

Abstract
How large is the volume of sequence space that is compatible with a given protein structure? Starting from random sequences, low free energy sequences were generated for 108 protein backbone structures by using a Monte Carlo optimization procedure and a free energy function based primarily on Lennard–Jones packing interactions and the Lazaridis–Karplus implicit solvation model. Remarkably, in the designed sequences 51% of the core residues and 27% of all residues were identical to the amino acids in the corresponding positions in the native sequences. The lowest free energy sequences obtained for ensembles of native-like backbone structures were also similar to the native sequence. Furthermore, both the individual residue frequencies and the covariances between pairs of positions observed in the very large SH3 domain family were recapitulated in core sequences designed for SH3 domain structures. Taken together, these results suggest that the volume of sequence space optimal for a protein structure is surprisingly restricted to a region around the native sequence.
The sequences of naturally occurring proteins are shaped by a complex interplay of selective pressures. In addition to the overriding selective pressure on proper protein function, there is presumably selection for stability and solubility as well as random drift brought about by neutral mutations. Two questions of particular relevance for this paper are, first, the extent to which sequences are shaped by selection for protein stability, and second, the extent to which this selection process has converged—i.e., to what extent are sequences optimal for their structures? These questions can be addressed by searching sequence space [either experimentally (1, 2) or computationally (3–5)] for low free energy sequences for naturally occurring structures, and comparing these sequences to their naturally occurring counterparts. In this paper we use a computational protein design procedure (6) to carry out such a test.
There has been exciting recent progress with computer-based protein design. Highlights have included the design of a novel α-helical bundle protein with a right-handed superhelical twist and of a de novo sequence that adopts the zinc finger fold (7, 8). Interestingly, most protein redesign efforts that have used automated methods to pack side chains on the backbone of a naturally occurring protein have yielded sequences similar to the naturally occurring sequence. For example, in the redesign of the DNA-binding protein Zif268, four of the eight core residues were the same as the native amino acid, two were mutations from His to Phe, and one was a mutation from Phe to Tyr, and the structure of the redesigned protein showed that the conformations of the core side chains are very similar to the conformation of the native protein (7). In a different study, sequences were computed for the core residues of four proteins, and 51% of the amino acids were identical to the native amino acid (4). In another case whole sequences were generated for proteins and the resulting profile matrices were matched with the native sequence by the profilesearch technique (5).
Do these results indicate that the sequences of naturally occurring proteins are close to optimal for their structures, or are they a consequence of the structure-refinement process? The energy functions used to refine protein structures typically have many common features with the energy functions used for protein design. Therefore, it is possible that refinement builds a “memory” of the native sequence into the structure, and the design procedure to some extent reads this information out from the structure. To address this issue, we have undertaken a large-scale test of the design process with crystal structures of various resolutions. The atomic coordinates of high-resolution structures are less dependent on the energy functions used in refinement, and therefore if the design process is just reversing the refinement process, the designed sequences for the high-resolution structures should be less native-like.
Methods
All amino acids, except for cysteine, were considered at each sequence position. Amino acid side chains were restricted to the conformations contained in the backbone-dependent rotamer library (a total of ≈150 rotamers for all amino acids at a given site) of Dunbrack and Cohen (9). Rotamers rarely seen in the Protein Data Bank (PDB), <3%, were not included. Backbone coordinates were held constant and sequence space was searched by using a simple Metropolis Monte Carlo procedure in which a move consists of exchanging one rotamer for another at a randomly chosen position (≈1 million substitutions per run of Monte Carlo) (10). Unlike the more widely used dead-end elimination-based methods (11, 12), Monte Carlo does not guarantee a globally optimal solution. Convergence was addressed by starting multiple runs for a given structure with different random sequences; in almost all cases the sequences obtained were nearly identical in the core and had similar energies. The lowest-energy sequence from 5 different runs of Monte Carlo was used for comparisons with the native sequence. The advantage of the Monte Carlo procedure is that it can be very fast: a typical sequence/rotamer search for an 80-residue protein takes ≈5 min on an Intel 450-MHz processor.
