

Abstract
Prior investigations of traumatic axonal injury (TAI), and pharmacological treatments of TAI pathology, have focused exclusively on the role of myelinated axons, with no systematic observations directed towards unmyelinated axon pathophysiology. Recent electrophysiological evidence, however, indicates that unmyelinated axons are more vulnerable than myelinated axons in a rodent model of experimental TAI. Given their susceptibility to TAI, the present study examines whether unmyelinated axons also respond differentially to FK506, an immunophilin ligand with well-established neuroprotective efficacy in the myelinated fiber population. Adult rats received 3.0 mg/kg FK506 intravenously at 30 min prior to midline fluid percussion injury. In brain slice electrophysiological recordings, conducted at 24 h postinjury, compound action potentials (CAPs) were evoked in the corpus callosum, and injury effects quantified separately for CAP waveform components generated by myelinated axons (N1 wave) and unmyelinated axons (N2 wave). The amplitudes of both CAP components were suppressed postinjury, although this deficit was 16% greater for the N2 CAP. While FK506 treatment provided significant neuroprotection for both N1 and N2 CAPs, the drug benefit for the N2 CAP amplitude was 122% greater than that for the N1 CAPs, and improved postinjury strength-duration and refractoriness properties only in N2 CAPs. Immunocytochemical observations, of TAI reflected in intra-axonal pooling of amyloid precursor protein, indicated that FK506 reduced the extent of postinjury impairments to axonal transport and subsequent axonal damage. Collectively, these studies further substantiate a distinctive role of unmyelinated axons in TAI, and suggest a highly efficacious neuroprotective strategy to target this axonal population.
Keywords: Traumatic axonal injury, Traumatic brain injury, FK506, Compound action potentials, Corpus callosum, Neuroprotection
1. Introduction
Investigations of traumatic brain injury (TBI) reveal a multifaceted pathology, with the consistent observation that axons are among the most vulnerable cellular components to the forces of injury (Maxwell et al., 1997; Povlishock, 1992; Smith and Meaney, 2000). Axonal damage has been described across the spectrum of brain injury ranging from mild to severe and is assumed to underlie much of the resulting morbidity and mortality (Adams et al., 1999; Fitzpatrick et al., 1998). Despite progress in characterizing traumatic axonal injury (TAI), questions remain regarding whether all axons undergo essentially similar responses to injury, or rather distinct populations of axons incur injuries which differ in type or degree (Marmarou and Povlishock, 2006). This issue also has implications for studies of neuroprotective compounds, because optimal pharmacologic strategies may be those which target specific agents to identified axonal subpopulations. Thus, greater knowledge of how axonal phenotype influences the course of TAI would benefit both the fundamental understanding of axonal injury and the search for effective therapies.
The presence or absence of myelin is one phenotypic dimension on which axons may be classified into discrete populations. Recent findings from our laboratory indicated that the unmyelinated axon population sustained more severe functional and structural impairment than myelinated axons, in a rat model of experimental TBI (Reeves et al., 2005). The specific factors underlying an enhanced vulnerability of unmyelinated axons are, at present, not entirely clear. It is likely, however, that the greater axolemmal exposure of these fibers constitutes a target for pathological processes, such as those operative in TBI, which disrupt or degrade plasma membranes through multiple routes, including mechanical poration (Pettus and Povlishock, 1996), lipid peroxidation (Evans, 1993), and disturbances of transmembrane ionic fluxes and subsequent proteolysis (review by Maxwell et al., 1997). An elevated vulnerability, of unmyelinated axons, assumes added significance in view of stereological counts showing unmyelinated fibers comprise at least 76% of axons in lateral subcortical white matter (Partadiredja et al., 2003), and approximately 80% of corpus callosum axons (Gravel et al., 1990). Despite the evidence that unmyelinated fibers constitute the numerical majority of many CNS pathways, nearly all prior studies of TAI pathobiology have focused exclusively on myelinated axons. Accordingly, progress towards a comprehensive description of injury and treatment mechanisms in TAI will necessitate an assessment of the unique role of the unmyelinated axon population. No prior studies have specifically compared the effects of neuroprotectants on unmyelinated versus myelinated axons.
This issue is of more than academic interest in that the focus on myelinated axons has also extended to preclinical laboratory studies exploring various therapeutic approaches to attenuate myelinated axonal damage. In these studies, the immunophilin ligands, cyclosporin-A (CsA) and tacrolimus (FK506), have proved effective in blunting TAI pathology via attenuation of mitochondrial permeability transition and calcineurin activation, respectively (Buki et al., 1999; Marmarou and Povlishock, 2006; Singleton et al., 2001). In view of these findings in the myelinated fiber population, it was reasonable to determine if some of these protective effects translated to the unmyelinated axon population, and to provide further insight into the therapeutic and potentially mechanistic actions of the immunophilin ligands.
To pursue these issues, the present study evaluated the neuroprotective effects of FK506 on the function of both myelinated and unmyelinated corpus callosum axons following TBI in adult rats. This compound inhibits the activity of calcineurin (CaN), a Ca2+/calmodulin-dependent protein phosphatase which is highly localized in the central nervous system. While CaN is known to dephosphorylate diverse substrates (Morioka et al., 1999), the enzyme has specific activities which potentially influence axonal structure and function: modulating microtubule assembly through the dephosphorylation of MAP2, tubulin, and tau protein (Goto et al., 1985), and regulating the phosphorylation state of neurofilament proteins (Eyer and Leterrieir, 1988). Prior studies have demonstrated FK506 to significantly attenuate posttraumatic axonal swelling and impairments to axonal transport (Marmarou and Povlishock, 2006; Singleton et al., 2001). The current study found that systemic administration of FK506 exerted a strong neuroprotective effect on the function of unmyelinated axons in the corpus callosum, significantly alleviating posttraumatic suppression of evoked compound action potentials (CAPs), and provided a more moderate degree of functional protection in callosal myelinated axons. Qualitative immunocytochemical observations, using the intra-axonal β-amyloid precursor protein (APP), also documented the protective effects of FK506 in the corpus callosum and adjacent subcortical white matter.
2. Results
This study evaluated the efficacy of FK506 to provide neuroprotection against midline fluid percussion TBI. The present results describe the effects of administering FK506 (3 mg/kg) 30 minutes prior to TBI, which ensured that the drug was present at the time of injury. Because this study evaluated functional and structural effects of FK506 administration on axonal injury, it was also essential to ensure that injury levels did not differ significantly between drug- and vehicle-treated groups. For this purpose, consistency in injury severity was assured by including only animals with injury magnitudes in the range 2.0 ± 0.1 atmospheres. In addition, the duration of suppression of the righting reflex, used as an index of traumatic unconsciousness, was compared between injured groups receiving FK506 and vehicle. The overall mean latency to regain the righting reflex, for all animals given fluid percussion injury, was 8.02 ± 0.39 minutes. None of the injury groups used in the study differed significantly from this overall mean righting latency (F(1,12)=0.90; p=0.362).
The primary functional measure used to evaluate the effects of TBI, and FK506 treatment, was amplitude of evoked CAPs. However, posttraumatic changes in CAP amplitude may reflect alterations in the size of action potentials of individual axons, or in the total number of axons recruited into the CAP field potential. Accordingly, assessments of CAP amplitude were supplemented with measurements of refractoriness and strength-duration properties, both of which reflect intrinsic functional properties of axons and are less sensitive to the absolute numbers of responsive fibers. Refractoriness evaluates the discharge-recovery cycle of axons, while strength-duration analyses address response properties of axons at low, near-threshold, levels of stimulation. Together, this set of electrophysiological outcome measures provide critical functional correlates to the well established structural alterations and ionic disturbances of TBI.
2.1 The effects of injury on CAP amplitude
CAPs evoked in the corpus callosum of in vitro brain slices, in the present study, exhibited biphasic waveforms consistent with prior investigations (Preston et al., 1983; Reeves et al., 2005; Swanson et al., 1998). An early waveform component is generated predominantly by relatively fast-conducting myelinated axons, and a later-occurring component reflects mainly slower unmyelinated axons. The present study used recording conditions which were optimized for separate quantification of the myelinated (‘N1’) and the unmyelinated (‘N2’) CAP components. An example CAP waveform in Fig. 1 illustrates the amplitude measurement for the myelinated (‘N1’) and the unmyelinated (‘N2’) components (Fig 1, C.,D.).
