

Abstract
This study examines the adhesion, spreading, and migration of human umbilical vein endothelial cells on crosslinked films of artificial extracellular matrix (aECM) proteins. The aECM proteins described here were designed for application in small-diameter grafts and are composed of elastin-like structural repeats and fibronectin cell-binding domains. aECM-RGD contains the RGD sequence derived from fibronectin; the negative control protein aECM-RDG contains a scrambled cell-binding domain. Covalent attachment of poly(ethylene glycol) (PEG) to aECM substrates reduced nonspecific cell adhesion to aECM-RDG-PEG but did not preclude sequence-specific adhesion of endothelial cells to aECM-RGD-PEG. Variation in ligand density was accomplished by mixing aECM-RGD-PEG and aECM-RDG-PEG prior to crosslinking. Increasing the density of RGD domains in crosslinked films resulted in more robust cell adhesion and spreading but did not affect cell migration speed. Control of cell-binding domain density in aECM proteins can thus be used to modulate cell adhesion and spreading, and will serve as an important design tool as these materials are developed further for use in surgery, tissue engineering and regenerative medicine.
Introduction
A central goal of research in tissue engineering and regenerative medicine is the design of biomaterials that can be used to control critical aspects of cellular behavior. Such materials might guide seeded cells toward the phenotypes and architectures needed to restore tissue function, or induce cells from surrounding tissue to infiltrate implanted matrices. An important step toward these goals has been taken through grafting of RGD and other cell adhesion sequences to polymeric matrices.1 Cell proliferation, adhesion, spreading, migration, and differentiation are influenced by the overall density of matrix-bound RGD peptides2-5 as well as by nanoscale ligand clustering.6-9 Biochemical gradients have also been shown to govern haptotaxis, cell distribution, and cell alignment.10-12
Genetic engineering of proteins offers a straightforward route to materials that exhibit some of the most important chemical and physical properties of the extracellular matrix (ECM). Structural and functional domains derived from ECM proteins can be incorporated easily into engineered proteins, and many artificial protein-based materials have been developed for use in tissue engineering applications.13-21
The artificial extracellular matrix (aECM) proteins described in this work were designed for use in small-diameter vascular grafts.22-30 Although poly(ethylene terephthalate) and expanded poly(tetrafluoroethylene) have been successful in large-diameter grafts, their use in small-diameter grafts has been problematic.31-34 Synthetic grafts are thought to fail because of (i) the absence of a confluent endothelial layer and (ii) a compliance mismatch between the graft and surrounding tissue that leads to intimal hyperplasia and thrombosis. To address these issues, aECM proteins were designed with elastin-like repeats to confer elastomeric properties and with cell-binding domains to promote endothelialization (Figure 1). By crosslinking through lysine residues within the elastin-like domains, we have varied the elastic moduli of aECM protein films from ca. 0.1 to 1.0 MPa, an appropriate range for many soft-tissue applications.23 Furthermore, the design allows for facile incorporation of different cell-binding domains, and previous work has elucidated cellular responses to the fibronectin-derived RGD and CS5 domains in adsorbed aECM proteins.24, 26, 28, 29 aECM proteins thus allow good control over both biophysical and biochemical cues; however, because many applications of aECM proteins will require the use of crosslinked matrices, it is essential to understand cellular responses to crosslinked aECM films.
Figure 1.

Amino acid sequences of aECM proteins. aECM-RGD contains the RGD cell-binding domain. aECM-RDG is a negative control protein in which the cell-binding domain has been scrambled. Both proteins contain a T7 tag, a heptahistidine tag, an enterokinase cleavage site, and elastin-like domains with lysine residues that serve as crosslinking sites.
In this study, we present a method for preparing crosslinked aECM films suitable for cell studies, and we demonstrate that cells recognize the RGD sequence within crosslinked, PEGylated protein films. We vary the density of adhesion ligands by mixing aECM-RGD and aECM-RDG prior to crosslinking, and we examine the role of RGD density in modulating the adhesion, spreading, and migration of human umbilical vein endothelial cells.
Materials and Methods
Protein Expression and Purification
aECM-RGD and aECM-RDG were expressed in Escherichia coli and purified via temperature cycling as described previously.26 The purity and molecular weight of the proteins were verified by SDS-PAGE gels, Western blots with an anti-T7 tag horseradish peroxidase-conjugated antibody (Novagen, San Diego, CA), amino acid analysis, and matrix-assisted laser desorption ionization-mass spectrometry (MALDI-MS).