The free energy function was a linear combination of the following terms: (i) the attractive portion of a standard 12–6 Lennard–Jones potential with van der Waals radii and well depths from the CHARMM19 parameter set (13) except that the van der Waals radii were multiplied by 0.95; (ii) a repulsive term that connects with the 12–6 potential at E = 0 and then ramps linearly up to a value of 10.0 kcal/mol when the two atoms are 0 Å apart (this is less repulsive than a 12–6 potential and compensates to some extent for the use of a fixed backbone and rotamer set); (iii) backbone-dependent internal free energies of the rotamers estimated from PDB statistics [ln P(rot|φ, ψ) (ref. 9)]; (iv) the solvation energy computed using the Lazaridis–Karplus implicit solvation model (14); (v) an approximation to electrostatic interactions in proteins based on PDB statistics [ln P(pair) in ref. 15]; (vi) the side-chain–main-chain hydrogen bond term of Gordon et al. (16); and (vii) reference values for each amino acid that are summed to approximate the free energy of the denatured state. The weights on these terms and the 20 reference energies were determined by maximizing the product of exp(−E(aaobs))/(∑ exp(−E(aai))) over a training set of 30 proteins by using a conjugate-gradient-based optimization method, where E(aaobs) is the energy of the native amino acid at a position, and the partition function in the denominator is over all 20 amino acids at each position. In this process only one residue was changed at a time and all other residues were kept in their native conformation. Subsequently the parameters were refined slightly based on the results of complete redesign calculations on the training set proteins. The weights and reference values as well as a more complete description of the energy function are given in supplementary material at www.pnas.org. All results reported in this paper were on an independent protein test set not used in the determination of the parameters in the model.
For simulations with SH3 domains, sequences were generated for 11 SH3 domains (1abo, 1ad5, 1bb9, 1cka, 1csk, 1fmk, 1lck, 1pht, 1sem, 1shf, 1shg, and 1ycs). Eleven core residues were varied: 4, 6, 10, 18, 20, 26, 28, 39, 41, 50, and 55 (residue number in 1fmk-82). Covariance between pairs of positions in the designed sequences was computed with the measure of covariance used by Larson and coworkers (S. F. Larson, A. A. Di Nardo, and A. R. Davidson, personal communication):
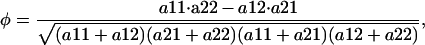 |
where a11 is the number of times both amino acids are at the residues of interest, a22, neither amino acid is present, a12, only amino acid 1 is present, and a21, only amino acid 2 is present.
Results
To determine the extent to which native sequences are optimal for their structures, low free energy sequences were computed for a test set of 108 proteins with less than 30% sequence identity with each other and crystal structures with resolutions better than 3.0 Å. Remarkably, 51% of the core residues in the designed sequences were identical to the naturally occurring residue, and 27% of all of the designed residues were identical to the native amino acid (Table 1 and Figs. 1 and 2). It must be emphasized that the design procedure has no prior knowledge of the native sequence; the native-like sequences emerge because they have the lowest free energy according to the Lennard–Jones and solvation terms, which dominate the potential function used in the design process. The similarity is particularly remarkable because the procedure is expected to fail in cases where function has been optimized at the expense of protein stability (for example in protein active sites).
Table 1.