Figure 1.
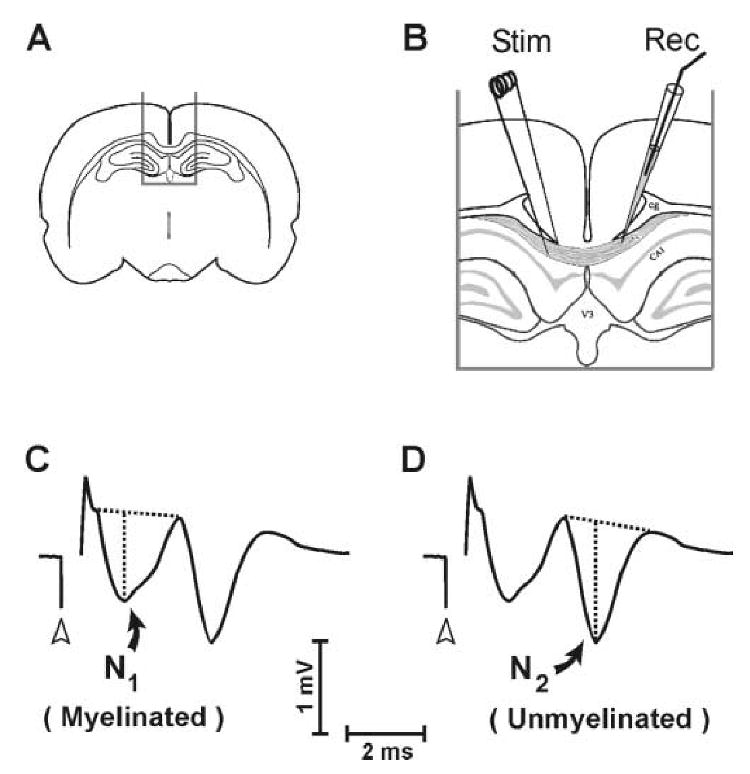
Placement of electrodes for callosal CAP recording, and measurement technique for N1 and N2 CAP components. A., B. Stimulating and recording electrodes were positioned in midline corpus callosum of brain slices separated by approximately 1.0 mm. C. Quantification of the myelinated (N1) wave component. D. Quantification of the unmyelinated (N2) wave component. In (C.) and (D.) CAP amplitude was measured as vertical distance from the local negative peak to a tangent joining preceding and following positivities. Open arrowheads indicate time of stimulus onset.
Fluid percussion injury suppressed callosal CAP amplitudes when measured at 24 h postinjury. Examples of CAP waveforms, evoked using graduated stimulus levels in input-output testing, are shown in Fig. 2A for vehicle- and FK506- treated sham and injured rats. Sample waveforms from a vehicle-treated sham and TBI rat (left side of Fig. 2A) are representative of the profound response reductions observed postinjury. The mean CAP amplitudes are plotted for each experimental group in Fig. 2B, as curves relating evoked CAP amplitude to normalized stimulus current. Statistical comparisons among these curves, assessing injury- or FK506-induced effects, were confined to CAPs evoked at a stimulus range of 30-100% of maximum. CAPs evoked using the lowest stimulus currents were analyzed during strength-duration testing (see below). Fig. 2B shows that TBI significantly suppressed both myelinated (‘N1’) and unmyelinated (‘N2’) CAP components: the TBI(vehicle) curve was significantly lower than the Sham(vehicle) curve for both the N1 (F(1,12)=28.07; p<0.001) and the N2 (F(1,12)=25.89; p<0.001) CAP amplitude.
Figure 2.
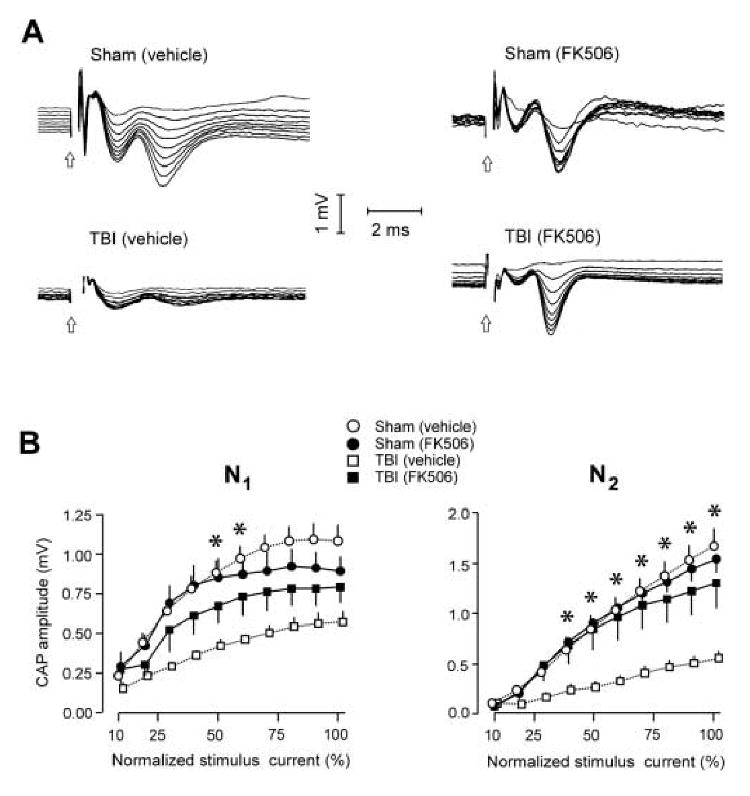
Differential effects of FK506 treatment on postinjury suppression of callosal N1 and N2 CAP amplitudes. A. Samples of CAP field potentials recruited with graduated series of stimulus pulses (current stepped from 10% to 100% of maximum) for all experimental groups. Open arrow = time of stimulus onset. B.[Left panel] Average amplitude of N1 over the full range of stimulation current used, for all groups. N1 amplitude was significantly decreased by TBI, but FK506 treatment provided significant neuroprotection only at selective stimulus intensities (50-60% of maximum): *p<0.05, comparing TBI(vehicle) vs. TBI(FK506) groups. [Right panel] Average amplitude of N2 over the full stimulus range, for all groups. TBI produced a more severe suppression of the N2 CAP amplitude, at a greater range of stimulus intensities (40-100%): *p<0.05, comparing TBI(vehicle) vs. TBI(FK506) groups.
To address whether TBI functionally impaired unmyelinated axons to a greater extent than myelinated axons, a statistical analysis compared the relative postinjury percent decreases in N1 and N2 amplitudes. Again considering CAPs evoked with stimulus currents of 30-100% maximum, the average N1 CAP amplitude measured in vehicle-treated TBI rats was 49.05±12.73% of that measured in vehicle-treated sham rats. The N2 CAP amplitudes were more severely reduced in amplitude following injury, to 32.84±15.14% of the vehicle-treated sham level, and this differential injury effect was significant (F(1,6)=11.64; p<0.05). Thus, at 24 h the injury effect to N2 was approximately 16% more severe than for N1. These present measurements of posttraumatic CAP suppression, and the relatively greater injury effect in unmyelinated than in myelinated fibers, replicated our prior measurements at 24h, obtained during a time-course study spanning 3 h to 7 d postinjury (Reeves et al., 2005), where the injury effect to N2 was 22% more severe than for N1.
