

Abstract
IL-13 is known to affect many processes that contribute to an asthmatic phenotype, including inflammation, fibrosis, and mucus production. Members of the aquaporin (AQP) family of transmembrane water channels are targets of regulation in models of lung injury and inflammation. Therefore, we examined AQP mRNA and protein expression in allergen and IL-13-induced mouse models of asthma. Lungs from ovalbumin sensitized and ovalbumin challenged (OVA/OVA) and IL-13 treated mice showed airway thickening, increased mucus production, and pulmonary eosinophilia. Pulmonary function tests showed a significant increase in methacholine-induced airway hyperreactivity in OVA/OVA and IL-13-treated mice as compared with controls. Quantitative PCR analysis revealed differential regulation of AQPs in these two models. AQP1 and AQP4 mRNA expression was downregulated in the OVA/OVA model, but not in the IL-13 model. AQP5 mRNA was reduced in both models, whereas AQP3 was upregulated only in the IL-13 model. Western analysis showed that diminished expression of an apically localized aquaporin, (AQP5), and concomitant upregulation of a basolateral aquaporin (AQP3 or AQP4) are characteristic features of both inducible asthma models. These results demonstrate that aquaporins are common targets of gene expression in both allergen and IL-13 induced mouse models of asthma.
Keywords: IL-13, aquaporin, AHR, asthma, lung, inflammation
1. Introduction
Allergic asthma is a complex disorder, which involves a significant contribution of environmental stimuli for the manifestation of symptoms and degree of clinical severity. The salient features of allergic asthma involve inflammation, increased mucus production, airway remodeling, temporary bronchoconstriction, and airway hyperresponsiveness to bronchoconstrictive agents [1]. Experimentally, the model of intraperitoneal (i.p.) ovalbumin sensitization followed by intranasal (i.n.) or intratracheal (i.t.) ovalbumin challenge in mice generates an allergen-induced asthmatic phenotype, which is characterized by IgE and T cell inflammatory responses, similar to what is seen clinically in human patients [2]. Several previously published studies have established a role for IL-13 in asthma [3,4,5]. Specifically, intranasal IL-13 instillation produces airway hyperreactivity (AHR), mucus hypersecretion, subepithelial fibrosis, and eosinophilia in mice [6,7]. Moreover, studies performed in a variety of different human subpopulations have identified genetic associations between polymorphisms in a number of inflammatory response genes involving the IL-4/IL-13 pathway with atopic asthma [8,9,10,11]. A number of experimental approaches have examined the neutralization of IL-13 as a novel therapeutic strategy for allergic asthma [7,12]. While the exact mechanism is not clear, evidence suggests that alterations in ion and fluid homeostasis may be involved in IL-13 induced AHR [13, 14]. IL-13 treatment is known to alter ion transport along the apical and basolateral membranes of bronchiolar epithelium, thereby functionally converting this cell layer from an absorptive to secretory tissue [14,15]. Moreover, the mucus hypersecretion that accompanies the IL-13 mediated asthma phenotype is characterized by the upregulation of mucin genes and a calcium activated chloride channel, further implicating changes in epithelial dependent fluid handling as a contributor to the asthmatic phenotype [13].
Aquaporins (AQPs) are a family of transmembrane water channels that mediate physiological and cellular responses to changes in fluid volume and osmolarity. AQPs are required for normal secretory and absorptive functions of many tissues, including the eye, lung, salivary gland, sweat glands, and kidney and provide essential systemic regulation of water homeostasis in whole organisms [16,17]. Four aquaporins are expressed in the lung [18,19,20]. AQP1 is expressed in the apical and basolateral membranes of peribronchiolar and alveolar endothelia, and in the visceral pleura. Studies of AQP1 null mice revealed a reduction of osmotic water permeability in capillary endothelium [21] and AQP1 deficient or Colton null human patients exhibit decreased vascular permeability [22], implicating its importance in water transport across the endothelium. AQP3 and AQP4 are expressed in the epithelium from the upper and lower respiratory tract, respectively [19]. AQP3/AQP4 double knockout mice showed moderate reduction in osmotic water permeability in upper airways but with little effect on salt content of airway surface liquid [20]. AQP5 is expressed at high level in type I pneumocytes in the distal lung but moderately in epithelia from upper airway [23]. Functionally, AQP5 is important for regulating airspace-capillary osmotic water permeability [24] and MUC5AC mucin secretion in airway submucosal glands [25,26]. A reduction in human AQP5 expression has been shown to be correlated with an increase in mucus production and compromised lung function in patients with chronic obstructive pulmonary disease [27]. In addition, AQP5 function is critical in systemic pulmonary physiology where it facilitates the maintenance of airway tone. Mice deficient in AQP5 are hyperresponsive to cholinergic stimulation, and exhibit a significant increase in total lung resistance with an accompanying decrease in dynamic compliance [23]. The expression of AQPs is altered in various models of lung infection [28], inflammation [29,30], acute and chronic injury [31,32], and fibrosis [33], indicating a role in these pathological conditions. Therefore, in this study, we used two different mouse models of inducible asthma to investigate AQP mRNA and protein expression in mouse lung. The data presented here show that AQP gene expression in mouse lung is differentially regulated in allergen and IL-13 induced mouse models of asthma, suggesting that alterations in fluid and ion handling in the lung may serve as important mechanistic features of airway pathophysiology in these inducible asthma models.
2. Materials and Methods
2.1 Animal Models
Eight-week-old female BALB/c mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA) and housed in a pathogen-free environment. The Institutional Animal Care and Use Committee review board of Wyeth Pharmaceuticals approved all experiments involving the use of live mice.
Antigen induced airway inflammation and hyperresponsiveness (AHR)
40 female BALB/c mice were randomly assigned to 2 groups. Mice were immunized by i.p. injection with 20 μg of ovalbumin (OVA) emulsified in 2.25 mg alum (AlumImject; Pierce, Rockford, IL, USA) in a total volume of 200 μl on days 0 and 14. To induce an allergic reaction within the lung, animals were challenged via the airways with OVA (5% in PBS; OVA/OVA animals) or PBS (OVA/PBS animals) for 30 min on days 26, 27 and 28 by ultrasonic nebulization. AHR measurement and sample collection were performed on day 30 (48 h after the last challenge).