Results from redesigning 108 small proteins
| Residue | Core residues (>20 Cβ atoms within 10 Å)
|
All residues
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. correct | No. native | No. designed | No. correct/ No. native | No. correct/ No. designed | No. correct | No. native | No. designed | No. correct/ No. native | No. correct/ No. designed | |
| Ala | 78 | 114 | 137 | 0.68 | 0.56 | 226 | 645 | 779 | 0.35 | 0.29 |
| Asp | 0 | 9 | 2 | 0.00 | 0.00 | 84 | 465 | 538 | 0.18 | 0.16 |
| Glu | 0 | 11 | 1 | 0.00 | 0.00 | 121 | 607 | 902 | 0.20 | 0.13 |
| Phe | 43 | 76 | 103 | 0.57 | 0.42 | 103 | 286 | 361 | 0.36 | 0.29 |
| Gly | 35 | 48 | 39 | 0.73 | 0.90 | 389 | 555 | 560 | 0.70 | 0.69 |
| His | 0 | 11 | 5 | 0.00 | 0.00 | 3 | 174 | 34 | 0.02 | 0.10 |
| Ile | 63 | 112 | 128 | 0.56 | 0.49 | 135 | 482 | 360 | 0.28 | 0.37 |
| Lys | 3 | 20 | 9 | 0.15 | 0.33 | 109 | 642 | 788 | 0.17 | 0.14 |
| Leu | 92 | 131 | 174 | 0.70 | 0.53 | 263 | 675 | 667 | 0.39 | 0.39 |
| Met | 3 | 21 | 6 | 0.14 | 0.50 | 7 | 167 | 34 | 0.04 | 0.20 |
| Asn | 4 | 19 | 5 | 0.21 | 0.80 | 63 | 395 | 277 | 0.16 | 0.23 |
| Pro | 12 | 14 | 26 | 0.86 | 0.46 | 208 | 359 | 455 | 0.58 | 0.46 |
| Gln | 1 | 15 | 5 | 0.07 | 0.20 | 20 | 392 | 114 | 0.05 | 0.17 |
| Arg | 1 | 15 | 5 | 0.07 | 0.20 | 33 | 471 | 320 | 0.07 | 0.10 |
| Ser | 2 | 24 | 7 | 0.08 | 0.29 | 80 | 502 | 439 | 0.16 | 0.18 |
| Thr | 4 | 21 | 10 | 0.19 | 0.40 | 110 | 457 | 563 | 0.24 | 0.19 |
| Val | 83 | 139 | 153 | 0.60 | 0.55 | 187 | 568 | 466 | 0.33 | 0.40 |
| Trp | 8 | 19 | 16 | 0.42 | 0.50 | 26 | 94 | 114 | 0.28 | 0.23 |
| Tyr | 10 | 51 | 39 | 0.20 | 0.26 | 65 | 282 | 447 | 0.23 | 0.15 |
| Total | 444 | 870 | 870 | 0.51 | 0.51 | 2219 | 8218 | 8218 | 0.27 | 0.27 |
No. correct is the number of residue positions that have the same amino acid in the designed and native sequence. No. native and no. designed are the number of times an amino acid appears in the native and designed sequences, respectively. Cysteines were not varied in this study and were kept in their native conformation during design.
Figure 1.

Sequence identity between designed and native sequences for core residues as a function of crystal structure resolution. The average sequence identity to native for sequences generated for a set of 88 NMR structures is shown as a square.
Figure 2.

Sequence alignments between designed (DES) and wild-type (WT) sequences for four proteins. A black background indicates identical amino acids and a gray background indicates similar amino acids. The following PDB files were used: Hpr (1poh), CI2 (1ypc), CspB (1csp), and Fyn (1avz).
The fraction of core residues correctly predicted for each protein is displayed in Fig. 1 as a function of the resolution of the crystal structure. For most proteins, at least a third of the core residues were identical to those in the native sequence, and for many of the proteins, the fraction was much higher. The lack of dependence on crystallographic resolution suggests that the design process is not simply recapitulating the structure refinement process. To investigate this point further we redesigned four proteins with high-resolution crystal structures (2igd, 1rgg, 1rb9, and 1iro) that were refined without the use of noncovalent energy terms (17). The results are similar to those seen with the larger test set: 35% of all residues and 46% of the core residues were replaced with the native amino acid. When NMR structures are used as a template, native amino acids are recovered less frequently. In sequences designed for 88 NMR structures of small proteins (60–100 residues), 25% of the core residues were identical to the naturally occurring amino acid and 16% of all of the residues were the native amino acid. The first structure in the PDB file was used in each case.
In multiple sequence alignments of naturally occurring sequences, some positions are more strongly conserved than others because of structural constraints, particularly in the core. Considerable variation in residue conservation was also observed in the designed sequences: when several sequences were generated for one protein, some positions had little preference for any one amino acid, whereas other positions almost always replaced the same amino acid. Interestingly, there was a correlation between the degree of conservation of a residue in the design process and the level of identity to the native sequence. At core sites that were highly conserved in the design procedure (low sequence entropy) the native amino acid was selected more than 90% of the time, whereas at core sites with high sequence entropy the native amino acid was selected less than 20% of the time (Fig. 3). Furthermore, sites conserved in the natural evolutionary process were often conserved in the design procedure: the sequence entropy in the multiple sequence alignments was correlated with the sequence entropy in the designed sequences (Fig. 3).