Cell Culture
Human umbilical vein endothelial cells (HUVECs) were purchased from Cambrex BioSciences (Walkersville, MD) and maintained in a 37°C, 5% CO2 humidified environmental chamber. Cells were grown in endothelial growth medium-2 (EGM-2, 2% serum, Cambrex BioSciences); passages 2–7 were used. Near confluent HUVEC cultures were non-enzymatically detached by treatment with 0.61 mM EDTA.
Substrate Preparation
A 12 mm base-cleaned glass coverslip was covered with 20 μL of an aqueous aECM protein solution (3.6 mg/mL) containing the bifunctional crosslinker bis(sulfosuccinimidyl) suberate (BS3, Pierce Biotechnology, Rockford, IL). The molar ratio of activated ester to protein-bound amine was 1:4. To slow the rate of crosslinking, protein solutions and coverslips were kept on ice. Coverslips were spin-coated at 4000 rpm for 45 s on a Specialty Coating Systems model P-6000 spin coater and stored overnight in a humidified chamber at 4°C. To covalently attach polyethylene glycol (PEG) to aECM films, 50 μL of a 50 mM methoxy-PEG-succinimidyl propionate (mPEG-SPA, MW 5000, Nektar Therapeutics, San Carlos, CA) solution in water was placed on parafilm and the coverslips were placed protein-side down in the PEG solution. The coverslips were incubated for 2 h at room temperature, rinsed three times with water, sterilized with 95% ethanol for 1 h, and rinsed three times with water.
Substrate Characterization
To ensure uniformity, crosslinked protein films were examined via fluorescence microscopy. Films were blocked with 10% bovine serum albumin (BSA, fraction V, Sigma, St. Louis, MO) and incubated with an anti-T7 monoclonal antibody (Novagen) at a dilution of 1:2000 at room temperature overnight. After three water rinses, films were incubated with a Cy2-conjugated affinity-purified goat anti-mouse antibody (Chemicon, Temecula, CA) at a dilution of 1:10 for 1 h at room temperature. Coverslips were rinsed four times with water and examined on a Zeiss Axioplan II fluorescence microscope (Thornwood, NY) equipped with a monochrome Axiocam and AxioVision 3.1 software.
Crosslinked protein films were scratched with a razor blade to reveal the underlying glass substrate. Height was measured by imaging over the scratch with an AutoProbe M5 atomic force microscope (Park Scientific Instruments, Woodbury, NY) in constant-force contact mode, using pyramidal tips (0.58 N/m, Veeco DNP-S).
An M-probe surface spectrometer (Thermo VG Scientific, Waltham, MA) with monochromatic Al Kα x-rays (1486.6 eV) was used for x-ray photoelectron spectroscopy (XPS). X-rays at an incident angle of 35° from the surface illuminated a 250 × 1000 μm elliptical spot. A charge neutralizer was used because the samples were non-conductive. Ten detailed peak scans were collected with an instrument resolution of 1 eV and a step size of 0.1 eV. The ESCA 2000 analysis software v. 102.04 (Service Physics, Bend, OR) was used for peak integration. Three spots were analyzed on each substrate, and at least three substrates were examined for each condition.
Cell viability on crosslinked aECM proteins was measured by monitoring the cleavage of the tetrazolium salt WST-1 (Roche, Penzberg, Germany). After 24 h in serum-containing medium (EGM-2), there were no differences in viability between cells grown on aECM-RGD-PEG and those grown on fibronectin. The viability of cells grown on the negative control protein aECM-RDG-PEG was 43 ± 10% of the viability of cells grown on fibronectin. Three independent experiments were performed, each in triplicate.