2.2. FK506 is neuroprotective against posttraumatic CAP suppression
FK506 treatment, administered 30 min prior to the fluid percussion injury, produced a marked protection against postinjury decreases in N2 CAP amplitude, and a more moderate level of protection for N1 amplitudes. FK506 effects were dependent on the level of stimulus current, and the drug effects were evaluated at specific levels of normalized stimulus intensity. N2 CAP amplitudes recorded in FK506-treated TBI rats were significantly elevated above those in vehicle-treated TBI rats (F(1,11)=8.22; p<0.05). Comparisons at specific levels of stimulus intensity showed the FK506 protection of the N2 CAPs to be significant at stimulus levels of 40-100% (see asterisks in Fig. 2B, comparing TBI (vehicle) vs. TBI (FK506)). In contrast, FK506 treatment led to significant N1 CAP protection only at two stimulus intensities (50% and 60%, Fig. 2B), and an omnibus F test covering the full analysis range (30-100% of maximum) narrowly missed significance (F(1,11)=4.49; p=0.058).
The preceding analysis demonstrated that the drug elevated the N2 CAP amplitudes over a greater range of stimulus intensities than was found for the N1 amplitudes. An additional index of neuroprotection was the percent increase in CAP amplitudes attributable to drug administration. In this respect, FK506 administration produced an average 177.1 ± 11.3% increase in N2 CAP amplitudes, compared to a 55.2 ± 5.1% increase in N1 amplitudes. Collectively, these observations are consistent with a differentially larger degree of FK506 protection for unmyelinated axons (N2 generators) than for myelinated axons (N1 generators). While the preceding indices reveal an unequal magnitude of drug benefit, it should be emphasized that the FK506 protective effect was not exclusive to, but only relatively greater for, the N2 CAP. For example, while CAPs recorded from vehicle-treated TBI rats were significantly suppressed by injury, CAPs in FK506-treated TBI rats did not differ from vehicle-treated sham levels for the N1 (F(1,11)=3.14; p=0.104) or the N2 (F(1,11)=0.26; p=0.623) CAP components.
Inspection of the mean N1 amplitude curves in Fig. 2B suggests that FK506 exerts a complex effect on myelinated fibers. In injured rats, FK506 provided functional protection only at intermediate stimulus intensities, and this was not observed at higher stimulus levels. In sham control rats given FK506, the mean N1 CAP amplitudes appeared to decrease below the level of vehicle-treated shams at higher stimulus intensities (Fig. 2B), although this trend was not statistically significant (F(1,11)=0.38; p=0.551). Despite the fact that the quantitative analyses did not show a significant effect of FK506 on CAPs in uninjured (sham) rats, the plots of mean N1 amplitude (Fig. 2B) suggest that the drug treatment may lower the current levels at which the N1 component of evoked CAPs reaches asymptote. Consistent with this possibility, visual inspection of CAPs acquired during input-output stimulation (right side of Fig. 2A) also provide a qualitative indication that the N1 reaches a maximum amplitude rapidly in FK506 treated rats.
2.3 Refractoriness testing
The refractoriness of the callosal fibers was analyzed by quantifying the suppression of the second CAP response in paired stimulus trials. This analysis was completed for a subset of 22 rats as follows: sham (vehicle) (n=6), sham (FK506) (n=6), TBI (vehicle) (n=5), and TBI (FK506) (n=5). Fig. 3A shows example series of the second response evoked in paired stimulus presentations, at the indicated interpulse intervals (IPI, 1.5 to 6.0 ms), after subtracting out the responses to the conditioning pulse, for a representative slice from each experimental condition recorded at 24 h postinjury. Fig. 3B plots the CAP amplitude elicited by the second pulse in each paired stimulation (C2) divided by the CAP amplitude to single pulse stimulation (C1). These C2/C1 ratios are averaged for each analytic group and plotted for N1 (left panel of Fig. 3B) and for N2 (right panel). These mean values were fitted to Boltzmann sigmoid curves (lines in Fig. 3B), with average adjusted R2 values for N1 = 0.869 and for N2 = 0.928. Rightward shifts in these curves correspond to increases in the refractory recovery cycle in the callosal axons, indicative of axonal damage. Analyses of the refractory results showed that the fluid percussion injury produced significant rightward shifts for both the N1 (Z=2.23; p<0.05) and N2 (Z=2.49; p<0.05) CAP components (comparing sham (vehicle) vs. TBI (vehicle) in Fig. 3B). However, FK506 treatment significantly ameliorated this deficit only for the N2 CAP, the refractory curve for which was not different from that of vehicle-treated sham rats (Z=1.50; p=0.134: comparing sham (vehicle) vs. TBI (FK506) in Fig. 3B). In TBI rats treated with FK506 the refractory curve for the N1 CAP remained significantly shifted from that of vehicle-treated sham rats (Z=3.33; p<0.01). The computed sigmoid functions were used to estimate the interpulse interval at which the CAP2, evoked by the second pulse in paired stimulus trials, achieved 50% of the amplitude of a single pulse presentation (graphically corresponding to curve intersection with C2/C1 = 50% in Fig. 3B). Accordingly, these curves show that C2/C1 = 50% at 3.7 ms for N1, and 4.4 ms for N2, for the vehicle-treated sham group. Although the absolute magnitudes of injury-induced rightward shifts were modest, equaling 0.55 ms for N1 and 0.73 ms for N2, the significance of the curve shifts indicated that TBI altered fundamental activation properties of the axons, slowing the recovery cycle times of both the myelinated and unmyelinated axon populations. FK506 treatment reduced refractory changes only for the N2 CAP, strongly indicating a differential therapeutic effect for the unmyelinated axon population, as regards this functional property.
Figure 3.
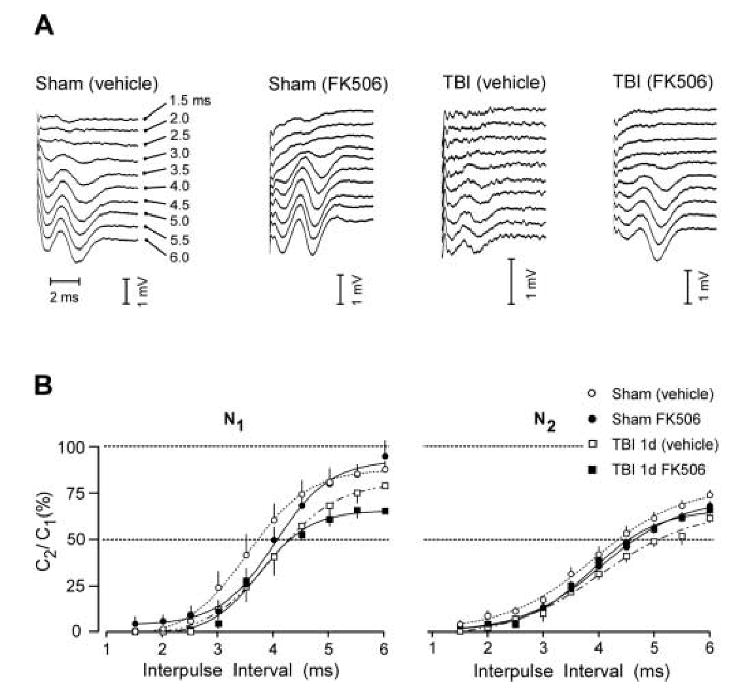
Effect of TBI and FK506 treatment on callosal CAP refractoriness. A. Example waveforms show second potentials in paired responses (interpulse intervals = 1.5 to 6.0 ms), after subtraction of response to the conditioning pulse, of representative cases from each experimental group. B. Plots of mean CAP amplitude elicited by the second pulse in each paired stimulation (C2) divided by the CAP amplitude to single pulse stimulation (C1), for all groups. Average C2/C1 ratios were fitted to Boltzmann sigmoid curves, and showed TBI-induced increases in refractoriness (rightward curve shifts) which was significant for N1 and N2. FK506 prevented this injury effect only for the N2 CAP component.
2.4 Strength-duration properties
An assessment of axonal excitability, which included an evaluation of the parameters of rheobase and the strength-duration time constant, was conduced on a subset of 17 rats as follows: sham (vehicle) (n=5), sham (FK506) (n=5), TBI (vehicle) (n=4), and TBI (FK506) (n=4). The current threshold data conformed well to the hyperbolic function given by Weiss' law (Bostock et al., 1983; Burke et al., 2000), and goodness-of-fit R2 values were high, with mean values ranging from 0.931 to 0.994.