Figure 3.

Sequence conservation in designed sequences correlates with sequence identity to the native sequence and sequence conservation in protein families. When the design program shows a strong preference for a particular amino acid at a sequence position, it more often prefers the native amino acid, and the residue is likely to have low sequence variability in naturally occurring sequences. Each position in each redesigned protein was assigned to a bin (x axis) based on the sequence entropy (∑frequency(aai)⋅ln(frequency(aai)) summed over all 20 amino acids, aai) at the position in a large set of sequences generated by the Monte Carlo search procedure (the numbers of residues in bins 1–7 are, respectively, 86, 91, 91, 126, 107, 79, and 94; higher sequence entropy is to the right). The left y axis indicates the percentage of residue positions that had the native amino acid in the designed sequences. The right y axis indicates the average sequence variability observed in naturally occurring sequences as derived from multiple sequence alignments (MSAs). The MSAs were taken from HSSP files (21). Results are shown for core residues. Only residue positions that had at least 10 sequences in the MSA were used (60 proteins total).
What features of proteins are the energy function recognizing that makes the native amino acid easily identified for some core positions? Given that the design program relies primarily on sterics and packing to choose good sequences for the core residues, there are probably more packing constraints at the low entropy sites. Indeed, the residues in the low entropy sites make on average 20% more contacts (atoms within 6 Å) than the residues in the high entropy sites. Of the hydrophobic amino acids, tyrosine and methionine were the least conserved in the core (Table 1). Methionine is unfavorable in part because the attractive portion of the Lennard–Jones potential is weak for sulfur atoms in the CHARMM19 parameter set. Tyrosine is unfavorable in the core because the energy function penalizes the burial of hydroxyl groups. Glycine was frequently recovered at positions with positive φ angles.
One limitation of the automated design procedure is that the backbone is held fixed throughout the simulation and therefore some sequences, which require only small movements in the backbone to be good, may be excluded. To test whether the native sequence is still preferred when small backbone motions are allowed, we used NMR structures as a source of alternative backbone structures and designed sequences for nine proteins for which there are crystal and NMR structures available (Fig. 4). In most cases the lowest-energy sequences were obtained for the crystal structure, and as found above they were native-like. It appears that as the backbone varies from the generally more accurate crystal structure, nonnative sequences do become more preferred but their energies are not as good as the native sequence–structure pair. In the cases where the energies for the NMR structures were comparable to the energy of the crystal structure, the sequences were native-like.
Figure 4.

Sequence design for alternative backbone conformations. Sequences were designed for 9 proteins for which there is a NMR and crystal structure available. The free energy of a sequence (in kcal/mol) is plotted against sequence identity (over all residues) to the native sequence. The results for the crystal structures are shown as open squares (for Cl2 and fyn, two independently determined crystal structures were used). The following PDB files were used: CI2 (1ypc, 2ci2, 3ci2), ubiquitin (1d3z, 1ubq), CspB (1csp, 1nmf), fyn (1a0n, 1avz, 1efn), Hpr (1hdn, 1poh), protein L (2ptl, (J. O'Neill and K. Zhang, personal communication)), tendamistat (1brn, 2ait), interleukin (1icw, 1il8), and barstar (1a19, 1abt).
The results described thus far demonstrate that the optimization procedure can to some extent recover the sequence of a protein from its structure. To determine whether the variation in sequence in a large protein family could be recapitulated by the design procedure, we chose the SH3 domain, which includes over 400 naturally occurring proteins. One thousand sequences were generated for 11 different SH3 crystal structures with the identities of 11 core residues varied. The final 11,000 sequences were then used to generate an amino acid profile at each sequence position. These profiles were compared with profiles derived from a MSA of 233 SH3 domains (S. M. Larson and A. R. Davidson, personal communication). There is a good match between the profiles (Fig. 5). Thus, it appears that evolution has sampled most of the sequence space compatible with the SH3 structural core, and has to some extent reached equilibrium.
