

Abstract
Previous studies have demonstrated that traumatic brain injury (TBI) causes brain edema via aquaporins (AQPs), the water transporting proteins. In the present study, we determined the role of hypoxia inducible factor-1α (HIF-1α), which is a transcription factor in response to physiological hypoxia, in regulating expression of AQP4 and AQP9.
Adult male Sprague-Dawley rats (400–425g) received a closed head injury using the Marmarou weight drop model with a 450 g weight and survived for 1, 4, 24 and 48 hours. Some animals were administered 30 minutes after injury with 2-Methoxyestradiol (2ME2), a naturally occurring metabolite of estradiol which is known to post-transcriptionally down-regulate HIF-1α expression, and sacrificed 4 hours after injury. Real-time PCR and Western blot were used, respectively, to detect gene and protein expressions of manganese superoxide dismutase (MnSOD, showing hypoxic stress), HIF-1α, AQP4, and AQP9.
ANOVA analysis demonstrated a significant (p<0.05) increase in gene expression of MnSOD, HIF-1α, AQP4, and AQP9, starting at 1 hour after injury through 48 hours. Western blot analysis further indicated a significant (p<0.05) increase in protein expression of these molecules at the same time points. Pharmacological inhibition of HIF-1α by 2ME2 reduced the up-regulated levels of AQP4 and AQP9 after TBI.
The present study suggests that hypoxic conditions determined by MnSOD expression after closed head injury contribute to HIF-1α expression. HIF-1α, in turn, up-regulates expression of AQP4 and AQP9. These results characterize the pathophysiological mechanisms, and suggest possible therapeutic targets for TBI patients.
Keywords: close head injury, brain edema, MnSOD, hypoxia, 2ME2
Introduction
Brain edema leading to an expansion of brain volume has a crucial impact on morbidity and mortality following traumatic brain injury (TBI) as it increases intracranial pressure, impairs cerebral perfusion and oxygenation, and contributes to additional ischemic/hypoxic injuries [40]. Recent data have demonstrated a pivotal role of Aquaporins (AQPs) in inducing brain edema after ischemic stroke and TBI [13,14,19,30,31,33,39,40]. AQPs are water-transporting proteins which have been identified as the main water channels to provide a key route for water movement in the brain [13,30,31,40]. AQP4 is expressed in astrocyte foot processes near capillaries and in ependymal cells lining the ventricles-key sites for water movement between the cellular, vascular, and ventricular compartments. AQP4 expression is markedly altered in experimental models and patients with brain injury [3,14,19,38,39]. Transgenic mice lacking AQP4 are partially protected from brain swelling in response to ischemic stroke [23]. There is also evidence that the expression of AQP9 can be found in astrocytic end feet and may play a crucial role in aggravation of traumatic brain edema [4,33]. Although the detrimental effect of AQPs in brain edema, at early stage, has been reported, the upstream pathway by which AQPs were up-regulated after TBI has not been charted to date.
Hypoxia inducible factor-1α (HIF-1α) is a protein normally scarce in cells, but greatly upregulated during ischemia/hypoxia [41]. It is believed that HIF-1α which has been shown to be harmful in cerebral ischemia [9], is a key component of the cellular response to pathophysiologic conditions [16]. HIF-1α accumulation could lead to angiogenesis, glycolysis, erythropoiesis, or cell death mediated by different target genes [37]. A recent study in rats demonstrated that under hypoxic conditions, up-regulation of HIF-1α was associated with AQP4 increase in the brain [18]. However, to our knowledge, whether TBI causes an increase in HIF-1α expression by inducing hypoxic stress, and whether HIF-1α expression affects AQP’s up-regulation after TBI, have not been studied. In the present study, we determined the key role of HIF-1α in regulating expression of AQP4 and AQP9. We determined expression of the marker manganese superoxide dismutase (MnSOD) to indicate hypoxic conditions after TBI.
Materials and Methods
Subject
A total of 32 adult male Sprague-Dawley rats (400–425 g, Charles River, Wilmington, MA) were used. Animals were divided into one control group (n=5) and five TBI groups. TBI groups include animals sacrificed 1 (n=5), 4 (n=5), 24 (n=5), and 48 (n=5) hours after trauma. Additionally, one TBI group was given 2-methoxyestradiol (2ME2) 30 minutes after TBI (n=6) and sacrificed 4 hours after injury.
