem. 2007;50:3153–3157. doi: 10.1021/jm070120i. [DOI] [PubMed] [Google Scholar]
- (xvi).(a) Zhou A, Hanson P. Org Lett. 2008;10:2951–2954. doi: 10.1021/ol8009072. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jiménez-Hopkins M, Hanson P. Org Lett. 2008;10:2951–2954. doi: 10.1021/ol8009072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (xvii).For the use of α-halo arylsulfonamides in synthesis of sultams, for Heck reactions, see: Grigg R, York M. Tetrahedron Lett. 2000;41:7255–7258.Evans P, McCabe T, Morgan BS, Reau S. Org Lett. 2005;7:44–46. doi: 10.1021/ol0480123.Vasudevan A, Tseng P-S, Djuric SW. Tetrahedron Lett. 2006;47:8591–8593.Paquette LA, Dura R, Fosnaugh N, Stephanian M. J Org Chem. 2006;71:8483–8445. doi: 10.1021/jo061404y.For radical cyclization Bressy C, Menant C, Piva O. Synlett. 2005;4:577–582.for alkyne 6-endo cyclizations Barange DK, Nishad TC, Swamy K, Bandameedi V, Kumar D, Bukkapattanam RS, Vyas K, Pal M. J Org Chem. 2007;72:8547–8550. doi: 10.1021/jo701470h.
- (xviii).Sultam 5 could not be separated from by-products and isolated for characterization. The crude reaction mixture was taken through crude and subjected to ozonolysis to afford sultam 11.
- (xix).The Mitsunobu reaction of unsubstituted benzene sulfonamides did not occur under standard conditions. Plausibly, the presence of the electron-withdrawing α-bromide group reduces the pKa of the sulfonamide sufficiently to enable Mitsunobu alkylation.
- (xx).(a) Ritleng V, Sirlin C, Pfeffer M. Chem Rev. 2002;102:1731–1769. doi: 10.1021/cr0104330. [DOI] [PubMed] [Google Scholar]; (b) Kakiuchi F, Chatani N. Adv Synth Catal. 2003;345:1077. [Google Scholar]
- (xxi).Ames DE, Opalko A. Tetrahedron. 1984;40:1919–1925. [Google Scholar]
- (xxii).Ley SV, Thomas WW. Angew Chem Int Ed. 2003;42:5400–5449. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]
- (xxiii).Yin Y, Ma W, Zhuo C, Zhao G. J Org Chem. 2007;72:5731–5736. doi: 10.1021/jo070681h. [DOI] [PubMed] [Google Scholar]; (b) Müller TE, Beller M. Chem Rev. 1998;98:675–703. doi: 10.1021/cr960433d. [DOI] [PubMed] [Google Scholar]
- (xxiv).Lane C, Snieckus V. Synlett. 2000;9:1294–1296. [Google Scholar]
- (xxv).During the Sonogashira reaction, we observed 22 along with tandem Sonogashira and N-H addition to TMS alkyne, similar to 24 with TMS intact.
- (xxvi).Rolfe Alan, Young K, Hanson PR. Eur J Org Chem. 2008:5254–5262. doi: 10.1002/ejoc.200800651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (xxvii).El-Rayyes NR, Bahtiti NH. J Heterocyclic Chem. 1989;26:209. [Google Scholar]
- (xxviii).Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]