IL-13 induced airway inflammation and hyperresponsiveness (AHR)
BALB/c mice were challenged via the airways by i.n. administration of 5 μg of recombinant murine IL-13 in a total volume of 50 μl on days 0, 1 and 2, whereas vehicle-control animals were given 50 μl of PBS (n=40 per group) in a chronic exposure inflammatory model. Recombinant murine IL-13 was expressed in E. coli, purified and refolded at Wyeth Research. A three day course of administration of 5 μg of recombinant IL-13 has been shown to produce consistent AHR and induce lung remodeling [5]. Ten animals from each group were used for bronchoalveolar fluid (BALF) cell collection to evaluate lung inflammation and the remaining 10 animals were used to study lung function on day 3 post-IL-13 instillation. Animals were sacrificed by CO2 asphyxiation. The lungs were fixed or snap frozen for histochemical or expression analyses respectively. Measurements of AHR and sample collection for quantitative real-time PCR, Western blots, and histochemical analyses were performed on day 3 (72 hrs). To examine potential dose responsiveness, mice were challenged via the airways by i.n. administration of 0.5 or 10 μg of recombinant murine IL-13 in a total volume of 50 μl on days 0, 1 and 2. Lungs were harvested on day 3 (after 72 hours) and used for real time PCR and Western blotting. Control groups included untreated and PBS-treated mice.
To examine the effect of acute inflammation on aquaporin mRNA expression, mice were treated with a single dose of 10 μg of mIL-13 by i.n. administration. Lung tissues were collected at 6, 24, 48 and 72 hr after mIL-13 instillation, and used for quantitative real-time PCR analysis.
2.2 Lung tissue preparation for histochemical staining
The lungs were excised, inflated, and fixed immediately with 700 μl with 4% paraformaldehyde in PBS administrated via the trachea. Each sample was immersed in 4% paraformaldehyde in PBS for 24 hours to complete the fixation process. Fixed tissues were embedded in paraffin wax within 48 hr for sectioning using an embedding unit (Leica model: EG1160). Five-micron transverse serial sections were cut for both histochemical and immunohistochemical staining. For histology, tissue samples were stained with hematoxylin-eosin (H&E), Massons Trichrome (MT) or periodic acid-Schiff (PAS) using standard methodologies. Images from samples were viewed and captured by using Nikon microscope (Eclipse E800, Nikon Melville, NY, USA)) equipped with a CCD camera (Spot RT Slider, Diagnostic Instrument Inc., Sterling Heights, MI, USA).
2.3 Measurement of inflammatory cell influx
The lungs were excised from mice and BALF was collected by flushing the lungs via the trachea 3X with 700 μl of PBS. Total BALF cellular influx was quantified using a CellDyn coulter counter (Cell-Dyn3500, Abbott Diagnostics, Abbott Park, IL, USA). Cell differential was quantified using a Becton Dickenson Vantage SE Cell Sorter (Becton Dickenson, Franklin Lake, NJ, USA) configured with a 488/10 filter plus a polarizing filter in front of the FL1 detector.
2.4 Measurement of lung resistance (RL) & dynamic compliance (cDyn)
Airway resistance (R) and dynamic compliance (cDyn) were evaluated as described previously [34]. Anesthetized, tracheostomized mice were mechanically ventilated at a rate of 150 breaths/min and a tidal volume of 200 μl. Resistance computations were derived from the tracheal pressure and airflow signals, using an algorithm of covariance. Aerosolization was performed by a nebulizer in-line with the inspiratory tubing from the ventilator. Aerosol was administered in narrow “puffs”, each puff a small part of each inspiration. A dose consisted of twenty ventilator breaths. Increases in total resistance were analyzed over a three-minute period after the last puff. Increasing methacholine concentrations within the nebulizer provided the successive steps of the dose response.
2.5 Quantitative real-time PCR (Taqman) analysis
Real-time PCR was used to evaluate lung aquaporin mRNA expression in the allergen OVA sensitization/OVA challenge model (OVA/OVA; n=6), the OVA/PBS group (n=6), and naïve controls (n=5). Real-time PCR was also used to evaluate aquaporin expression following a 3 day course (72 hr) of 5 μg i.n. administration of IL-13, as well as potential dose responsiveness at 0.5 and 10 μg doses in a chronic exposure inflammatory model (described above). The kinetics of AQP mRNA expression in mouse lung over a 72 hour time course was examined using the acute inflammatory model described above in which mice were administered a single dose of 10 μg of IL-13. For all conditions, total RNA was extracted from frozen lung samples (n= 5 per group) using the RNeasy mini kit and manufacturer's protocol (QIAGEN). Two Hundred and fifty ng total RNA in a 50 μl reaction mix was used for each reverse-transcription/PCR with the one-step protocol (Reverse Transcriptase qPCR Master Mix; EUROGENTEC) as follows: 48°C × 30 min (reverse transcription); 95°C × 10 min (hot starting); 95°C × 15 sec/60°C×1 min for 40 cycles. Cycle number at Threshold (CT) was normalized to CT value for GAPDH gene. All Taqman assays were performed on the ABI prism 7700 sequence detector (Applied Biosystems, Foster City, CA, USA). The primers used for AQP1, AQP3, AQP4, AQP5, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Genbank accession numbers NM_007472, NM_016689, NM_009700, NM_009701, M32599, respectively) are shown in Table 1. Each result was fit to a standard curve and cycle number at threshold values were normalized to GAPDH. The averages of the normalized AQP1, 3, 4, and 5 mRNA levels from the OVA/PBS and OVA/OVA groups were expressed as percent change in expression vs. naïve controls. The averages of the normalized AQP1, 3, 4, and 5 mRNA levels of the five animals from the IL-13 treated were expressed as percent change in expression vs. controls.
Table 1. Oligonucleotide and probe sequences of AQPs and GAPDH for qPCR (Taqman Assay).
| Primer | Probe | |
|---|---|---|
| CGCCACGGCCATTCTC | CGGGCATCACCTCCTCCCTAGTCG | |
| Mu AQP1 | Reverse: TTGCGGCCAAGTGAATTG | |
| Forward: CAATTCTGGCTATGCCGTCAA | CCTGCCCGTGACTTTGGACCTCG | |
| Mu AQP3 | Reverse: AGCCAGGGCGGTGAAGAG | |
| Forward: TCTGGCCACGCTTATCTTTGT | TGGTGGATCCCACACCGAGCAA | |
| Mu AQP4 | Reverse: TTCTGAGCCACCCCAGTTT | |
| Forward: CCCAGCCCGATCTTTCG | ACCGATTCATGACCACCGCAGGG | |
| Mu AQP5 | Reverse: TCCTACCCAGAAGACCCAGTGA | |
| Forward: CGTGTTCCTACCCCCAATGT | CGTGGATCTGACGTGCCGCC | |
| Mu GAPDH | Reverse: GTCATCATACTTGGCAGGTTTCTC |
Probes were labeled at the 5′ end with FAM (fluorescent dye) and 3′ end with TAMRA (quencher).