Close Head Trauma Model
To produce TBI, Marmarou’s rat acceleration impact model [24] was used. Unlike other TBI models that directly injure the brain cortex (fluid percussion and cortical impact), the Marmarou model is a closed-head rather than an open-head TBI model, so it is more representative of actual concussive TBI, which rarely involves penetration of the brain [32]. Briefly, the anesthetized rats were placed prone on a foam-covered platform. A 450 g weight was first aligned with the surface of a steel helmet which was directly attached onto the skull over the sagittal suture and between the bregma and lambda sutures, and then dropped directly onto it from a height of 2 meters. The helmet was placed there to ensure that the skull would not be fractured during the trauma procedure, so the brain would not be directly impacted. Control animals were anesthetized and had the helmet attached to them, but they did not receive the traumatic brain injury.
Inhibition of HIF-1α with Administration of 2ME2
Some animals (n=6) were administered with intravenous injections 30 minutes after closed head injury with 2ME2 (Sigma Aldrich, St. Louis, MO) at a dose of 2.5 mg/kg. 2ME2 is a naturally occurring metabolite of estradiol, which is known to post-transcriptionally down-regulate the expression of HIF-1α[22]. Those animals were sacrificed 4 hours after trauma.
Gene Expression of MnSOD, HIF-1α, AQP4 and AQP9
The traumatized rats were sacrificed at 1, 4, 24, and 48 hours after TBI. Samples from the whole brain of each animal were homogenized and prepared for both mRNA and protein processing. A sensitive real-time reverse transcription (RT) polymerase chain reaction (PCR) technique [11,12] was used to determine expression of genes encoding MnSOD, HIF-1α, AQP4 and AQP9. The total RNA from half the amount of samples containing whole brain was isolated by using a RNA STAT-60 kit according to the manufacturer’s instructions (Invitrogen, Gaithersburg, MD). Random Primers from Promega were used to create First-strand DNA synthesis using SuperScript RNase H Reverse Transcriptase kit (Invitrogen). The cDNA was then amplified using an ABI Prism 7900HT sequencing detection system for real-time PCR with SYBR Green PCR Master Mix from Applied Biosystems. The gene specific rat primers for MnSOD, HIF-1α, AQP4 and AQP9 were designed or obtained from previous studies (Table 1). For internal PCR control, the rat ribosomal protein L32 (rpL32) was used as a housekeeping gene for each sample. Reactions were performed in a 20 μl volume with 0.5 μM primers. PCR cycles consist of an initial denaturation step at 95°C for 9 min, followed by 45 cycles of a 95° C denaturation for 10 seconds, 60°C annealing for 5 seconds, and 72°C extension for 15 seconds. The relative mRNA levels of gene expression were determined using the threshold cycle (CT) and arithmetic formulas [11,12]. Subtracting the CT of the housekeeping gene from the CT of target gene yields the ΔCT in each group (control and experimental groups), which was entered into the equation 2−ΔCT and calculated for the exponential amplification of PCR. The mean amount of gene from control group was arbitrarily assigned a value of 1 to serve as a reference. The expression of the target gene from experimental groups therefore represents the fold-difference expression relative to the reference gene.
Table 1.
Sequences of primers
| Gene | Primer/probe | Sequence |
|---|---|---|
| MnSOD | Primer (forward): | 5′-GTGGTGGAGAACCCAAAGGA-3′ |
| Primer (reverse): | 5′-GCGTGCTCCCACACATCAAT-3′ | |
| HIF-1α | Primer (forward): | 5′-ACTTGGACGCTCTGCCTATG-3′ |
| Primer (reverse): | 5′-TTGCGGGGGTTGTAGA-3′ | |
| AQP4 | Primer (forward): | 5′-TTGGACCAATCATAGGCGC-3′ |
| Primer (reverse): | 5′-GGTCAATGTCGATCACATGC-3′ | |
| AQP9 | Primer (forward): | 5′-GATGCCTTCTGAGAAGGACG-3′ |
| Primer (reverse): | 5′-AGAGAGCCATCACGACTGC-3′ | |
| rpL32 | Primer (forward): | 5′-TGTCCTCTAAGAACCGAAAAGCC-3′ |
| Primer (reverse): | 5′-CGTTGGGATTGGTGACTCTGA-3′ |
Protein Expression
To investigate protein synthesis of MnSOD, HIF-1α, AQP4 and AQP9, Western blot analysis was used [20]. Samples containing both cerebral hemispheres were processed in lysis buffer including protease inhibitors on ice. Equal volumes (10 ul) of tissue extracts normalized by protein concentration were mixed with sodium dodecyl sulfate (SDS) sample buffer. The samples were separated by electrophoresis through 10% polyacrylamide gel (Bio-Rad, Hercules, CA) and then transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA). Supernatants were used as whole-tissue lysates and protein concentration was determined using the Bradford assay (Bio-Rad, CA, USA). Four different primary antibodies were used: polyclonal rabbit anti-MnSOD (1:1,000, Santa Cruz), polyclonal rabbit anti-HIF-1α(1:2,000, Santa Cruz), polyclonal rabbit anti-AQP4 (1:500, Santa Cruz), and polyclonal anti-AQP9 (1:2000, Santa Cruz). Protein equal loading was confirmed by intracellular protein β-actin (goat polyclonal anti-β-actin antibody, dilution 1:1,000, Santa Cruz). These antibodies were incubated with the membrane at 25° C for 1 hour. After 3 wash cycles, the membrane was then incubated with secondary antibody conjugated to horseradish peroxidase (Sigma) for 30 minutes. Finally, the targeted antigens were visualized by using the standard chemical luminescence methods (ECL, Amersham Parmacia Biotech). To quantify the relative levels of target protein expression, blot images were analyzed using an image analysis program (ChemiGeniusQ System) and the expression intensity of the proteins from different groups was statistically compared.