Strength-duration analyses demonstrated an FK506 neuroprotective effect only for the N2 CAP, with the drug treatment preventing an injury-induced excitability alteration. In contrast, the N1 CAP component showed more minor changes in strength-duration parameters, and these were associated only with FK506 treatment, not reaching significance for injury effects. Curves plotting mean threshold current as a function of stimulus duration are shown for N1 (Fig. 4A[left panel]) and for N2 (Fig. 4B[left panel]). A significant TBI-induced rightward curve shift, denoting reduced axonal excitability, was seen for the N2 CAP (Z=2.03; p<0.05: comparing sham (vehicle) vs. TBI (vehicle) in Fig. 4B[left panel]). This loss of N2 excitability was prevented by FK506 treatment, as there was no significant difference between the vehicle-treated sham group and the FK506-treated TBI group (Z=1.015; p=0.310). Regarding the N1 CAPs, the curves for FK506-treated shams (Z=2.075; p<0.05) and for FK506-treated TBI rats (Z=1.987; p<0.05) were significantly shifted away from the vehicle-treated sham curve, although the magnitude of these N1 changes were minor.
Figure 4.
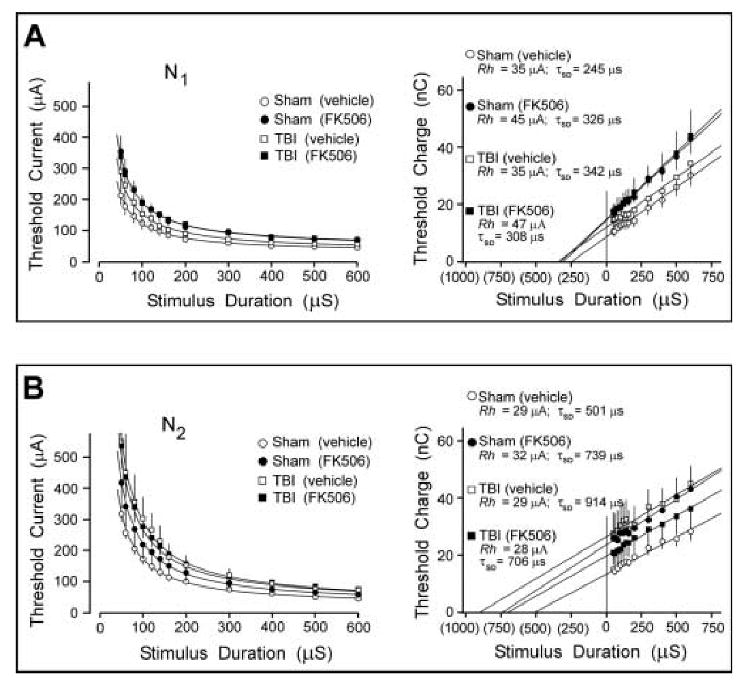
Effects of TBI and FK506 on strength-duration and charge-duration curves for N1 and N2 CAP components. A.[Left panel] Threshold data is plotted for N1 waves, and fitted to Weiss' formula (curves). [Right panel] Charge-duration transformation of the same data provides a graphical estimate of the strength-duration time constant (τSD) as an extrapolation of the regression line to the zero charge axis. Rheobase (Irh) is estimated as the slope of the regression line. τSD and Irh are listed for each analytic group above regression lines. B. Equivalent analysis of N2 threshold data. Overall, these analyses indicated that strength-duration properties of the N2 CAP were relatively more vulnerable to injury, and showed more neuroprotection following FK506 treatment, than was the case for the N1 CAP. TBI induced a significant shift in the N2 threshold curve, which was prevented by FK506. This corresponded to an 82% postinjury increase in the N2τSD, which was attenuated to 40% by FK506. In contrast, the N1 CAP showed minor threshold shifts associated more with FK506 than with TBI, and no significant changes in τSD.
The strength-duration data were transformed to demonstrate the relationship of threshold stimulus charge to stimulus pulse duration. This provided estimates of rheobasic current (Irh, slope of the regression line) and strength duration time constant,τSD, as the (negative) x-intercept where threshold charge is zero (Fig. 4A,B[right panels]). The computed values of the parameters Irh and τSD are listed for all experimental groups above the regression lines in Fig. 4A,B. In this analysis changes in rheobase current were modest, with the largest departure from the sham-vehicle level being a 34% elevation of the N1Irh in FK506-treated rats. The parameter τSD appeared more sensitive to TBI, and FK506 treatment, with the N2 CAPs from vehicle-treated rats showing an 82% increase in τSD (914 ms), above that of sham-vehicle rats (501 ms). FK506 treatment reduced this injury-induced shift, with the N2τSD of drug-treated TBI rats (706 ms) elevated only 40% above that of sham-vehicle rats.
2.5 Immunocytochemical findings
APP immunoreactive swellings appeared in the corpus callosum and adjacent subcortical white matter at 24h postinjury (Fig. 5 C and D). These APP positive foci were similar to those previously correlated with sites of axonal injury, swelling and disconnection (Stone et al., 2000). While quantitative assessments were not performed, qualitative comparisons between the drug-treated and vehicle groups revealed consistent and striking differences. FK506-treated animals showed dramatic reduction of immunoreactive axonal swellings in all regions sampled. This finding, as noted above, is consistent with reduced axonal damage and improved fiber conductance within the corpus callosum, although these immunocytochemical studies could not discriminate between unmyelinated and myelinated damage.
Figure 5.
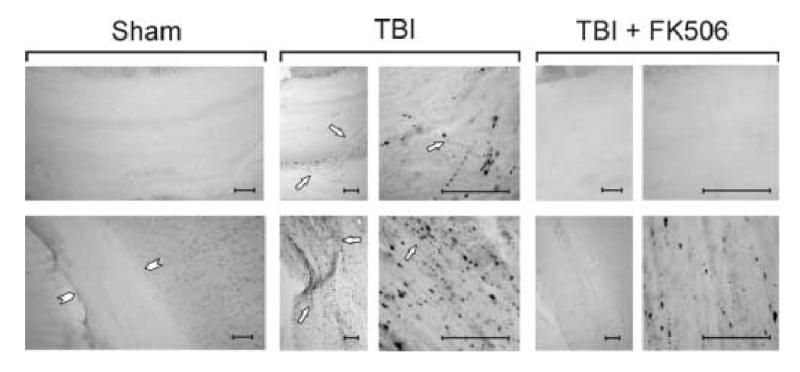
FK506 effects on postinjury increases in APP labeled axons within the corpus callosum and subcortical white matter adjacent to electrophysiological recording at 24 h postinjury. Top row: Representative sections show immunohistochemical detection of APP labeled damaged axons in mid-dorsal corpus callosum of sham-injured, vehicle- and FK506-treated TBI conditions. As previously observed, APP staining of injured axons was absent in the sham control callosum, but visible at discrete foci after TBI (arrows). With FK506 treatment APP profile was similar to that of sham control. Bottom row: Representative sections show APP label in subcortical white matter of sham-injured, vehicle-and FK506-treated TBI conditions. As in callosum, sham control cases showed no APP staining of white matter axons (arrowheads). After TBI, significant axonal damage is detected with APP in the white matter (arrows). FK506 treatment reduced APP staining. Calibration bars = 50 μm.
3. Discussion
This is the first study to address the specific issue of differential therapeutic efficacy in unmyelinated and myelinated axons, in response to a neuroprotectant administration. As appropriate for the initial studies of drug effects in specific fiber populations, a pretreatment strategy was implemented, ensuring that the drug was present at the time of injury. The efficacy of postinjury FK506 administration, on these axon populations, remains a topic for subsequent studies. The present study provided evidence for differential treatment sensitivities for the two axon subtypes in the corpus callosum. The findings of the study also serve to emphasize the necessity of accounting for the distinctive role of the unmyelinated axon population in the pathology, and treatment, of axonal injury.