Cell Resistance to Detachment
Experiments to measure cell resistance to detachment by normal forces were adapted from a previously described method.26 Briefly, a fibronectin solution (10 μg/mL) was adsorbed onto control wells in a black 24-well Visiplate (Perkin Elmer, Wellesley, MA) at 4°C overnight. All wells were blocked with 0.2% heat-inactivated BSA. Vacuum grease was applied to the undersides of dry coverslips to adhere them securely to the 24-well plate. After cells were fluorescently labeled with calcein acetoxymethyl ester (Molecular Probes, Carlsbad, CA), 1 mL of a cell suspension (2.67 × 105 cells/mL in serum-free medium) was added to each well and incubated for 30 min on crosslinked protein films. The relative numbers of cells were measured by fluorescence (excitation at 485 nm; emission at 538 nm). Each well was filled with 1 mL of Percoll (21% w/w in PBS, Sigma), and the plates were centrifuged upright for 10 min at 100g. Because Percoll has a higher density (1.123 g/ml) than the cells (∼1.07 g/ml), a buoyancy force is exerted on the cells.8, 35 By using Archimedes' theorem, we estimated the detachment force applied to each cell to be 26 pN. The liquid and non-adherent cells were removed, and the remaining cells were quantified by fluorescence. The fraction of cells retained in each well was calculated by dividing the fluorescence of the remaining cells by the fluorescence of the cells before centrifugation. A cell adhesion index (CAI) was calculated as the fraction of cells retained in a test well divided by the fraction of cells retained on fibronectin subjected to 1g (0.26 pN). Error bars represent standard deviations of three or more independent experiments, each performed in triplicate. A one-tailed two-sample t-test that assumed equal variances was applied to determine statistical significance.
Cell Spreading
HUVECs in serum-free medium were seeded at a concentration of 4.8 × 104 cells per well in a 6-well plate. Cells on crosslinked aECM films were imaged at 15 min intervals by using a 10× phase contrast objective on a Nikon Eclipse TE300 inverted microscope. Images were manually scored for the number of spread (i.e., dark) versus non-spread (i.e., bright and refractive) cells. Three independent experiments were performed.
Cell Migration
Cells in EGM-2 were added to each well of a 6-well plate at densities of 1.6 × 104 to 6 × 104 cells per well. The cells were allowed to adhere for 2 h, after which they were imaged using a Nikon Eclipse TE300 inverted phase contrast microscope surrounded by a 37°C incubation box. To maintain physiological pH, a humidified gas mixture of 5% CO2, 20% O2, and 75% N2 was continuously bubbled through the 6-well plate. Teflon tape was used to seal the plate and a thin layer of mineral oil (embryo-tested, Sigma) was added to the top of the medium to prevent evaporation. Cells at various locations were recorded every 15 min for 24 h using a motorized stage and the MetaMorph Basic Imaging Software (Molecular Devices, Downingtown, PA).
The image sequences were imported into ImageJ 1.30v software (U.S. National Institutes of Health, Bethesda, MD) and saved as Quicktime files. Dynamic Image Analysis Software (DIAS) 3.2 (Solltech, Oakdale, IA) was used to analyze the Quicktime movies. The images were thresholded to automatically trace cells. The outlines were then manually edited to erase incorrectly outlined areas and to adjust some tracings. Only cells that were well-spread, isolated, and tracked for at least 8 h were included in the analysis. At least three independent experiments were performed with a minimum of 80 cells tracked in total for each substrate.
Results and Discussion
Substrate Characterization
Protein films were prepared with a molar ratio of activated ester to protein-bound amine of 1:4. Under these conditions, quantitative intermolecular aminolysis of BS3 would yield 4.25 crosslink sites per protein. Several molar ratios were tested to ensure that crosslinking produced coherent films containing residual amine sites for subsequent coupling to poly(ethyleneglycol). Film thickness was typically 8–10 nm as measured by atomic force microscopy (AFM) after dehydration. Fluorescence imaging was used to verify film uniformity. When washed with water or with 0.05% sodium dodecyl sulfate (SDS), crosslinked protein films remained uniform and coherent; spin-coated protein films prepared in the absence of BS3 did not.
PEGylation of Crosslinked aECM Films
Grafting of poly(ethyleneglycol) (PEG) was used to reduce nonspecific adhesion of cells to crosslinked aECM films. The extent of PEGylation was assessed by X-ray photoelectron spectroscopy (XPS) (see Supporting Information). After incubation of films with mPEG-SPA (MW 5000) for 2 h, XPS indicated an average grafting density of ca. 1.8 PEG molecules per protein chain. Longer reaction times yielded no further increase in grafting density. When a PEG variant with no reactive ends (MW 4600) was used, the amount of PEG detected on the surface was equivalent to ca. 0.2 PEG molecules associated with each protein strand. Covalent attachment through lysine residues is necessary for effective PEGylation of aECM films.