Statistical analysis
All the data was described as mean ± SE. Statistical analysis was performed with SPSS for Windows, version 13.0 (SPSS, Inc.). The differences among multiple groups was assessed using one-way analysis of variance (ANOVA) with a significance level at p<0.05. Post-hoc comparison between groups was further detected using the least significant difference (LSD) method.
Results
ANOVA and Post-hoc analysis demonstrates the significant increase in MnSOD [F(4,21)=4.840, p<0.01], HIF-1α [F(4,21)=15.148, p<0.01], AQP4 [F(4,21)=3.864, p<0.01] and AQP9 [F(4,21)=6.974, p<0.01] expression at the transcription level as soon as 1 hour post-TBI, and persisting for 48 hours (Figure 1). AQP4 mRNA expression peaked at 4 hours, while AQP9 mRNA expression continued to rise, peaking at 48 hours, mirroring similar results with protein levels. Figure 2 shows a significant elevation of MnSOD [F(4,21)=35.407, p<0.01] and HIF-1α [F(4,21)=2.645, p<0.05] as well as AQP4 [F(4,21)=3.862, p<0.05] and AQP9 [F(4,21)=17.043, p<0.01] protein determined by Western blot after brain trauma, which start as early as 1 hours post-TBI and persist at 4 through 48 hours. Again, AQP4 protein expression peaked at 4 hours, followed by AQP9 mRNA expression, peaking at 48 hours. Since AQP4 expression peaked at 4 hours post-TBI, 2ME2 was administered to the TBI rats at this time point. We demonstrated mRNA and protein expression of AQP4 and 9 at control, 4 hours post-TBI levels, and 4 hours post-TBI with injection of 2ME2 levels in Figure 3. ANOVA analysis demonstrated a significant reversal of AQP4 [F(2,14)=4.334, p<0.05] and AQP9 [F(2,14)=22.384, p<0.01] up-regulation caused by TBI when 2ME2 was administered to the rats. Gene expression of AQP4 and AQP9 returned to near control levels. Correspondingly, protein expression of AQP4 and AQP9 were significantly [F(2,14)=9.179, p<0.01; F(2,14)=24.330, p<0.01] increased. Post-hoc analysis indicates that intravenous injection of 2ME2 30 minutes after TBI significantly (p<0.05) reversed those changes and kept AQP4 and AQP9 expression at or below control levels. The data suggest a key role of HIF-1α n inducing AQP expression.
Figure 1.