This study demonstrated that midline fluid percussion injury strongly suppressed callosal CAP amplitude, which was consistent with prior reports (Baker et al., 2002). The present finding, that TBI produced a more severe functional deficit for the N2, than for the N1, CAP amplitude was also in agreement with our prior observations (Reeves et al., 2005). Multiple lines of evidence indicate that the two CAP components are generated by distinct populations of faster-conducting myelinated fibers (N1) and slower unmyelinated fibers (N2) (Preston et al., 1983; Swanson et al., 1998). The biphasic negative CAP indicates an underlying bimodal distribution of conduction velocities, and not a unitary population of axons varying along a single continuous dimension. Inhibition of potassium channels with 4-AP prolongs the duration of only the N2 CAP (Reeves et al., 2005), consistent with an unmyelinated status in fibers generating that field component. Results from several ultrastructural and electrophysiological investigations in the corpus callosum, including assessments of axon caliber, myelination status, and conduction velocity, accord well with the identification of the N1 and N2 CAP components with myelinated and unmyelinated fiber populations, respectively (Preston et al., 1983; Sturrock, 1980; Swadlow et al., 1980; Swanson et al., 1998).
Considering evidence from the present electrophysiological and morphological observations, it is likely that postinjury functional changes reflect both the damage and disconnection of some number of axons, rendering them incapable of being recruited into the CAP field potential, and sublethal changes within other axons which remain responsive in the aftermath of injury, but with altered functional properties. It is likely that the neuroprotective activity of FK506 involves an attenuation of both categories of fiber pathology, leading to reductions in the numbers of damaged and disconnected axons, as suggested by our qualitative morphological observations, and improvements in the functional profile of axons which remain responsive postinjury. Unfortunately, the definitive resolution of this issue of axonal damage and disconnection versus functional perturbation and recovery will require the use of serial section EM analyses with stereological assessments. While we have initiated these studies, these are highly labor intensive and complex and are beyond the scope of the current communication. However, despite this fact, our current physiological findings do shed important insight into this issue. TBI-induced decreases in CAP amplitude may reflect the numerical loss of axons or a partial functional degradation within viable and responsive fibers. In contrast, electrophysiological observations of threshold-level CAPs, in the strength-duration protocol, and of refractoriness are less sensitive to the absolute numbers of responsive fibers, and are more reflective of the functional properties of individual active fibers. The principal charge carrier in the CAP signal is flux through voltage-gated Na+ channels, and both strength-duration and refractoriness depend critically on Na+ conductances. τSD appears to be strongly dependent on a non-inactivating Na+ conductance due to ‘persistent’ Na+ channels (Bostock and Rothwell, 1997), and refractoriness has long been recognized to depend largely on recovery of Na+ channels from inactivation (Hodgkin and Huxley, 1952). The present observations, regarding FK506 effects on strength-duration and refractory properties, indicated that the neuroprotective benefit was primarily to the N2 CAP. Together, these factors suggest a feature of FK506 neuroprotection may be a prevention of postinjury pathology affecting Na+ channels in unmyelinated axons, perhaps analogous to the Na+ channel subunit pathology described in small caliber axons by Iwata et al (2004), following in vitro axonal injury.
FK506 may exert a neuroprotective effect through multiple pathways, due to the diversity of substrates dephosphorylated by calcineurin. In addition to interacting with modulators of the axonal cytoskeleton, the compound may also operate on other cellular targets to impact white matter pathophysiology. FK506 binds to FKBP12, an integral part of the IP3 and ryanodine receptor complexes, and appears to alter cytosolic Ca2+ gated through these channels (Brillantes et al., 1994; Cameron et al., 1995). In this context, growing evidence supports a pathogenic role of ionic dysregulation involving Ca2+ channels on axonal endoplasmic reticulum (Stys, 2005). It is conceivable that FKBP12 expression is different in unmyelinated and myelinated axons, accounting for the distinctive effect of the compound on these two fiber subtypes, although this has not, to our knowledge, been systematically studied. An additional protective role of FK506 may involve inhibition of the postinjury activity of BAD, a member of the bcl-2 gene family, preventing formation of the proapoptotic BAD/Bcl-x(L) complex (Noto et al., 2004; Springer et al., 2000). Non-neuronal cells may also be sites of protective action in white matter, as evidenced by reduced oligodendrocyte cell death, following FK506 treatment, in a spinal cord contusion injury (Nottingham et al., 2002).
Irrespective of the mechanism(s) of FK506 neuroprotection, the immunophilin ligand has proved effective in multiple injury models. FK506 treatment alleviated posttraumatic cerebral edema following controlled cortical impact (Stover et al., 2001), reduced the number of APP immunoreactive axons after impact acceleration injury (Singleton et al., 2001), and decreased axonal damage associated with rapid rewarming from postinjury hypothermia (Suehiro et al., 2001). In models of spinal cord injury, FK506 treatment reduced caspase-3 activation and apoptosis (Springer et al., 2000), improved neurological function (Madsen et al., 1998), preserved axons following partial transection of dorsal spinal columns (Bavetta et al., 1999), and facilitated the recovery of CAP amplitudes in an in vitro model of hypoxic injury to spinal cord white matter (Mobeley et al., 2003). These varied applications show the generality of the neuroprotective effects observed after FK506 administration, and support its use in the current study to address differential therapeutic efficacy for unmyelinated axons.
The increased vulnerability of unmyelinated axons to TBI may result from their relatively greater axolemmal exposure to membrane-targeting properties of TBI pathology. Axons are the neuronal component with the highest membrane-to-cytoplasm ratio, and this ratio is proportionately higher in small diameter than larger fibers. Based on estimates of median diameter for unmyelinated (∼0.15 μm) and myelinated (∼0.43 μm) axons in cerebral white matter (Partadiredja et al., 2003), and modeling the two classes of axons as equal-length cylinders, unmyelinated axons have a computed axolemma-to-axoplasm ratio which is over 2.5 fold larger than myelinated axons. Consequently, unmyelinated axons are predicted to be preferentially impacted by membrane-specific TBI pathomechanisms including a collapse of transmembrane potential, lipid peroxidation, and ion channel degeneration (e.g., review by Stys, 2004), and proteolysis of spectrin and other cytoskeletal components closely subjacent to plasma membrane (Saatman et al., 2003).
The high membrane content, and lack of insulating myelin, may also underlie the greater sensitivity of unmyelinated fibers to FK506 treatment. While structural features of unmyelinated axons may increase their vulnerability to TBI pathology, as noted above, their greater axolemmal exposure may also facilitate uptake of neuroprotective compounds, such as FK506. An additional corollary, of the close contact of unmyelinated axolemma and the extracellular environment, is that both injury and drug effects are likely to reflect axolemmal interactions with the extracellular matrix (ECM) and regulatory matrix metalloproteinases (MMPs). In this regard, growing evidence indicates that MMP expression is upregulated following TBI (Kim et al., 2005; Phillips and Reeves, 2001), and that posttraumatic inhibition of MMP activity can support recovery of function (Falo et al., 2006). Ongoing ultrastructural and molecular studies in our laboratories are now addressing these issues.
A preferential vulnerability, of unmyelinated axons, may relate importantly to the cognitive and memory deficits which are commonly observed to persist in head injured patients (Levin, 1996). It has long been recognized that the distribution of axon subtype, along the anteroposterior axis of the corpus callosum, is in register with projections from functionally and cytoarchitectonically distinct neocortical areas in rhesus monkey (Lamantia and Rakic, 1990) and human (Aboitiz et al., 1992), and callosal regions with the largest proportion of unmyelinated axons interconnect association cortex. It is conceivable that a postinjury loss of unmyelinated axons disproportionately impacts fibers subserving associative memory functions, with a relative sparing of sensorimotor-dedicated axons.
4. Experimental Procedures
The procedures for this study followed all national guidelines for the care and use of experimental animals, and the experimental protocol was approved by the Medical College of Virginia Animal Research Committee. Male Sprague-Dawley rats (n = 34) weighing 300-350 g, at the start of the study, were used in these experiments. Animals were housed in individual cages in a temperature-(22° C) and humidity-controlled (50% relative) animal facility on a 12-h light/dark cycle. Rat chow and water were continually available.