2.6 Western blot analysis
Rabbit polyclonal antibodies were generated against a synthetic peptide corresponding to the rat AQP1 (NH2-CEEYDLDADDINSRVEMKPK-COOH) and AQP5 (NH2-CEPEEDWEDHREERKKTIELTAH-COOH) C-terminal residues and conjugated to keyhole limpet hemocyanin (KLH; Zymed laboratories, San Francisco, CA, USA). The AQP1 and AQP5 antibodies were affinity purified from serum with sulfolink coupling gel (Pierce, Rockford, IL, USA), conjugated with 2 mg of synthetic peptide. A pre-absorption control was performed with 1 mg of AQP1 or AQP5 peptide with 0.2 mg/ml of antibody pre-incubated overnight at 4°C. Pre-immune controls were conducted with 0.2 mg/ml of IgG purified pre-immune serum from each rabbit. Rabbit polyclonal antibodies directed to amino acids 1-80 of human AQP3 (sc-20811) or amino acids 244-323 of human AQP4 (sc-20812) which cross react with mouse AQP3 or AQP4 respectively were purchased from Santa Cruz Biotechnology. For the allergen exposure model, total membrane protein fractions from the lungs of naïve (n=5), OVA/PBS (n=6), and OVA/OVA (n=6) mice were isolated. For the IL-13 model, total membrane protein fractions from the lungs of control (PBS) mice (n=10), and mIL-13 treated mice (n=10) were isolated. For all conditions, the total membrane preparations were quantified, size fractionated by SDS-PAGE, and subjected Western analysis as previously described [35]. Densitometry was used to quantify both Coomassie and India ink stained bands to confirm equivalent loading on membranes as described [28]. AQP protein abundance was expressed as a percentage of control group expression.
2.7 Statistical Analysis
All data was reported as mean ± SE. The significance of the differences between the groups was determined by either a two-tailed student t-test with equal variance or a two-way ANOVA. A P- value of < 0.05 was considered significant.
3. Results
3.1 mIL-13 intranasal instillation results in changes in lung architecture
To assess airway remodeling in the mIL-13 induced asthma model, lung sections from mice treated with a three-day course of intranasally administered IL-13 (5 μg per dose) or PBS (controls), were examined for histological changes in airway architecture. Hematoxylin and eosin (H&E) staining of lung sections from PBS control mice showed normal lung histology (Figure 1A) whereas lung sections from the mIL-13 treated mice confirmed the presence of airway thickening due to epithelial cell hyperplasia/metaplasia (Figure 1C). Moreover, periodic acid-Schiff (PAS) staining revealed hypersecretion of mucus in lungs of the mIL-13 treated mice (Figure 1D), as compared to the PBS controls (Figure 1B). Massons trichrome stain revealed no subepithelial fibrosis or collagen deposition (data not shown). In addition, only the lung samples from OVA sensitized and OVA challenged (OVA/OVA) (Figure 1G & Figure 1H) but not the OVA sensitized and PBS challenged (OVA/PBS) (Figure 1E & Figure 1F) mice showed thickening of basement membrane and mucus hypersecretion, similar to previously published data from the classic antigen-dependent model of asthma [36]. These data indicate that a three-day course of i.n. mIL-13 administration caused alteration in lung structure by thickening of epithelium and mucus hypersecretion.
Figure 1. IL-13- and OVA- induced lung remodeling.
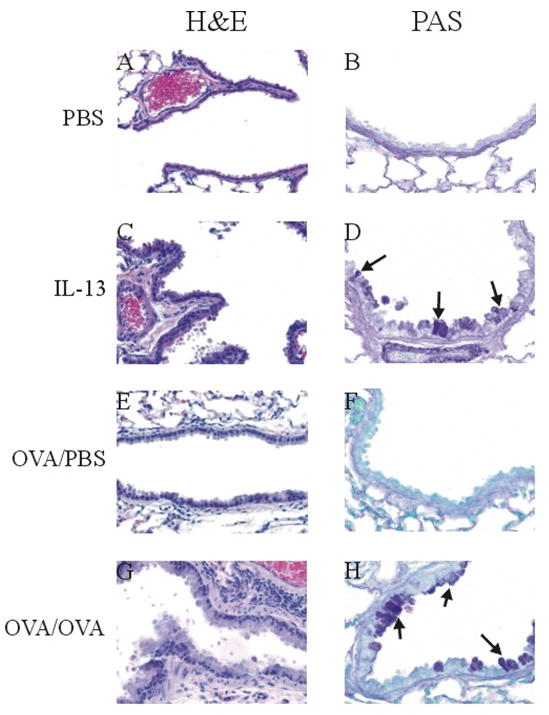
Photomicrographs of paraffin-embedded sections of typical lung tissue 1 day after the last i.n administration of PBS or mIL-13 (1 dose/day × 3 days) and 2 days after the last i.n. challenge of PBS or OVA. (A) and (E) Terminal bronchi stained with H & E show single layer of airway epithelium in PBS control animal (40X); (C) mIL-13 treated mice and (G) OVA/OVA mice show epithelial hyperplasia/metaplasia and thickening of basement membrane in the airway (40X). (B) and (F) Minimal mucus production is observed in small airway of the PBS control animal with PAS staining (60X). (D) mIL-13 treated mice and (H) OVA/OVA mice show goblet cells hyperplasia and increases mucus production with PAS staining (60X). Arrows indicate PAS positive goblet cells.
3.2 OVA sensitization/OVA challenge and mIL-13 instillation result in lung inflammation and reduced lung function
The degree of inflammation that occurred in the lungs of OVA/OVA and mIL-13 treated mice was compared to their respective PBS controls using bronchoalveolar lavage (BAL) total cell counts as well as differential eosinophil counts. Total BAL cell counts were significantly higher in the OVA/OVA group as compared to the OVA/PBS control group, with 87×104 ± 7.1×104 cells from the OVA challenged animals in comparison with 5.4×104 ± 0.9×104 cells from the PBS challenged animals (Figure 2A; p≤0.001; n=10). Similarly, BAL cell counts were increased in mIL-13 dosed mice with 30.5×104 ± 1.6×104 total cells collected in comparison with 8.5×104 ± 1.6×104 cells with PBS control (Figure 2C; p≤0.001; n=10). Additionally, eosinophil cell counts from the BAL were also significantly higher in both models in comparison with the respective PBS controls (Figure 2B and 2D): 563×103 ± 51.6×103 cells with OVA challenged animals in comparison with 3.5×103 ± 0.5×103 cells with PBS challenge (p≤0.001; n=10), and 51.1 × 103 ± 11.2 × 103 cells with mIL-13 treated animals in comparison with 1.7 × 103 ± 0.4 × 103 cells with PBS controls (p≤0.001; n=10). These results confirm that both OVA/OVA and intranasal mIL-13 administration generate pulmonary inflammation and eosinophilia, consistent with previously reported results using similar models [4].
Figure 2. OVA and mIL-13 enhance lung inflammation.