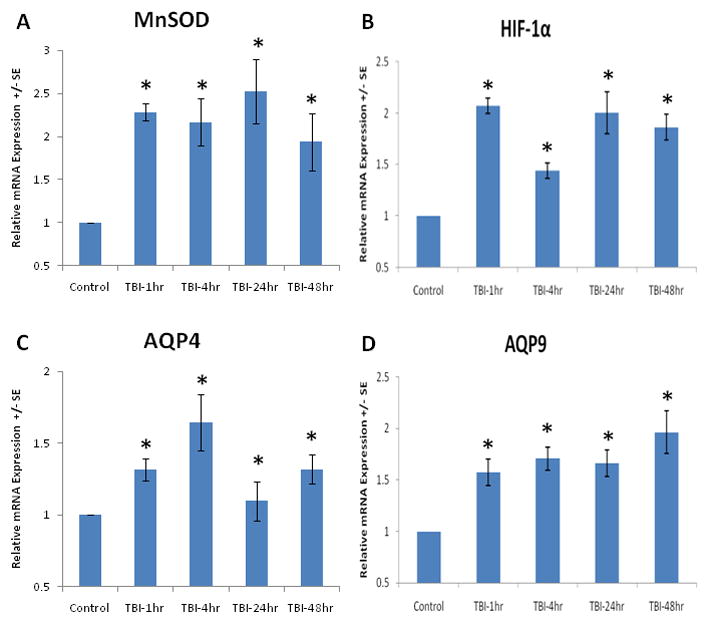
Real time PCR analysis demonstrates the significant (p<0.05, indicated by *) increase in MnSOD (A), HIF-1α (B), AQP4 (C) and AQP9 (D) gene expression as soon as 1 hour post-TBI, and persisting for 48 hours. AQP4 mRNA expression peaked at 4 hours, followed by AQP9 mRNA expression, peaking at 48 hours. Values are mean +/− SE.
Figure 2.

Expression of MnSOD (A) and HIF-1α (B) as well as AQP4 (C) and AQP9 (D) protein determined by Western blot after brain trauma. A significant (p<0.05, indicated by *) elevation of the target proteins start as early as 1 hours post-TBI and persist at 4 through 48 hours. Again, AQP4 protein expression peaked at 4 hours, followed by AQP9 expression, peaking at 48 hours. Protein equal loading was confirmed by intracellular protein β-actin. Representative immunoblots are presented. Values are mean +/− SE.
Figure 3.

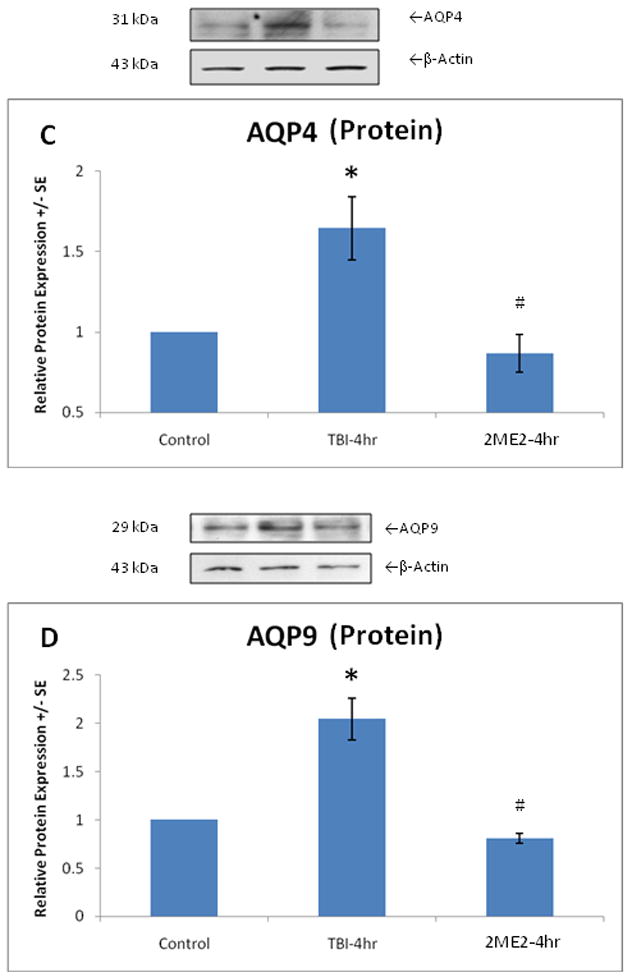
We demonstrated mRNA and protein expression of AQP4 and 9 at control, 4 hours post-TBI, and 4 hours post-TBI with injection of 2ME2 levels. Real time PCR analysis demonstrated a significant reversal (p<0.05, indicated by #) of AQP4 (A) and AQP9 (B) gene up-regulation due to TBI (p<0.05, indicated by *) when 2ME2 is administered to the rats. Transcription of AQP4 and AQP9 returned to near control levels. Protein expression of AQP4 (C) and AQP9 (D) are significantly (p<0.05, indicated by *) increased, but intravenous injection of 2ME2 30 minutes post-TBI significantly (p<0.05, indicated by #) reversed those changes and kept AQP4 and AQP9 expression at or below control levels. Protein equal loading was confirmed by intracellular protein β-actin. Representative immunoblots are presented. Values are mean +/− SE.