4.1 Fluid Percussion TBI Procedure and FK506 Administration
The model used for injury was the same as that described by Dixon et al. (1987). Briefly, all animals were surgically prepared under nose-cone isoflurane anesthesia (2% in carrier gas of 70% N2O, 30% O2). Rats were positioned in a stereotaxic frame, and a 4.8 mm skull craniotomy was prepared over the midline, centered between bregma and lambda. A modified Leur-Loc syringe hub (2.6 mm i.d.) was then positioned over the exposed dura and bonded in place with cyanoacrylate adhesive, and the entire complex was anchored by two steel screws and dental acrylic. The open hub was packed with Gelfoam prior to scalp suturing and Bacitracin applied. Once animals had recovered from anesthesia, they were returned to their home cages.
Twenty-four hours after hub implantation, rats were administered intravenous FK506 using a dosing paradigm previously shown to attenuate structural correlates of TAI after impact acceleration brain injury (Singleton, et al., 2001). Rats were randomly assigned to receive FK506 (n=14) or vehicle (n=20). While under nose-cone isoflurane anesthesia (described above), the central penile vein was catheterized via insertion of a sterile 25 g butterfly needle. A single 3 mg/kg bolus of FK506 was administered by direct infusion over a period of 2 minutes, with the drug diluted with 0.9% saline to a final volume of 0.5 ml. This dosage was based on previous work demonstrating that 3 mg/kg FK506 delivered intravenously crosses the blood-brain barrier and establishes therapeutic levels of brain parenchymal concentration (Singleton et al., 2001) that correlate with near maximum inhibition of calcineurin (Ochiai et al., 1989; Butcher et al., 1997) consistent with neuroprotection. Vehicle groups were administered 0.9% saline (0.5 ml) via the same injection method.
At 30 minutes following FK506 or vehicle administration, rats received fluid percussion injury or a sham injury. Rats were anesthetized with isoflurane (4% in carrier gas of 70% N2O and 30% O2), connected to a fluid percussion device through the exposed hub, and injured at 2.0 ± 0.05 atmospheres. The injury device is described in greater detail elsewhere (Dixon et al., 1987). In brief, the device consisted of a 60 × 4.5 cm Plexiglas water-filled cylinder, fitted at one end with a piston mounted on O-rings, with the opposite end housing a pressure transducer (Entran Devices, Inc.; EPN-0300A). At the time of injury, the Leur-Loc fitting, filled with 0.9% saline, was attached to the transducer housing. The injury was produced by a metal pendulum that struck the piston, injecting a small volume of saline into the cranial cavity, briefly deforming the brain tissue (20 ms pulse duration). The resulting pressure pulse was recorded extracranially by the transducer and expressed in atmospheres of pressure. Injury magnitude was controlled by setting the height from which the pendulum was released. Following injury all animals were promptly ventilated with room air until spontaneous breathing resumed. The duration of suppression of the righting reflex was used as an index of traumatic unconsciousness. For control rats administered sham injuries, TBI procedures were as above except the intracranial pressure pulse was not injected. Once consciousness was regained, animals were placed in recovery chambers, monitored for any signs of distress (bleeding, respiratory difficulty), and then returned to their home cages.
4.2 Electrophysiological Recording Procedures
Following the fluid percussion injury, rats were allowed to survive for 24 hours prior to electrophysiological recording. A total of 25 rats underwent recording, in subgroups as follows: vehicle-treated sham (n=7); FK506-treated sham (n=5); vehicle-treated TBI (n=7); and, FK506-treated TBI (n=6). Each rat was decapitated under isoflurane anesthetized and the brain rapidly removed. Coronal slices of 450 μm thickness were cut in ice cold (4 to 6°C) artificial cerebrospinal fluid (ACSF) with a vibrating-knife microtome (Electron Microscopic Sciences model OTS-3000-03). Those slices were saved which contained midline-crossing segments of the corpus callosum, overlying the mid-dorsal hippocampus, which yielded 3 to 5 slices per brain. Slices were then transferred to a holding chamber containing oxygenated ACSF at room temperature, and were allowed to equilibrate under these conditions for at least 1 h prior to recording. For recording, a slice was transferred to a submersion-type chamber, and perfused at a rate of 1-2 ml / min with ACSF at 23 degrees C. The ACSF contained (in mM): NaCl 124, KCl 5, NaH2PO4 1.25, NaHCO3 26, MgSO4 1.3, CaCl2 2, glucose 10; pH 7.4; saturated with a 95% O2 / 5% CO2 gas mixture. A bipolar stimulating electrode (teflon-insulated tungsten; 0.3-0.4 mm intertip distance) was lowered into the corpus callosum, at approximately 0.5 mm lateral to midline, and a recording electrode (ACSF-filled glass micropipette; resistance 6 - 8 MΩ) was also placed in the corpus callosum of the contralateral hemisphere at a distance of approximately 1.0 mm from the stimulating electrode (Fig. 1A,B). The initial depth of electrodes was 200 μm below the surface of the slice, however, fine adjustments were made in the depths of both stimulating and recording electrodes to optimize the signal amplitude. Evoked callosal CAP field potentials were amplified (bandpass = DC to 10 kHz), digitized at 25 kHz, and stored on disk for offline analysis.
The callosal CAP has previously been characterized as a biphasic waveform with an early component generated predominantly by relatively fast-conducting myelinated axons, and a later-occurring component reflecting mainly slower unmyelinated axons (Preston et al., 1983; Reeves et al., 2005; Swanson et al., 1998). The present study used recording conditions which were optimized for separate quantification of the myelinated and unmyelinated CAP components. In particular, the selection of the recording temperature in the present study (23 degrees C) was based on a previous systematic comparison of CAPs recorded at temperatures ranging from 23-36 degrees C (Reeves et al., 2005).
Stimulation used for evoked CAPs was constant current stimulus-isolated square wave pulses, with stimulus pulse duration and intensity adjusted for specific recording protocols. For analyses of TBI-related changes in CAP amplitude, standardized input-output functions were generated, for each slice, by varying the intensity of stimulus pulses (200 μs duration, delivered at 0.1 Hz) in ten equal steps from approximately threshold level to an asymptotic maximum for the N1 CAP component. This method of current standardization was adopted from our previous observation that the N2 CAP component required more current to reach asymptote than was necessary for N1 (Reeves et al., 2005). This protocol results in conservative levels of stimulus currents, avoiding extensive stimulation at supramaximal levels for only one of the axonal subpopulations (i.e., the faster conducting N1 generating fibers), yet still permits measurement of the linear portion of the N2 input-output range. In refractoriness testing, pairs of pulses were presented with interpulse interval increased, in 0.5 ms steps, from 1.5 ms to 6.0 ms, using the maximum current level established during input-output testing for that slice. For strength-duration analyses, stimulus pulses were presented using a graded series of durations (in μs): 50, 60, 80, 100, 120, 140, 160, 200, 300, 400, 500, and 600. At each duration, stimulus current was adjusted to evoke a response which was 30% of the maximum response amplitude observed in input-output testing.
4.3 Statistical analysis
All quantitative electrophysiological analyses were conducted on waveforms which were the average of four successive sweeps, for enhanced signal-to-noise ratio. The amplitude of the two CAP components (N1 and N2) were routinely quantified as the maximum negative deflection measured relative to a tangent connecting the preceding and following positivities (see Fig. 1C and 1D for graphical illustrations of these measurements). For quantitative evaluation of injury or drug treatment effects, the unit of analysis was the rat, and not the slice. Accordingly, if more than one slice was used from a single rat, those multiple-slice data were averaged for that rat and used for the analysis. The effects of injury and FK506 on callosal CAP amplitude were evaluated using mixed-design ANOVA (SPSS v11.5) with injury condition as between-subjects factor and normalized stimulus intensity as repeated-measures factor. Comparisons involving specific stimulation levels (or interpulse intervals) were implemented using the simple main effects functions of SPSS MANOVA syntax, which also controlled for experimentwise error rate. In analyses of refractoriness, ratios of CAP2 / CAP1 (obtained in paired stimulus presentations) were plotted as a function of interpulse interval. Measures of CAPs evoked in strength-duration trials were summarized as curves relating threshold current to stimulus duration. The significance of injury- or drug-related curve shifts, in both refractory and strength-duration analyses, were evaluated with the Mann-Whitney U statistic. The significance level, α = 0.05, was used for all inferential statistics, and averaged values are expressed as mean ± SEM.