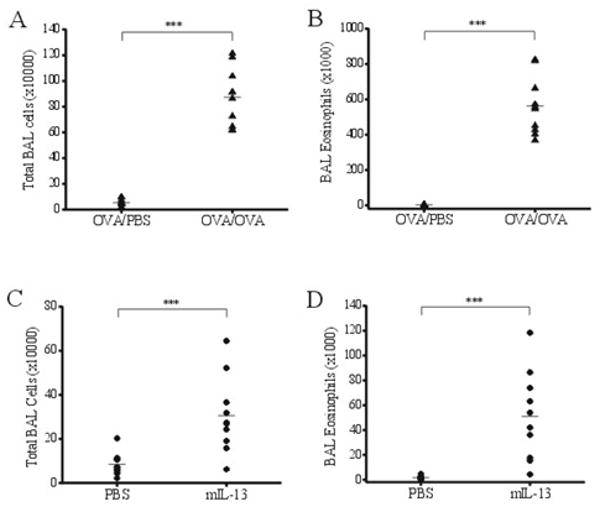
Influx of immune cells to BALF is used as a measure of lung inflammation. (A) OVA/OVA group shows a significant increase in total BAL cells as compared to the control OVA/PBS group (p≤0.01; n=10). (B) Number of BAL eosinophils is significantly elevated in OVA/OVA mice, compared with identically sensitized but PBS challenged animals (p≤0.001; n=10). (C) Similar increase in total BAL cells is also induced by mIL-13 instillation but not by PBS (p≤0.01; n=10). (D) mIL-13 instillation causes significant influx of eosinophils to the lung (p≤0.01; n=10).
To further characterize the antigen and IL-13 induced asthma models, measures of total lung resistance (RL) and dynamic compliance (cDyn) in response to methacholine challenge were used to assess AHR in treated mice as compared to the PBS-treated group. Mice subjected to OVA/OVA challenge as well as i.n. administration of mIL-13 showed a significant increase in RL (OVA/OVA Figure 3A; p≤0.05; n=7 ANOVA; mIL-13 Figure 3B; p≤0.01; n=10 ANOVA) and decrease in cDyn (OVA/OVA Figure 3C; p≤0.05; n=7 ANOVA; IL-13 Figure 3D; p≤0.05; n=10 ANOVA) respectively, as compared to the PBS challenged mice, in response to increasing doses of inhaled methacholine. These data demonstrate that AHR is induced in both OVA/OVA challenged and mIL-13 instilled mice.
Figure 3. AHR in animals treated with OVA or mIL-13.
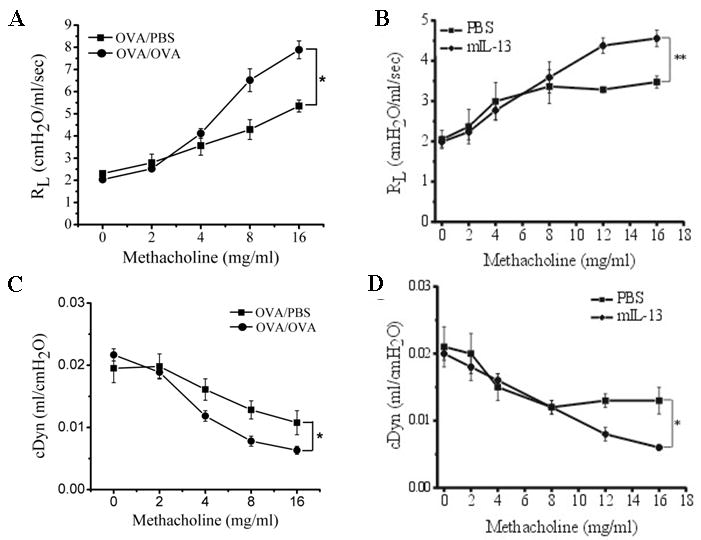
Airway responsiveness to methacholine is used to assess AHR development in animals with different treatments. (A) OVA/OVA animals show significant increase in lung resistance (RL) (p≤0.05; n=7 ANOVA) and (C) a decline in dynamic compliance cDyn (p≤0.0.05; n=7 ANOVA) as compared to the PBS control. (B) mIL-13 treated animals display a significant increase in RL (p≤0.01; n=10 ANOVA) and (D) a decline in cDyn (p≤0.05; n=10 ANOVA) as compared to the PBS control.
3.3 AQP mRNA and protein expression in OVA sensitized (OVA/PBS) and OVA challenged (OVA/OVA) mouse lung
Aquaporin mRNA expression was examined using quantitative real time PCR (Taqman) on lung samples taken from mice sensitized with OVA (PBS), and those sensitized and subsequently challenged with OVA (OVA), and compared to expression in naïve control mice. The levels of AQP1, 4, and 5 mRNA expression were similarly decreased by >75 % in the OVA challenged mice as compared to naïve controls (Figure 4; p≤0.001, n=6 each). AQP3 mRNA expression was not altered in either the PBS- or OVA-treated mice. Examination of protein expression by immunoblotting revealed a concomitant decrease in AQP1 and AQP5 protein abundance in the OVA challenged mice as compared to controls (Figure 5A and Figure 5B), consistent with the observed reduction in mRNA expression. AQP1 expression significantly decreased by 27% as compared to control animals (p≤0.001), whereas AQP5 expression was reduced by ∼64% that of controls (p≤0.0001). However, while no change in AQP3 mRNA expression was observed, AQP3 protein expression decreased by ∼92% (p≤0.05). Furthermore, contrary to the reduction of AQP4 mRNA expression seen in the lungs from OVA challenged mice, AQP4 protein abundance increased by 65% as compared to controls (p≤0.01).
Figure 4. mRNA expression of AQPs 1, 4, and 5 is significantly reduced in response to OVA challenge.
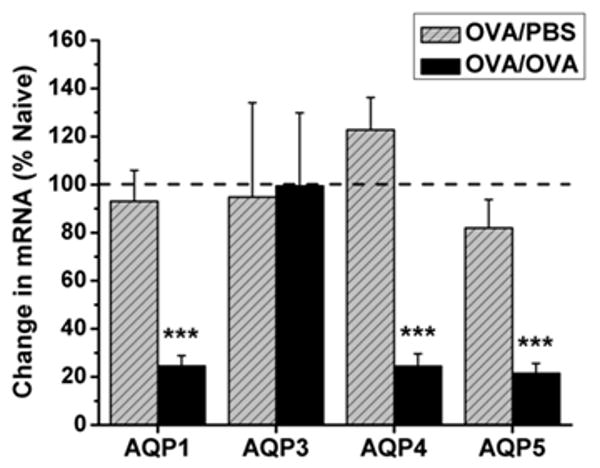
Lungs from mice subjected to OVA sensitization followed by OVA challenge (OVA/OVA; black bars) exhibited a 75% reduction in AQP1 mRNA expression (p≤0.001; n=6) 76% reduction in AQP4 mRNA expression (p≤0.001, n=6), and 79% reduction in AQP5 (p≤0.001, n=6) mRNA expression as compared to naïve animals. Lungs from mice sensitized with OVA and challenged with PBS (OVA/PBS; hatched gray bars) showed no change in AQP expression as compared to naïve controls. All values are reported as a percent of naïve AQP expression (100%).
Figure 5. Reduction in AQPs 1, 3, and 5 protein expression in OVA-challenged (OVA/OVA) mouse lungs.