We showed previously that cell adhesion to adsorbed (uncrosslinked) aECM proteins is dependent upon presentation of authentic cell-binding domains.26 In contrast, in the absence of PEGylation, crosslinked protein films exhibit significant nonspecific adhesion. HUVECs were incubated on crosslinked substrates for 30 min and then subjected to a 26 pN normal detachment force for 10 min. On films without PEG modification, aECM-RDG retained nearly as many cells as aECM-RGD (Figure 2). On PEGylated films, nearly 4-fold more cells were retained on aECM-RGD as compared to aECM-RDG. PEGylated films are designated aECM-RGD-PEG and aECM-RDG-PEG, respectively. The high cell adhesion index (CAI) for aECM-RDG (76.9 ± 6.0%) indicates significant nonspecific adhesion in the absence of an authentic cell-binding domain. The CAI was reduced to 21.2 ± 7.8% when cells were seeded on aECM-RDG-PEG. All further studies were conducted with PEGylated crosslinked films.
Figure 2.

PEGylation reduces nonspecific cell adhesion on aECM films. See text for definition of cell adhesion index. Data represent three experiments, each performed in triplicate. Error bars represent one standard deviation. * represents p < 0.01.
HUVEC Resistance to Detachment Forces
Crosslinked films prepared from mixtures of aECM-RGD-PEG and aECM-RDG-PEG were used to determine the role of RGD density in controlling cell adhesion. For films containing 0–1% aECM-RGD-PEG, the CAI was low (16–21%), but significantly higher than the CAI on the negative control protein BSA (2.0 ± 3.0%). As the fraction of aECM-RGD-PEG was raised from 0.05 to 1.0, the CAI increased from 51.1 ± 28.8% to 96.0 ± 21.3%. Films containing aECM-RGD-PEG fractions greater than 0.25 were not statistically different (p ≤ 0.5) from fibronectin controls (CAI 91.3 ± 27.0%).
Based on the density of elastin (1.31 g/mL)36 and a polymer weight fraction of 0.56,27 the concentration of cell-binding domains in a 10 nm-thick near-surface layer of a hydrated, crosslinked aECM-RGD film is estimated as 3.8 × 105 per μm2 (CBD/μm2). Previous studies have shown that ligand densities ranging from 102 to 104 CBD/μm2 are adequate to support cell adhesion.8, 37, 38 The results shown in Figure 3 suggest that roughly 104 CBD/μm2 are required for significant adhesion to aECM-RGD-PEG. Houseman and Mrksich have examined the role of peptide microenvironment in modulating cell adhesion; for RGD peptides appended to oligo(ethylene glycol) monolayers, cell adhesion was found to be increasingly sensitive to ligand density as the oligomers were extended from three EO units to six.38 The PEG chains used in the present work are much longer (> 100 units), and our assumption of an accessible thickness of 10 nm is arbitrary. The effective ligand densities in aECM-RGD-PEG films therefore may be significantly lower than our estimates; nevertheless, it appears that the ligand density required for cell adhesion on such films falls near the high end of the range of values reported previously.
Figure 3.

HUVEC resistance to detachment forces. By raising the concentration of aECM-RGD-PEG, the number of adherent cells can be increased. Crosslinked films were made by mixing aECM-RGD-PEG and aECM-RDG-PEG. Data represent three experiments, each performed in triplicate. Error bars represent one standard deviation.
HUVEC Spreading on Crosslinked aECM Films
Cell spreading on crosslinked aECM films is dependent on the density of RGD domains. HUVECs plated on PEGylated films were monitored at 15 min intervals by phase contrast microscopy and categorized as dark (spread) or bright (rounded). On films containing 50–100% aECM-RGD-PEG, half of the cells spread within 60–90 min (Figure 4). When compared to the positive control, fibronectin, the percentage of well-spread cells on 100% aECM-RGD-PEG was statistically lower at all time points (p-values < 0.03). Reducing the aECM-RGD-PEG content resulted in significant reduction in the extent of cell spreading; fewer than 1% of cells spread on aECM films containing no authentic cell-binding sequences. Comparison of the percentages of well-spread cells on 50-100% aECM-RGD-PEG films versus those on 0% aECM-RGD-PEG gave p-values < 0.05 at 30-120 min. Similar comparisons for cells on 25% and 10% aECM-RGD-PEG films gave p-values < 0.05 at time points greater than or equal to 45 or 105 min, respectively. No significant differences between 5% aECM-RGD-PEG and negative control films were observed.
Figure 4.