Strength-duration data were fitted to Weiss' formula (Bostock et al., 1983; Burke et al., 2000):
where Q is the stimulus charge, Irh is the rheobasic current, τSD the strength-duration time constant, and T is stimulus duration. In Weiss' formulation, there is a linear relationship between stimulus charge (equal to threshold stimulus current multiplied by its duration) and stimulus duration. Accordingly, Irh and τSD were estimated by performing a linear regression of Q on T, wherein τSD (corresponding to chronaxie) is given by the negative intercept of the regression line on the duration axis. Rheobase (Irh) is the threshold current for an infinitely long stimulus and is estimated by the slope of the regression line.
4.4 Immunocytochemical analysis
A subset of 9 rats were evaluated for the histological correlates of FK506 treatment, as follows: sham (vehicle) (n=3), TBI (vehicle) (n=3) and TBI (FK506) (n=3). To assess the effect of FK506 on overall axonal pathology in the corpus callosum, brain slices adjacent to those subjected to electrophysiological recording were used to examine the prevalence of amyloid precursor protein (APP)-labeled axons. APP serves as a commonly utilized marker for impaired axonal transport and axonal swelling (Stone et al., 2000). Selected slices were fixed by immersion in a solution of 4% paraformaldehyde + 0.1 M sodium phosphate buffer. Blocked tissues were then embedded in agar and 40 μm vibratome sections generated. Sections were rinsed 3 × 10 min in PBS, and endogenous peroxidase activity was blocked with 0.3% H2O2 in PBS for 30 min. Tissue was preincubated for 60 min with 0.2% Triton X (Sigma Chemical Co., St. Louis,MO) in 10% normal goat serum (NGS)/PBS prior to incubation overnight in rabbit C-terminus APP antibody at a dilution of 1:2500 (Zymed Laboratories) in 1%NGS/PBS. Sections were then washed 3 × 10 min in PBS containing 1% NGS and incubated in a 1:200 dilution of biotinylated goat anti-rabbit immunoglobulin (diluted 1:200 in 1% NGS/PBS; Vector, Burlingame, CA) for 60 min followed by 3 × 10 min rinses in PBS. After incubation in an avidin–biotin peroxidase complex (ABC standard Elite kit, Vector, Burlingame, CA) at a dilution of 1:200 for 60 min and rinsing in PBS and 0.1 M phosphate buffer 3 × 10 min, the sections were processed for visualization of the immunohistochemical complex using 0.05% diaminobenzidine (DAB) (Sigma Chemical Co., St. Louis, MO), 0.01% H2O2, 0.3% imidazole in 0.1 M PBS. Next, the sections were mounted on gelatin-coated glass slides, dehydrated and cover slipped.
Acknowledgments
The authors wish to thank Susan Walker, Lesley Harris, and Raiford Black for technical assistance. Supported by Virginia Commonwealth Neurotrauma Initiative 04-091 (TMR), HD055813 (JTP), and NS44372 (LLP).
Abbreviations
- APP
β-amyloid precursor protein
- CAP
compound action potential
- CaN
calcineurin
- FK506
(tacrolimus)
- Irh
rheobasic current
- N1
myelinated CAP waveform
- N2
unmyelinated CAP waveform
- TAI
traumatic axonal injury
- TBI
traumatic brain injury
- τSD
strength-duration time constant
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aboitiz F, Scheibel AB, Fisher RS, Zaidel E. Fiber composition of the human corpus callosum. Brain Res. 1992;598:143–153. doi: 10.1016/0006-8993(92)90178-c. [DOI] [PubMed] [Google Scholar]
- Adams JH, Jennett B, McLellan DR, Murray LS, Graham DI. The neuropathology of the vegetative state after head injury. J Clin Pathol. 1999;52:804–806. doi: 10.1136/jcp.52.11.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker AJ, Phan N, Moulton RJ, Fehlings MG, Yucel Y, Zhao M, Liu E, Tian GF. Attenuation of the electrophysiological function of the corpus callosum after fluid percussion injury in the rat. J Neurotrauma. 2002;19:587–599. doi: 10.1089/089771502753754064. [DOI] [PubMed] [Google Scholar]
- Bavetta S, Hamlyn PJ, Burnstock G, Lieberman AR, Anderson PN. The effects of FK506 on dorsal column axons following spinal cord injury in adult rats: Neuroprotection and local regeneration. Exp Neurol. 1999;158:382–393. doi: 10.1006/exnr.1999.7119. [DOI] [PubMed] [Google Scholar]
- Bostock H, Rothwell JC. Latent addition in motor and sensory fibres of human peripheral nerve. J Physiol. 1997;498:277–294. doi: 10.1113/jphysiol.1997.sp021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostock H, Sears TA, Sherratt RM. The spatial distribution of excitability and membrane current in normal and demyelinated mammalian nerve fibers. J Physiol. 1983;341:41–58. doi: 10.1113/jphysiol.1983.sp014791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Buki A, Okonkwo DO, Povlishock JT. Postinjury cyclosporin A administration limits axonal damage and disconnection in traumatic brain injury. J Neurotrauma. 1999;16:511–521. doi: 10.1089/neu.1999.16.511. [DOI] [PubMed] [Google Scholar]
- Burke D, Bartley K, Woodforth IJ, Yakoubi A, Stephen JPH. The effectws of a volatile anaesthetic on the excitability of human corticospinal axons. Brain. 2000;123:992–1000. doi: 10.1093/brain/123.5.992. [DOI] [PubMed] [Google Scholar]
- Butcher SP, Henshall DC, Teramura Y, Iwasaki K, Sharkey J. Neuroprotective actions of FK506 in experimental stroke: in vivo evidence against an antiexcitotoxic mechanism. J Neurosci. 1997;17:6939–6946. doi: 10.1523/JNEUROSCI.17-18-06939.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron AM, Steiner JP, Sabatini DM, Kaplin AI, Walensky LD, Snyder SH. Immunophilin FK506 binding protein associated with inositol 1,4,5-triphosphate receptor modulates calcium flux. Proc Natl Acad Sci USA. 1995;92:1784–1788. doi: 10.1073/pnas.92.5.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, Young HF, Hayes RL. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- Evans PH. Free radicals in brain metabolism and pathology. Br Med Bull. 1993;49:577–587. doi: 10.1093/oxfordjournals.bmb.a072632. [DOI] [PubMed] [Google Scholar]
- Eyer J, Leterrier JF. Influence of the phosphorylation state of neurofilament proteins on the interactions between purified filaments in vitro. Biochem J. 1988;252:655–660. doi: 10.1042/bj2520655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falo MC, Fillmore HL, Reeves TM, Phillips LL. MMP3 expression profile differentiates adaptive and maladaptive synaptic plasticity induced by brain injury. J Neurosci Res. 2006;84:768–781. doi: 10.1002/jnr.20986. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick MO, Maxwell WL, Graham DI. The role of the axolemma in the initiation of traumatically induced axonal injury. J Neurol Neurosurg Psychiatry. 1998;64:285–287. doi: 10.1136/jnnp.64.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto S, Yamamoto H, Fukunaga K, Iwasa T, Matsukado Y, Miyamoto E. Dephosphorylation of microtubule-associated protein 2, tau factor, and tubulin by calcineurin. J Neurochem. 1985;45:276–283. doi: 10.1111/j.1471-4159.1985.tb05504.x. [DOI] [PubMed] [Google Scholar]