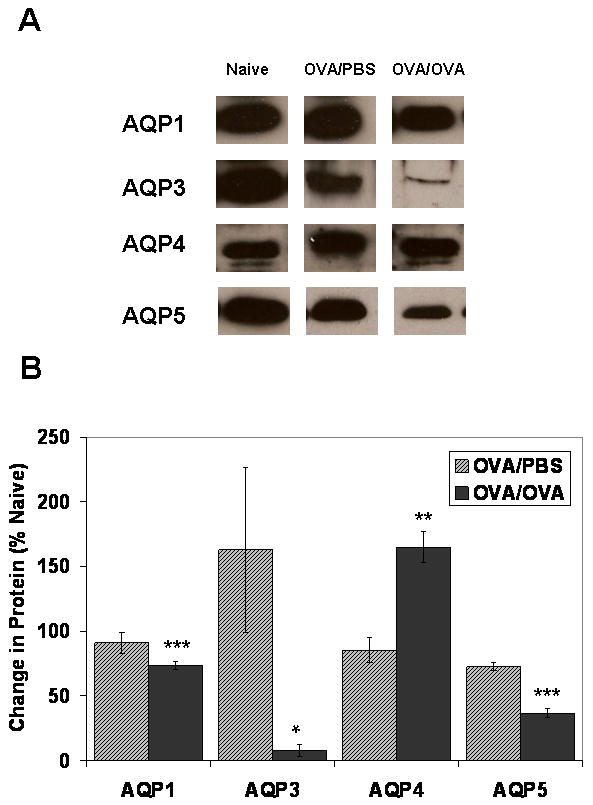
(A) Representative Western blot analysis of AQP1, AQP3, AQP4, and AQP5 on total membrane proteins isolated from OVA sensitized/PBS challenged (OVA/PBS) and OVA sensitized/OVA challenged (OVA/OVA) mouse lung (n=6 each group). (B) Normalized densitometric values for each immunoblot were expressed as fold change vs. naïve controls (n=5). AQP1 expression significantly decreased by 27% as compared to control animals (p≤0.001); AQP3 protein expression decreased by ∼92% (p≤0.05); AQP5 expression was reduced by ∼64% that of controls (p≤0.0001). AQP4 protein expression is increased by 65% as compared to controls (p≤0.01).
3.4 AQP mRNA and protein expression in mIL-13 induced models of asthma
In order to investigate potential kinetic regulation of AQP expression in the acute stages of IL-13 induced asthma, real time quantitative PCR (Taqman) was used to assess relative changes in AQP mRNA in mIL-13 treated lungs following a single dose of 10 μg of IL-13. A significant increase (250%), in total lung AQP3 mRNA was detected within 24 hr (Figure 6A) as a consequence of IL-13 administration to the lung. In contrast, a significant decline in IL-13 treated lung AQP5 mRNA expression level to ∼50% of the PBS treated control lung, was detected within 24 hr (Figure 6A). A partial recovery of both AQP3 and AQP5 mRNA expression, comparable to that seen in the PBS treated animals, was observed by 48 hours in the acute inflammation model. No significant changes in AQP1 and AQP4 mRNA expression were observed at 24 hr. AQP3 and AQP5 mRNA expression was also examined after a 3 day course of IL-13 administration (5 μg) in a chronic exposure model of IL-13 induced asthma. A ∼60% decrease in AQP5 expression was observed 24 hours after the last dose of IL-13 (p<0.01; Figure 6B), whereas as AQP3 expression was not significantly different than PBS controls in the chronic exposure model (Figure 6B). Additional experiments were conducted to examine potential dose-responsiveness of IL-13 induced changes in AQP3 and AQP5 mRNA expression. No significant changes in AQP mRNA expression were observed between 0.5 μg, 5 μg or 10 μg doses of IL-13 (data not shown).
Figure 6. AQP3 mRNA expression level is increased and AQP5 mRNA expression is decreased in response to mIL-13 administration.
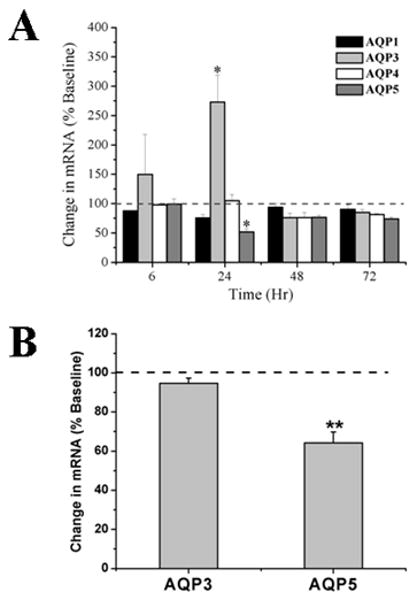
(A) The kinetics of AQP1, AQP2, AQP3, and AQP5 mRNA expression in the lung tissues was evaluated by quantitative real-time PCR in an acute inflammatory induced asthma model following the i.n. administration of a single dose of recombinant IL-13 (10μg), at 6, 24, 48 and 72 hr. AQP expression in each sample was normalized with the housekeeping gene GAPDH and results are reported as percent change relative to the PBS-treated group at time 0 hour. AQP3 and AQP5 mRNA was significantly changed (p≤0.05; n=3) twenty-four hours after IL-13 treatment. (B) AQP3 and AQP5 expression was subsequently examined in a chronic exposure model of IL-13 induced asthma (3 day course of 5 μg per dose). AQP5 mRNA was significantly reduced (p≤0.01; n=5) at 72 hours, whereas AQP3 mRNA was not different than the control PBS treated group.
To assess if the changes in AQP3 and AQP5 mRNA expression are accompanied by changes in protein levels, AQP protein expression in mIL-13 treated mice (3 doses of 5 μg IL-13) relative to PBS controls was examined by immunoblotting. At 72 hours, AQP3 protein was increased by ∼112% (p<0.05) as compared to PBS controls (Figure 7A and 7B). Consistent with IL-13 effects on AQP5 gene expression, densitometric analysis of Western blots of total membrane proteins showed an ∼60% reduction in AQP5 protein expression in lungs of mIL-13 treated mouse lung, as compared to PBS controls (p<0.05; Figure 7A and 7B). IL-13 treatment did not result in statistically significant changes in either AQP1 or AQP4 protein expression. Varying the dose of IL-13 (0.5 and 10 μg; data not shown) did not result in differences in the level of AQP expression as compared to that which was observed with the 5 μg IL-13.
Figure 7. Increase in AQP3 and decline of AQP5 protein expression in lungs from mIL-13 treated mice.