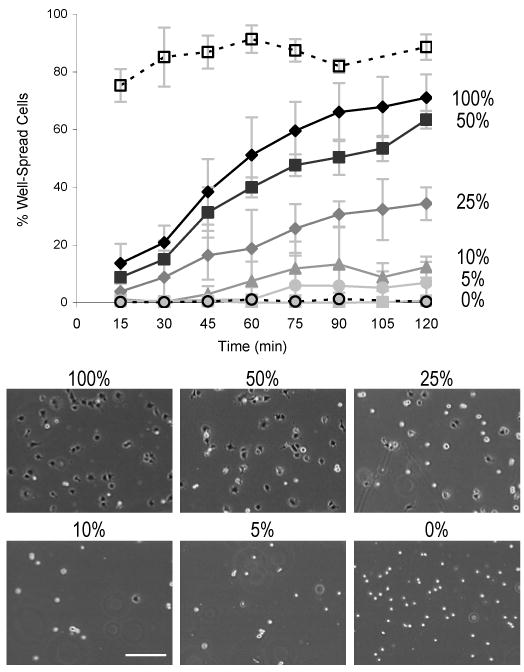
HUVEC spreading on crosslinked aECM films. Cell spreading kinetics can be modulated by varying the density of adhesion ligands. Fibronectin (□) and BSA controls (○) are represented by dotted lines. The percentages to the right of the traces indicate the percentage of aECM-RGD-PEG in each film. Phase contrast micrographs show that substrates with higher aECM-RGD-PEG content have a larger number of dark, well-spread cells and fewer bright, rounded cells after 90 min of incubation. Scale bar represents 200 μm. Data represent three experiments. Error bars represent one standard deviation.
HUVEC Migration Rates
A phase contrast microscope outfitted with a motorized stage and an environmental chamber was used to track cells every 15 min for a minimum of 8 h. Migration rate was calculated as the distance traveled divided by the tracking time. Measured HUVEC speeds ranged from 0.45 to 0.55 μm/min but did not depend on the concentration of cell-binding ligand. Published values for endothelial cells migrating on fibronectin or RGD peptides range from 0.25 to 0.67 μm/min;39-43 the migration speeds measured in this study fall within the upper half of reported values. Although previous studies have shown that cell migration rates can, under some circumstances, vary in a biphasic manner with substrate adhesiveness,9, 42, 44 cells on aECM-RGD-PEG films did not exhibit such behavior.
Conclusion
We report a simple method for making crosslinked aECM films suitable for cell studies. By mixing otherwise identical proteins that contain authentic and scrambled cell-adhesion ligands, we fix all of the physical and chemical properties of the films while varying only their biological information content. Crosslinked, PEGylated protein substrates show low levels of nonspecific cell adhesion but retain the ability to bind cells in a sequence-specific manner. Moreover, by varying the concentration of authentic cell-binding domains in the protein films, we were able to modulate cell adhesion and spreading, but not cell migration rate. Current work addresses the importance of ligand presentation and mechanical properties in modulating cellular responses to aECM proteins and the relevance of aECM proteins to clinical soft-tissue engineering.
Supplementary Material
Figure 5.

HUVEC migration on aECM substrates. Cell speed on aECM films does not correlate with aECM-RGD-PEG concentration. At least 80 cells were tracked every 15 min for at least 8 h. Error bars represent one standard deviation.
Acknowledgments
We thank Paul Nowatzki for performing the AFM study, Elizabeth Jones and David Koos for advice on cell migration studies, Marissa Mock for helpful discussions on spin-coating protein films, Scott Fraser for help with fluorescence microscopy, and the Molecular Materials Research Center of the Caltech Beckman Institute for help with XPS. This work was supported by a Whitaker graduate fellowship to J.C.L., by NIH grant EB1971, and by the NSF Center for the Science and Engineering of Materials at the California Institute of Technology.
Footnotes
Supporting Information Available. Supporting information includes 1) AFM thickness measurement on dry, crosslinked aECM-RGD films and 2) XPS data on crosslinked aECM-RGD, aECM-RGD-PEG, and aECM-RGD films containing PEG MW 4600 with no reactive ends. This information is available free of charge via the Internet at http://pubs.acs.org.
Bibliography
- 1.For review of RGD biomaterials: Hersel U, Dahmen C, Kessler H. Biomaterials. 2003;24:4385–4415. doi: 10.1016/s0142-9612(03)00343-0.