Figure 5.

Amino acid profiles for six core residues in SH3 domains. The empty bars are derived from an SH3 domain MSA (S. M. Larson and A. R. Davidson, personal communication) and the shaded bars, from 11,000 computed sequences generated by using the backbones from 11 separate SH3 domain structures.
It has been proposed that covariance in MSAs can be used for predicting contacting residues for use in structure prediction. Different studies have reached different conclusions on this issue; the major complication is distinguishing the covariances due to physical constraints from those due to lineage effects. Larsen et al. (S. F. Larson, A. A. Di Nardo, and A. R. Davidson, personal communication) recently found significant covariances in the SH3 family by using a method that reduced such lineage effects. We find that these covariances are to some extent reproduced in the designed sequences. Pairwise covariances were determined by calculating φ coefficients (see Methods) for each possible amino acid/residue pair. A positive covariance (positive φ coefficient) indicates that a particular pair of amino acids is often seen at the residue positions of interest, whereas a negative covariance (negative φ coefficient) indicates that the pair is rarely seen at the residue positions of interest. Almost all pairs of residues found to be positively correlated in the naturally occurring sequences were also positively correlated in the designed sequences, and likewise for the negative covariances (Fig. 6). These results suggest that the forces modeled by the design procedure, primarily Lennard–Jones packing, are responsible at least in part for the amino acid covariances in the SH3 domain family.
Figure 6.

Sequence covariances derived from an SH3 domain MSA compared with covariances derived from computer-generated sequences. Each point corresponds to one pair of covarying residues. φ values greater than 0 indicate a positive covariance (see Methods), whereas values less than 1 indicate a negative covariance. The covariances in the MSA were identified by Larson et al. (S. F. Larson, A. A. Di Nardo, and A. R. Davidson, personal communication).
Discussion
More than half of the core residues in the lowest free energy sequences generated by our design procedure are identical to the amino acids at the corresponding positions in the native protein sequences. As noted in the Introduction, one possible source of such similarity is a “memory” of the native sequence inscribed in the native backbone coordinates by the potential functions used in refining the structures that can be “read” by using similar potential functions in the design process. We find, however, that the level of sequence identity between native and designed sequences is independent of the extent to which noncovalent energy terms were used in the refinement of the structures, and in particular, that such sequence identity is observed even for structures refined without use of any such terms. The simplest interpretation of these results is that the lowest free energy sequences for a given structure resemble the native sequence, and that the energy function we use represents protein energetics well enough to identify sequences that are genuinely low in free energy. For clarity, we note that “native sequences are close to optimal for their structures” does not mean that the free energies of native sequences are optimal [protein stability can be significantly enhanced with just a few mutations (18, 19)], but that the lowest free energy sequences for a structure are likely to be similar to the native sequence.
Also, it is important to emphasize that the “close to optimal” result applies to specific protein structures, not to protein folds. There are numerous examples of pairs of naturally occurring proteins with little sequence similarity but similar folds. In addition, combinatorial mutagenesis experiments have shown that the core sequence of a protein can be highly varied, albeit with some loss of stability, without destroying the protein fold (1, 2). However, our finding that the optimal sequences for NMR-determined backbone conformations generally have higher free energies than the more native-like sequences found for crystal structures (Fig. 4) suggests that nonnative low free energy sequence–structure pairs are relatively rare.
True de novo protein design, that is of a novel backbone, relies on the assumption that a sequence that will fold into the target structure actually exists. Since there appear to be so few good sequences for a unique structure, the probability that there is any good sequence for any single novel backbone structure may be very small. Therefore, it is probably necessary to allow the backbone to shift during the design of novel protein structures (8). Our results with the NMR structural ensembles are encouraging because they suggest that a good potential function can select out “designable” backbone structures from an ensemble of structures.