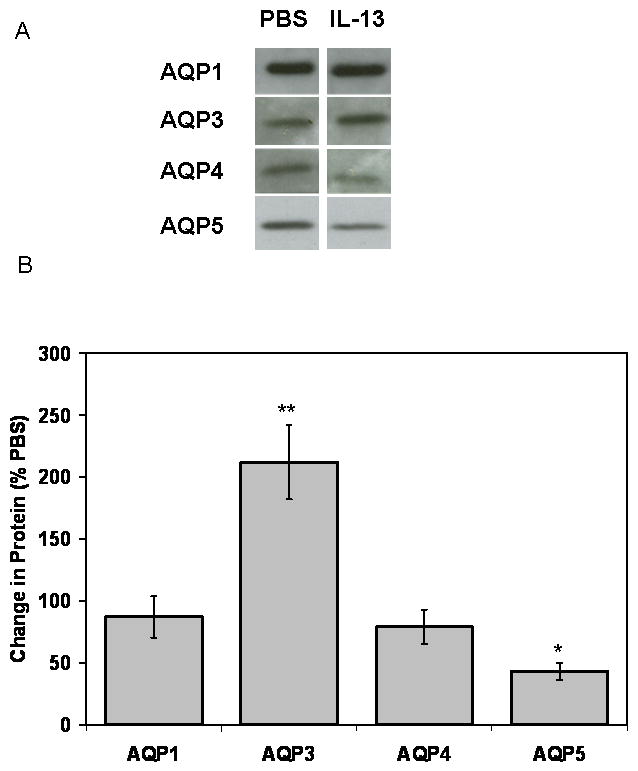
Instillation of mIL-13 leads to a reduction in AQP5 protein expression 72 hours after chronic IL-13 treatment. (A) Representative Western blot analysis of AQP1, AQP3, AQP4 and AQP5 on total membrane proteins isolated from IL-13 treated mice and PBS controls (n=10 each group). (B) Normalized densitometric values for each immunoblot were expressed as fold change vs. PBS controls. Densitometric analysis shows an approximate 112% increase in AQP3 protein expression and an ∼60% reduction in normalized AQP5 expression in mIL-13 treated lungs as compared to the control samples (p≤0.05; n=10). No significant change in AQP1 or AQP4 protein expression was observed.
4. Discussion
In characterizing the potential implications of therapeutic IL-13 blockade, it is important to elucidate the molecular mechanisms of IL-13 induced asthma pathogenesis and regulatory effects on AQPs. The data presented here identify AQPs as potential downstream targets of the inflammatory response to allergen and IL-13 induced asthma. Mice subjected to OVA sensitization/OVA challenge, or mIL-13 instillation, showed characteristic allergic asthma pathology: epithelial thickening, mucus hypersecretion and pulmonary eosinophilia accompanied by a significant increase in methacholine-induced AHR, as compared with control animals. Examination of AQP expression in these models revealed model-specific patterns of gene regulation. In the allergen model, OVA sensitization/OVA challenge resulted in a significant reduction in AQP1 and AQP5 lung mRNA and protein expression. While AQP3 mRNA level remained unchanged, AQP3 protein expression was diminished. To the contrary, AQP4 mRNA expression was significantly reduced, while AQP4 protein expression was in greater abundance as compared to the controls. The mechanisms regulating the observed gene expression are likely to be complex. It is likely that AQP3 mRNA expression levels have returned to normal levels, but were subject to early transcriptional regulation. This interpretation of the allergen model data is supported by the observed acute changes that occur in AQP3 mRNA expression which shows a trend as early as 6 hours, then peak at 24 hours, and subsequently return to normal levels by 48 hours in the IL-13 model. Thus it is possible that the disparity between AQP3 mRNA and protein levels in the OVA model is due to the rate of AQP3 protein translation and/or mRNA or protein turnover which may be differently regulated in part through allergen and/or cytokine (IL-13) mediated pathways. Similarly, it is also important to note that whereas AQP4 mRNA expression was downregulated in the OVA model, AQP4 protein expression was upregulated in the OVA model. These observations may also involve a potential complexity of post-transcriptional and post-translational regulation which remains to be discerned.
In the IL-13 instillation models, the regulation of mRNA expression of these four AQPs was more complex. AQP3 mRNA was acutely up-regulated by IL-13 in the single dosing regimen (10μg/kg), but was not affected in the chronic dosing (3× 5μg/kg) regimen, suggesting that AQP3 mRNA is subject to acute regulatory signals. AQP5 mRNA was consistently down-regulated in both IL-13 dosing regimens. A slightly different pattern of regulation was observed for AQP1, AQP3, and AQP4 in the chronic mIL-13 model, in comparison with the OVA/OVA model. In the mIL-13 chronic model, there was no change in either AQP1 or AQP4 protein expression, whereas AQP3 protein expression was increased approximately two fold as compared to control. Thus, it appears that AQP3 upregulation is an early event, which is followed by AQP3 protein upregulation as AQP3 mRNA abundance returns to steady state levels. Finally, as observed in the OVA model, AQP5 protein expression was also significantly reduced in the mIL-13 instilled mouse lungs.
Collectively, these data show that diminished expression of an apically localized aquaporin, (AQP5), and concomitant upregulation of a basolateral aquaporin (AQP3 or AQP4) are characteristic features of both inducible asthma models. Significant reductions in AQP5 mRNA and protein levels were observed in the allergen as well as the acute and chronic mIL-13 models. Thus, the downregulation of AQP5 may be a regulatory target for allergen-inducible asthma, and in IL-13 mediated AHR. AQP5 is the only known AQP expressed on the apical surface of airway and submucosal gland epithelium [23,24]. The in vivo AQP5 expression regulation reported here, confirms the in vitro observations reported recently where IL-13 treatment was shown to abolish AQP5 expression in primary cultures of human airway epithelial cells [15]. Therefore, the consistent downregulation of AQP5 (both mRNA and protein) in both models implies an important feedback mechanism for the diminished capacity of apical membrane water permeability in these models [15].
Coinciding with the down regulation of AQP5, an increase in AQP4 protein expression occurred in the OVA model, whereas AQP3 protein expression was increased upon IL-13 instillation. Both AQP3 and AQP4 are expressed on the basolateral membrane of epithelium from the upper and lower respiratory tract, respectively [19]. Therefore, the upregulation of an AQP on the basolateral membrane appears to be a consistent event in both models, and might be responsible for increase fluid flux across the epithelium in asthma.