- 2.Chua PH, Neoh KG, Kang ET, Wang W. Biomaterials. 2008;29:1412–1421. doi: 10.1016/j.biomaterials.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 3.Patel S, Tsang J, Harbers GM, Healy KE, Li S. J Biomed Mater Res Part A. 2007;83A:423–433. doi: 10.1002/jbm.a.31320. [DOI] [PubMed] [Google Scholar]
- 4.Rajagopalan P, Marganski WA, Brown XQ, Wong JY. Biophys J. 2004;87:2818–2827. doi: 10.1529/biophysj.103.037218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang V, Misra G, Amsden B. J Mater Sci Mater Med. 2008;19:2145–2155. doi: 10.1007/s10856-007-3306-0. [DOI] [PubMed] [Google Scholar]
- 6.Comisar WA, Kazmers NH, Mooney DJ, Linderman JJ. Biomaterials. 2007;28:4409–4417. doi: 10.1016/j.biomaterials.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 7.Hsiong SX, Carampin P, Kong HJ, Lee KY, Mooney DJ. J Biomed Mater Res Part A. 2008;85A:145–156. doi: 10.1002/jbm.a.31521. [DOI] [PubMed] [Google Scholar]
- 8.Koo LY, Irvine DJ, Mayes AM, Lauffenburger DA, Griffith LG. J Cell Sci. 2002;115:1423–1433. doi: 10.1242/jcs.115.7.1423. [DOI] [PubMed] [Google Scholar]
- 9.Maheshwari G, Wells A, Griffith LG, Lauffenburger DA. Biophys J. 1999;76:2814–2823. doi: 10.1016/S0006-3495(99)77435-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burdick JA, Khademhosseini A, Langer R. Langmuir. 2004;20:5153–5156. doi: 10.1021/la049298n. [DOI] [PubMed] [Google Scholar]
- 11.DeLong SA, Gobin AS, West JL. J Control Release. 2005;109:139–148. doi: 10.1016/j.jconrel.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 12.Kang CE, Gemeinhart EJ, Gemeinhart RA. J Biomed Mater Res Part A. 2004;71A:403–411. doi: 10.1002/jbm.a.30137. [DOI] [PubMed] [Google Scholar]
- 13.Girotti A, Reguera J, Rodriguez-Cabello JC, Arias FJ, Alonso M, Testera AM. J Mater Sci Mater Med. 2004;15:479–484. doi: 10.1023/b:jmsm.0000021124.58688.7a. [DOI] [PubMed] [Google Scholar]
- 14.Haider M, Cappello J, Ghandehari H, Leong KW. Pharm Res. 2008;25:692–699. doi: 10.1007/s11095-007-9282-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang J, Wong C, George A, Kaplan DL. Biomaterials. 2007;28:2358–2367. doi: 10.1016/j.biomaterials.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 16.Lim DW, Nettles DL, Setton LA, Chilkoti A. Biomacromolecules. 2008;9:222–230. doi: 10.1021/bm7007982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogiwara K, Nagaoka M, Cho CS, Akaike T. Biochem Biophys Res Commun. 2006;345:255–259. doi: 10.1016/j.bbrc.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 18.Rizzi SC, Ehrbar M, Halstenberg S, Raeber GP, Schmoekel HG, Hagenmuller H, Muller R, Weber FE, Hubbell JA. Biomacromolecules. 2006;7:3019–3029. doi: 10.1021/bm060504a. [DOI] [PubMed] [Google Scholar]
- 19.Urry DW, Pattanaik A, Xu J, Woods TC, McPherson DT, Parker TM. J Biomater Sci Polym Ed. 1998;9:1015–1048. doi: 10.1163/156856298x00316. [DOI] [PubMed] [Google Scholar]
- 20.Wu XY, Sallach R, Haller CA, Caves JA, Nagapudi K, Conticello VP, Levenston ME, Chaikof EL. Biomacromolecules. 2005;6:3037–3044. doi: 10.1021/bm0503468. [DOI] [PubMed] [Google Scholar]
- 21.Yang M, Tanaka C, Yamauchi K, Ohgo K, Kurokawa M, Asakura T. J Biomed Mater Res Part A. 2008;84A:353–363. doi: 10.1002/jbm.a.31348. [DOI] [PubMed] [Google Scholar]