The similarity between designed and naturally occurring sequences suggests that stability effects are the primary constraint in the evolution of core residues. A similar conclusion has been reached in an experimental study of the SH3 domain family: it was found that the stability changes produced by mutations in the core could be predicted from the amino acid frequencies and covariances observed in the SH3 domain family (ref. 20; A. R. Davidson, personal communication). Finally, the ability of the design procedure to reproduce the site-specific amino acid residue frequencies and covariances in a large protein family on the basis of structural information alone suggests that it could contribute to methods for remote homologue detection (5).
Supplementary Material
Acknowledgments
We thank Alan Davidson for generously sharing data and allowing us to make the comparisons of the designed sequence amino acid frequencies and covariances with those observed experimentally. We thank members of the Baker laboratory for helpful comments on the manuscript. B.K. was supported by the Cancer Research Fund of the Damon Runyon–Walter Winchell Foundation Fellowship, DRG-1533. This work was also supported by a grant from the National Institutes of Health to D.B.
Abbreviations
- MSA
multiple sequence alignment
- PDB
Protein Data Bank
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Sauer R T. Folding Des. 1996;1:R27–R30. doi: 10.1016/S1359-0278(96)00015-6. [DOI] [PubMed] [Google Scholar]
- 2.Plaxco K W, Riddle D S, Grantcharova V, Baker D. Curr Opin Struct Biol. 1998;8:80–85. doi: 10.1016/s0959-440x(98)80013-4. [DOI] [PubMed] [Google Scholar]
- 3.Saven J G, Wolynes P G. J Phys Chem B. 1997;101:8375–8389. [Google Scholar]
- 4.Desjarlais J R, Handel T M. Protein Sci. 1995;4:2006–2018. doi: 10.1002/pro.5560041006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koehl P, Levitt M. J Mol Biol. 1999;293:1183–1193. doi: 10.1006/jmbi.1999.3212. [DOI] [PubMed] [Google Scholar]
- 6.Ponder J W, Richards F M. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 7.Dahiyat B I, Mayo S L. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 8.Harbury P B, Plecs J J, Tidor B, Alber T, Kim P S. Science. 1998;282:1462–1467. doi: 10.1126/science.282.5393.1462. [DOI] [PubMed] [Google Scholar]
- 9.Dunbrack R L, Cohen F E. Protein Sci. 1997;6:1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voight C A, Gordon D B, Mayo S L. J Mol Biol. 2000;299:789–803. doi: 10.1006/jmbi.2000.3758. [DOI] [PubMed] [Google Scholar]
- 11.Desmet J, Maeyer M D, Hazes B, Lasters I. Nature (London) 1992;356:539–541. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- 12.Gordon D B, Mayo S L. J Comput Chem. 1998;19:1505–1514. [Google Scholar]
- 13.Neria E, Fischer S, Karplus M. J Chem Phys. 1996;105:1902–1921. [Google Scholar]
- 14.Lazaridis T, Karplus M. Proteins Struct Funct Genet. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 15.Simons K T, Ruczinski I, Kooperberg C, Fox B A, Bystroff C, Baker D. Proteins Struct Funct Genet. 1999;34:82–95. doi: 10.1002/(sici)1097-0134(19990101)34:1<82::aid-prot7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 16.Gordon D B, Marshall S A, Mayo S L. Curr Opin Struct Biol. 1999;9:509–513. doi: 10.1016/s0959-440x(99)80072-4. [DOI] [PubMed] [Google Scholar]
- 17.Network E-D V. J Mol Biol. 1998;276:417–436. doi: 10.1006/jmbi.1997.1526. [DOI] [PubMed] [Google Scholar]
- 18.Malakauskas S M, Mayo S L. Nat Struct Biol. 1998;5:470–475. doi: 10.1038/nsb0698-470. [DOI] [PubMed] [Google Scholar]
- 19.Perl D, Mueller U, Heinemann U, Schmid F X. Nat Struct Biol. 2000;7:380–383. doi: 10.1038/75151. [DOI] [PubMed] [Google Scholar]
- 20.Maxwell K L, Davidson A R. Biochemistry. 1998;37:16172–16182. doi: 10.1021/bi981788p. [DOI] [PubMed] [Google Scholar]
- 21.Dodge C, Schneider R, Sander C. Nucleic Acids Res. 1998;26:313–315. doi: 10.1093/nar/26.1.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.