Overall, the data presented here implicate alterations in water channel expression levels with abnormal airway responses in two distinct mouse models of inducible asthma, the OVA sensitized/OVA lung challenge model and a second model in which intranasal instillation of IL-13 leads to asthmatic changes. Both the allergen and cytokine induced asthma models lead to temporary and reversible bronchoconstriction, long term airway remodeling, and airway inflammation, which involve complex gene regulation [37]. These changes are related to alterations in the physiological mechanics of the lung. However, there are some important mechanistic differences between the models which generate different physiological responses. In the OVA model, several proinflammatory cytokines and chemokines are induced in the lung, including IL-13, IL-5, MCP-1, and TARC [38], resulting in an escalation of the immune response. IL-13 is a critical mediator of the respiratory changes in the OVA allergen sensitization/OVA challenge model [7]. In comparison, the mIL-13 instillation model results in an acute and localized cellular response driven by cytokine instillation [4]. Although many of the lung function changes induced by OVA immunization and challenge are IL-13-driven [4], the observed differences in AQP3 protein levels could simply explained by the difference in the time point chosen for analysis (24 hrs after last dose of IL-13 vs 48 hrs after the last OVA challenge). In the rat model of edema, animals treated with thio-urea show an increase in AQP3 protein within 4-10 hours [32], supporting that AQP3 protein are subjected to acute regulation. The rapid regulation of AQP3 might signify an important physiological feedback mechanism to acutely adjust fluid balance across epithelium. Alternatively, the difference in immune responses in these two models might explain the differential regulation of AQP3 by multiple cytokines. Down-regulation of AQP3 expression has been linked to mucus production in endobronchial biopsies of patients with bronchiectasis [39]. Similarly, we observed mucus production in both the OVA and IL-13 instillation models. Our observation of reduced AQP3 protein in the lungs of OVA-treated animals may reflects a change in cellular phenotype of epithelial cells from absorptive to secretory under prolong inflammatory condition. Future studies will explore the relationships of AQP3 expression and mucus production in the OVA model.
Regulatory alterations in the expression of aquaporin (AQP) mRNA and protein have been further documented in various models of lung injury and disease. A reduction in AQP1 protein expression was observed in response to lipopolysaccharide-induced acute lung injury [31]. In a thio-urea induced pulmonary edema model, AQP3 mRNA and protein levels were increased within 4 hours post-treatment, whereas AQP1, AQP4 and AQP5 mRNA levels were significantly reduced [32]. Moreover, AQP5 mRNA and protein expression were significantly reduced in association with pulmonary inflammation and edema resulting from adenoviral infection [28], bleomycin-induced lung fibrosis [33], and hyperoxia-induced lung injury [40]. Similarly, direct intratracheal instillation of pro-inflammatory agent phospholipase A2 caused a reduction in AQP5 expression in rat [29]. Furthermore, AQP5 mRNA and protein expression in a murine lung epithelial cell line (MLE-12) was also inhibited by the pro-inflammatory cytokine TNF-α through a nuclear-factor (NF) κB-dependent pathway [30]. Taken together, these published studies indicate that AQPs serve as common regulatory targets for altered gene expression during lung infection, inflammation, acute and chronic injury, airway remodeling and under pathophysiological conditions of altered lung function. The data presented here suggest that alterations of AQP3 and AQP5 gene expression may be a relatively early event in the lung's response to mIL-13 induced airway pathology. Though the physiological implications of this altered regulation are not currently defined, the identification of AQPs among the genes that are subject to differential gene expression in response to allergen and IL-13 induced asthma, indicates that AQPs may be an important new functional category of genes involved in this response. These data also suggest that further evaluation of AQP gene expression, post-translational regulation and function in other experimental models of asthma is warranted.
Acknowledgments
The authors would like to thank Delia Demers, Charlene DeClercq and Paul Wu (Wyeth research), and Julie Carroll, Anne Klapheke and Emily Kitchin (University Dayton undergraduate students) for technical assistance. This work was funded in part by NIH/NHLBI AREA research grant 1 R15 HL77290-01 to CMK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bochner BS, Busse WW. Allergy and Asthma. J Allergy Clin Immunol. 2005;115:953–9. doi: 10.1016/j.jaci.2005.02.032. [DOI] [PubMed] [Google Scholar]
- 2.Hamelmann E, Takeda K, Oshiba A, Gelfand EW. Role of IgE in the development of allergic airway inflammation and airway hyperresponsiveness--A murine model. Allergy. 1999;54:297–305. doi: 10.1034/j.1398-9995.1999.00085.x. [DOI] [PubMed] [Google Scholar]
- 3.Townley RG, Horiba M. Airway hyperrsponsiveness: A story of mice and men and cytokines. Clin Rev Allergy Immun. 2003;24:85–109. doi: 10.1385/CRIAI:24:1:85. [DOI] [PubMed] [Google Scholar]
- 4.Water DM, McIntire JJ, Berry G, McKenzie ANJ, Donaldson DD, DeKruyff RH, et al. Critical Role for IL-13 in the Development of Allergen-Induced Airway Hyperreactivity. J Immunol. 2001;167:4668–75. doi: 10.4049/jimmunol.167.8.4668. [DOI] [PubMed] [Google Scholar]
- 5.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, et al. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–61. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 6.Eum SY, Maghni K, Tolloczko B, Eidelman DH, Martin JG. IL-13 may mediate allergen-induced hyperresponsiveness independently of IL-5 or eotaxin by effects on airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2005;288:L576–84. doi: 10.1152/ajplung.00380.2003. [DOI] [PubMed] [Google Scholar]
- 7.Grunig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–63. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halapi E, Hakonarson H. Recent development in genomic and proteomic research for asthma. Curr Opin Pulm Med. 2003;10:22–30. doi: 10.1097/00063198-200401000-00005. [DOI] [PubMed] [Google Scholar]
- 9.Hoffjan S, Ober C. Present status on the genetic studies of asthma. Curr Opin Immunol. 2002;14:709–17. doi: 10.1016/s0952-7915(02)00393-x. [DOI] [PubMed] [Google Scholar]
- 10.Vercelli D. Genetics of IL-13 and functional relevance of IL-13 variants. Curr Opin Allergy Clin Immunol. 2002;2(5):389–93. doi: 10.1097/00130832-200210000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Wills-Karp M. The gene encoding interleukin-13: a susceptibility locus for asthma and related traits. Respir Res. 2000;1:19–23. doi: 10.1186/rr7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang G, Li L, Volk A, Emmell E, Petley T, Giles-Komar J, et al. Therapeutic dosing with anti-interleukin-13 monoclonal antibody inhibits asthma progression in mice. J Pharmacol Exp Ther. 2005;313(1):8–15. doi: 10.1124/jpet.104.076133. [DOI] [PubMed] [Google Scholar]