- 22.Carrico IS, Maskarinec SA, Heilshorn SC, Mock ML, Liu JC, Nowatzki PJ, Franck C, Ravichandran G, Tirrell DA. J Am Chem Soc. 2007;129:4874–+. doi: 10.1021/ja070200b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Zio K, Tirrell DA. Macromolecules. 2003;36:1553–1558. [Google Scholar]
- 24.Heilshorn SC, Di Zio K, Welsh ER, Tirrell DA. Biomaterials. 2003;24:4245–4252. doi: 10.1016/s0142-9612(03)00294-1. [DOI] [PubMed] [Google Scholar]
- 25.Heilshorn SC, Liu JC, Tirrell DA. Biomacromolecules. 2005;6:318–323. doi: 10.1021/bm049627q. [DOI] [PubMed] [Google Scholar]
- 26.Liu JC, Heilshorn SC, Tirrell DA. Biomacromolecules. 2004;5:497–504. doi: 10.1021/bm034340z. [DOI] [PubMed] [Google Scholar]
- 27.Nowatzki PJ, Franck C, Maskarinec SA, Ravichandran G, Tirrell DA. Macromolecules. 2008;41:1839–1845. [Google Scholar]
- 28.Panitch A, Yamaoka T, Fournier MJ, Mason TL, Tirrell DA. Macromolecules. 1999;32:1701–1703. [Google Scholar]
- 29.Richman GP, Tirrell DA, Asthagiri AR. J Control Release. 2005;101:3–12. doi: 10.1016/j.jconrel.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 30.Welsh ER, Tirrell DA. Biomacromolecules. 2000;1:23–30. doi: 10.1021/bm0002914. [DOI] [PubMed] [Google Scholar]
- 31.Isenberg BC, Williams C, Tranquillo RT. Circ Res. 2006;98:25–35. doi: 10.1161/01.RES.0000196867.12470.84. [DOI] [PubMed] [Google Scholar]
- 32.Kannan RY, Salacinski HJ, Butler PE, Hamilton G, Seifalian AM. J Biomed Mater Res Part B. 2005;74B:570–581. doi: 10.1002/jbm.b.30247. [DOI] [PubMed] [Google Scholar]
- 33.Vara DS, Salacinski HJ, Kannan RY, Bordenave L, Hamilton G, Seifalian AM. Pathol Biol. 2005;53:599–612. doi: 10.1016/j.patbio.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 34.Zilla P, Bezuidenhout D, Human P. Biomaterials. 2007;28:5009–5027. doi: 10.1016/j.biomaterials.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 35.Channavajjala LS, Eidsath A, Saxinger WC. J Cell Sci. 1997;110:249–256. doi: 10.1242/jcs.110.2.249. [DOI] [PubMed] [Google Scholar]
- 36.Lillie MA, Gosline JM. Biopolymers. 2002;64:115–126. doi: 10.1002/bip.10155. [DOI] [PubMed] [Google Scholar]
- 37.Neff JA, Tresco PA, Caldwell KD. Biomaterials. 1999;20:2377–2393. doi: 10.1016/s0142-9612(99)00166-0. [DOI] [PubMed] [Google Scholar]
- 38.Houseman BT, Mrksich M. Biomaterials. 2001;22:943–955. doi: 10.1016/s0142-9612(00)00259-3. [DOI] [PubMed] [Google Scholar]
- 39.Chon JH, Netzel R, Rock BM, Chaikof EL. Ann Biomed Eng. 1998;26:1091–1101. doi: 10.1114/1.139. [DOI] [PubMed] [Google Scholar]
- 40.Kouvroukoglou S, Dee KC, Bizios R, McIntire LV, Zygourakis K. Biomaterials. 2000;21:1725–1733. doi: 10.1016/s0142-9612(99)00205-7. [DOI] [PubMed] [Google Scholar]
- 41.Merzkirch C, Davies N, Zilla P. Anat Rec. 2001;263:379–387. doi: 10.1002/ar.1118. [DOI] [PubMed] [Google Scholar]
- 42.Shiu YT, Li S, Marganski WA, Usami S, Schwartz MA, Wang YL, Dembo M, Chien S. Biophys J. 2004;86:2558–2565. doi: 10.1016/S0006-3495(04)74311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fittkau MH, Zilla P, Bezuidenhout D, Lutolf M, Human P, Hubbell JA, Davies N. Biomaterials. 2005;26:167–174. doi: 10.1016/j.biomaterials.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 44.Palecek SP, Loftus JC, Ginsberg MH, Lauffenburger DA, Horwitz AF. Nature. 1997;385:537–540. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.