- Gravel C, Sasseville R, Hawkes R. Maturation of the corpus callosum of the rat: II Influence of thyroid hormones on the number and maturation of axons. J Comp Neurol. 1990;291:147–161. doi: 10.1002/cne.902910110. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata A, Stys PK, Wolf JA, Chen XH, Taylor AG, Meaney DF, Smith DH. Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. J Neurosci. 2004;24:4605–5613. doi: 10.1523/JNEUROSCI.0515-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Fillmore HL, Reeves TM, Phillips LL. Elevation of Hippocampal MMP-3 Expression and Activity During Trauma-Induced Synaptogenesis. Exper Neurol. 2005;192:60–72. doi: 10.1016/j.expneurol.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Lamantia AS, Rakic P. Cytological and quantitative characteristics of four cerebral commissures in the rhesus monkey. J Comp Neurol. 1990;291:520–537. doi: 10.1002/cne.902910404. [DOI] [PubMed] [Google Scholar]
- Levin HS. Neurobehavioral outcome of closed head injury: implications for clinical trials. In: Bandak FA, Eppinger RH, Ommaya AK, editors. Traumatic brain injury: bioscience and mechanics. Mary Ann Liebert; Larchmont, New York: 1996. pp. 105–114. [Google Scholar]
- Madsen JR, MacDonald P, Irwin N, Goldberg DE, Yao GL, Meiri KF, Rimm IJ, Stieg PE, Benowitz LI. Tacrolimus (FK506) increases neuronal expression of GAP-43 and improves functional recovery after spinal cord injury in rats. Exp Neurol. 1998;154:673–683. doi: 10.1006/exnr.1998.6974. [DOI] [PubMed] [Google Scholar]
- Marmarou CR, Povlishock JT. Administration of the immunophilin ligand FK506 differentially attenuates neurofilament compaction and impaired axonal transport in injured axons following diffuse traumatic brain injury. Exp Neurol. 2006;197:353–362. doi: 10.1016/j.expneurol.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Maxwell WL, Povlishock JT, Graham DI. A mechanistic analysis of non-disruptive axonal injury: a review. J Neurotrauma. 1997;14:419–440. doi: 10.1089/neu.1997.14.419. [DOI] [PubMed] [Google Scholar]
- Mobley LW, Sandeep MD, Agrawal K. Role of calcineurin in calcium-mediated hypoxic injury to white matter. Spine J. 2003;3:11–18. doi: 10.1016/s1529-9430(02)00442-4. [DOI] [PubMed] [Google Scholar]
- Morioka M, Hamada JI, Ushio Y, Miyamoto E. Potential role of calcineurin for brain ischemia and traumatic injury. Prog Neurobiol. 1999;58:1–30. doi: 10.1016/s0301-0082(98)00073-2. [DOI] [PubMed] [Google Scholar]
- Noto T, Ishiye M, Furuich Y, Keida Y, Katsuta K, Moriguchi A, Matsuoka N, Aramori I, Goto T, Yanagihara T. Neuroprotective effect of tacrolimus (FK506) on ischemic brain damage following permanent focal cerebral ischemia in the rat. Brain Res Mol Brain Res. 2004;128:30–38. doi: 10.1016/j.molbrainres.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Nottingham S, Knapp P, Springer J. FK506 treatment inhibits caspase-3 activation and promotes oligodendroglial survival following traumatic spinal cord injury. Exp Neurol. 2002;177:242–251. doi: 10.1006/exnr.2002.7975. [DOI] [PubMed] [Google Scholar]
- Ochiai T, Nakajima K, Sakamoto K, Nagata M, Gunji Y, Asano T, Isono K, Sakamaki T, Hamaguchi K. Comparative studies on the immunosuppressive activity of FK506, 15-deoxyspergualin, and cyclosporine. Transplant Proc. 1989;21:829–832. [PubMed] [Google Scholar]
- Partadiredja G, Miller R, Oorschot DE. The number, size, and type of axons in rat subcortical white matter on left and right sides: A stereological, ultrastructural study. J Neurocytol. 2003;32:1165–1179. doi: 10.1023/B:NEUR.0000021910.65920.41. [DOI] [PubMed] [Google Scholar]
- Pettus EH, Povlishock JT. Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res. 1996;722:1–11. doi: 10.1016/0006-8993(96)00113-8. [DOI] [PubMed] [Google Scholar]
- Phillips LL, Reeves TM. Interactive pathologies modify hippocampal plasticity. Restorative Neurol Neurosci. 2001;19:213–235. [PubMed] [Google Scholar]
- Povlishock JT. Traumatically induced axonal injury: Pathogenesis and pathobiological implications. Brain Pathol. 1992;2:1–12. [PubMed] [Google Scholar]
- Preston RJ, Waxman SG, Kocsis JD. Effects of 4-aminopyridine on rapidly and slowly conducting axons of rat corpus callosum. Exp Neurol. 1983;79:808–820. doi: 10.1016/0014-4886(83)90044-4. [DOI] [PubMed] [Google Scholar]
- Reeves TM, Phillips LL, Povlishock JT. Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Exp Neurol. 2005;196:126–139. doi: 10.1016/j.expneurol.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Abai B, Grosvenor A, Vorwerk CK, Smith DH, Meaney DF. Traumatic axonal injury results in biphasic calpain activation and retrograde transport impairment in mice. J Cereb Blood Flow Metab. 2003;23:34–42. doi: 10.1097/01.WCB.0000035040.10031.B0. [DOI] [PubMed] [Google Scholar]
- Singleton RH, Stone JR, Okonkwo DO, Pellicane AJ, Povlishock JT. The immunophilin ligand FK506 attenuates axonal injury in an impact-acceleration model of traumatic brain injury. J Neurotrauma. 2001;18:607–614. doi: 10.1089/089771501750291846. [DOI] [PubMed] [Google Scholar]
- Smith DH, Meaney DF. Axonal damage in traumatic brain injury. Neuroscientist. 2000;6:483–495. [Google Scholar]
- Springer JE, Azbill RD, Nottingham SA, Kennedy SE. Calcineurin-mediated BAD dephosphorylation activates the caspase-3 apoptotic cascade in traumatic spinal cord injury. J Neurosci. 2000;20:7246–7251. doi: 10.1523/JNEUROSCI.20-19-07246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JR, Singleton RH, Povlishock JT. Antibodies to the C-terminus of the beta-amyloid precursor protein (APP): a site specific marker for the detection of traumatic axonal injury. Brain Res. 2000;871:288–302. doi: 10.1016/s0006-8993(00)02485-9. [DOI] [PubMed] [Google Scholar]
- Stover JF, Schoning B, Sakowitz OW, Woiciechowsky C, Unterberg AW. Effects of tacrolimus on hemispheric water content and cerebrospinal fluid levels of glutamate, hypoxanthine, interleukin-6, and tumor necrosis factor-alpha following controlled cortical impact injury in rats. J Neurosurg. 2001;94:782–787. doi: 10.3171/jns.2001.94.5.0782. [DOI] [PubMed] [Google Scholar]
- Sturrock RR. Myelination of the mouse corpus callosum. Neuropathol Appl Neurobiol. 1980;6:415–420. doi: 10.1111/j.1365-2990.1980.tb00219.x. [DOI] [PubMed] [Google Scholar]
- Stys PK. White matter injury mechanisms. Curr Mol Med. 2004;4:113–130. doi: 10.2174/1566524043479220. [DOI] [PubMed] [Google Scholar]
- Stys PK. General mechanisms of axonal damage and its prevention. J Neurol Sci. 2005;233:3–13. doi: 10.1016/j.jns.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Suehiro E, Singleton SH, Stone JR, Povlishock JT. The immunophilin ligand FK506 attenuates the axonal damage associated with rapid rewarming following posttraumatic hypothermia. Exp Neurol. 2001;172:199–210. doi: 10.1006/exnr.2001.7765. [DOI] [PubMed] [Google Scholar]
- Swadlow HA, Waxman SG, Geschwind N. Small-diameter nonmyelinated axons in the primate corpus callosum. Arch Neurol. 1980;37:114–115. doi: 10.1001/archneur.1980.00500510072016. [DOI] [PubMed] [Google Scholar]
- Swanson TH, Krahl SE, Liu YZ, Drazba JA, Rivkees SA. Evidence for physiologically active axonal adenosine receptors in the rat corpus callosum. Brain Res. 1998;784:188–198. doi: 10.1016/s0006-8993(97)01323-1. [DOI] [PubMed] [Google Scholar]