- 13.Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z, et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nature Medicine. 2002;8:885–9. doi: 10.1038/nm734. [DOI] [PubMed] [Google Scholar]
- 14.Danahay H, Atherton H, Jones G, Bridges RJ, Poll RT. Interleukin-13 induces a hypersecretory ion transport phenotype in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2002;282:L226–36. doi: 10.1152/ajplung.00311.2001. [DOI] [PubMed] [Google Scholar]
- 15.Skowron-zwarg M, Boland S, Caruso N, Coraux C, Marano F, Tournier F. Interleukin-13 interferes with CFTR and AQP5 expression and localization during human airway epithelial cell differentiation. Exp Cell Res. 2007;313:2695–702. doi: 10.1016/j.yexcr.2007.02.035. [DOI] [PubMed] [Google Scholar]
- 16.Krane CM, Goldstein D. Comparative functional analysis of aquaporins/glyceroporins in mammals and anurans. Mamm Genome. 2007;18:452–62. doi: 10.1007/s00335-007-9041-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krane CM, Kishore BK. Aquaporins: The membrane water channels of the biological world. Biologist. 2003;50(2):81–6. [Google Scholar]
- 18.King LS, Nielsen S, Agre P. Aquaporin-1 water channel protein in lung: ontogeny, steroid induced expression, and distribution in rat. J Clin Invest. 1997;97:2183–91. doi: 10.1172/JCI118659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nielsen S, King LS, Christensen BM, Agre P. Aquaporins in complex tissues II: Subcellular distribution in respiratory and glandular tissues of rat. Am J Physiol. 1997;273:C1549–61. doi: 10.1152/ajpcell.1997.273.5.C1549. [DOI] [PubMed] [Google Scholar]
- 20.Song Y, Jayaraman S, Yang B, Matthay MA, Verkman AS. Role of aquaporin water channels in airway fluid transport, humidification, and surface liquid hydration. J Gen Physiol. 2001;117(6):573–582. doi: 10.1085/jgp.117.6.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bai C, Fukuda N, Song Y, Ma T, Matthay MA, Verkman AS. Lung fluid transport in aquaporin-1 and aquaproin-4 knockout mice. J Clin Invest. 1999;103:555–61. doi: 10.1172/JCI4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.King LS, Nielsen S, Agre P, Brown RH. Decreased pulmonary vascular permeability in aquaporin-1-null humans. Proc Natl Acad Sci USA. 2002;99(2):1059–63. doi: 10.1073/pnas.022626499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krane CM, Fortner C, Hand AE, McGraw DW, Lorenz JN, Wert SE, et al. Proc Natl Acad Sci USA. 24. Vol. 98. 2001. Aquaporin 5 Deficient Mouse Lungs are Hyperresponsive to Cholinergic-Stimulation; pp. 14114–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma T, Fukuda N, Song Y, Matthay MA, Verkman AS. Lung fluid transport in aquaporin-5 knockout mice. J Clin Invest. 2000;105(1):93–100. doi: 10.1172/JCI8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Z, Wang X, Gao L, Bai L, Zhu R, Bai C. Regulation of MUC5AC mucin secretion by depletion of AQP5 in SPC-A1 cells. Biochem Biophys Res Comm. 2006;342:775–781. doi: 10.1016/j.bbrc.2006.01.103. [DOI] [PubMed] [Google Scholar]
- 26.Chen Z, Zhu R, Bai L, Bai C. Downregulation of aquaporin 5 induced by vector-based short hairpin RNA and its effect on MUC5AC gene expression in human airway submucosal gland cells. Respir Physiol Neurobiol. 2006;152:197–203. doi: 10.1016/j.resp.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 27.Wang K, Feng YL, Wen FQ, Chen XR, Ou XM, Xu D, et al. Decreased expression of human aquaporin-5 correlated with mucus overproduction in airways of chronic obstructive pulmonary disease. Acta Pharm Sinica. 2007;28(8):1166–74. doi: 10.1111/j.1745-7254.2007.00608.x. [DOI] [PubMed] [Google Scholar]
- 28.Towne JE, Harrod KS, Krane CM, Menon AG. Decreased expression of aquaporin (AQP1) and AQP5 in mouse lung after acute viral infection. Am J Resp Cell Mol Biol. 2000;22(1):34–44. doi: 10.1165/ajrcmb.22.1.3818. [DOI] [PubMed] [Google Scholar]
- 29.Cher DN, Armugam A, Lachumanan R, Coghlan MW, Jeyaseelan K. Pulmonary inflammation and edema induced by phospholipase A2: Global gene analysis and effects on aquaporins and Na+/K+-ATPase. J Biol Chem. 2003;278(33):31352–60. doi: 10.1074/jbc.M302446200. [DOI] [PubMed] [Google Scholar]
- 30.Towne JE, Krane CM, Bachurski CJ, Menon AG. Tumor necrosis factor-alpha inhibits aquaporin expression in mouse lung epithelial cells. J Biol Chem. 2001;276(22):18657–64. doi: 10.1074/jbc.M100322200. [DOI] [PubMed] [Google Scholar]
- 31.Su X, Song Y, Jiang J, Bai C. The role of aquaporin-1 (AQP1) expression in a murine model of lipopolysaccharide-induced acute lung injury. Respir Physiol Neurobiol. 2004;142:1–11. doi: 10.1016/j.resp.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Sato K, Kobayashi K, Aida S, Tamai S. Bronchiolar expression of aquaporin-3 (AQP3) in rat lung and its dynamics in pulmonary oedema. Pflugers Arch Eur J Physiol. 2004;449:106–14. doi: 10.1007/s00424-004-1310-5. [DOI] [PubMed] [Google Scholar]
- 33.Gabazza EC, Kasper M, Ohta K, Keane M, D'Alessandro-Gabazza C, Fujimoto H, et al. Decreased expression of aquaporin-5 in bleomycin-induced lung fibrosis in the mouse. Pathol Int. 2004;54(10):774–80. doi: 10.1111/j.1440-1827.2004.01754.x. [DOI] [PubMed] [Google Scholar]
- 34.Jiao A, Fish SC, Mason LE, Schelling SH, Goldman SJ, Williams CMM. Endothelial selectins drive allergic responses but protect against silica-induced lung fibrosis and inflammation. Ann Allergy Asthma Immunol. 2007;98(1):83–8. doi: 10.1016/S1081-1206(10)60864-2. [DOI] [PubMed] [Google Scholar]
- 35.Krane CM, Towne JE, Menon AG. Cloning and characterization of murine Aqp5: evidence for a conserved aquaporin gene cluster. Mamm Genome. 1999;10:498–505. doi: 10.1007/s003359901030. [DOI] [PubMed] [Google Scholar]
- 36.Williams CMM, Galli SJ. Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in Mice. J Exp Med. 2000;192:455–62. doi: 10.1084/jem.192.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Follettie MT, Ellis DK, Donaldson DD, Hill AA, Diesl V, DeClercq C, et al. Gene expression analysis in a murine model of allergic asthma reveals overlapping disease and therapy dependent pathways in the lung. Pharmacogenet J. 2006;6:141–152. doi: 10.1038/sj.tpj.6500357. [DOI] [PubMed] [Google Scholar]
- 38.Kearley J, Robinson DS, Lloyd CM. CD4+CD25+ regulatory T cells reverse established allergic airway inflammation and prevent airway remodeling. J Allergy Clin Immunol. 2008;122(3):617–24. doi: 10.1016/j.jaci.2008.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsang KW, Leung JC, Tipoe GL, Leung R, Yan C, Ooi GC, et al. Down-regulation of aquaporin 3 in bronchiectatic airways in vivo. Respir Med. 2003;97(1):59–64. doi: 10.1053/rmed.2002.1413. [DOI] [PubMed] [Google Scholar]