Discussion
Marmarou’s model used in this study does not produce contusional focal lesions that directly cause nerve cell injury and death, but instead causes extensive diffuse axonal injury with little cell death [32]. The present study demonstrated that free radical production and oxidative stress indicated by MnSOD expression, which likely associates with brain hypoxia after TBI, are concurrent with HIF-1α expression. HIF-1α, in turn, up-regulates expression of AQP4 and AQP9.
Aquaporins (AQPs) are a family of water channel proteins that facilitate the diffusion of water through the plasma membrane [1,33]. In the rodent brain, three aquaporins, AQP1, AQP4, and AQP9, have been identified [2,5]. AQP1 was detected in epithelial cells of the choroid plexus [29], AQP4 in astrocytes with a polarization on astrocyte endfeet [28], and AQP9 in astrocytes of the white matter and in catecholaminergic neurons [4,6]. AQP1 and AQP4 are permeable to water and are presumed to be involved in cerebrospinal fluid formation and brain water homeostasis [25]. AQP9 is an aquaglyceroporin, a subgroup of the aquaporin family, and is permeable to water and also glycerol, monocarboxylates, and urea [6]. Studies have indicated that AQP4 and AQP9 mainly induce cytotoxic/cellular brain edema due to sustained intracellular water collection [31,42]. Studies also demonstrated that the AQPs involved in vasogenic edema due to blood–brain barrier (BBB) disruption resulting in extracellular water accumulation [13,14,33]. The present study provides a temporal expression pattern of AQP4 and −9 after TBI, showing an early expression of AQP4 followed by AQP9. This suggests that AQP4 up-regulation contributes to AQP9 expression.
Expression of an endogenous antioxidant enzyme, superoxide dismutase (SOD) is involved in oxidative stress under hypoxic/ischemic conditions [7,10,27]. There are three isoforms of SOD: mitochondrial manganese SOD (MnSOD), cytosolic copper–zinc SOD (CuZnSOD) and extracellular SOD (EcSOD). MnSOD production is increased during hypoxia or ischemia [7,10,27]. In this study, MnSOD was used to measure hypoxic stress in response to insult of TBI. The increase in MnSOD was found and associated with HIF-1α expression.
HIF-1α is a subunit of HIF-1, a heterodimeric transcription factor containing an inducible HIF-1α subunit and a constitutive HIF-1β subunit. HIF-1α is an oxygen sensor that plays a central role in the maintenance of oxygen homeostasis in body tissues [35,36]. Under normoxic conditions, the HIF-1α protein is expressed at very low levels due to rapid ubiquination and proteasomal degradation [17]. When oxygen is not present, though, the chemical pathway that degrades it cannot operate, so the protein rapidly accumulates [41]. HIF-1α was up-regulated under pathologic conditions such as hypoxia or ischemia due to the inhibition of degradation [21,26]. It is believed that oxygen tension plays a crucial role in the activation of HIF-1α [9,41]. In hypoxia, a previous study has shown that up-regulation of HIF-1α associated with disruption of BBB and increased permeability of blood vessels was linked to up-regulation of AQP4 which involved astrocytes in brain edema [18]. This present study further supports a notion that HIF-1α is up-regulated under hypoxic conditions determined by MnSOD production, which, in turn, leads to AQP4 and AQP9 over-expression after closed head TBI determined by administration of the HIF-1α inhibitor 2ME2. However, previous studies demonstrated that HIF-1 induces vascular endothelial cell growth factor (VEGF) expression under hypoxic stress [8,15], and that HIF-1α inhibition by 2ME2 may also inhibit VEGF activity, which up-regulates AQP4 mRNA and protein in brain [34] as well. Thus, a pathway via VEGF upregulation in AQP expressions and functions cannot be ruled out in our TBI model.
Although the focus of this study was to characterize the pathophysiological mechanisms of AQPs expression after TBI, it would be very likely that treatment with the HIF-1α inhibitor 2ME2 may benefit neurological outcomes by reducing water contents or brain swelling rates after brain trauma. Insight into the mechanisms by which brain AQPs are up-regulated could be of utmost clinical importance. Perturbed brain edema has been implicated in many neurological diseases including head trauma, stroke, and brain cancer, and water flow via AQP4 and AQP9 has a harmful effect in brain edema. Thus, our work suggests possible therapeutic targets along the HIF-AQP pathway for TBI patients with brain edema.
Acknowledgments
We are grateful to Miao Guo for technical assistance. This work was supported partially by NIH NS 39860 to José A. Rafols.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCE LIST
- 1.Agre P, King LS, Yasui M, Guggino WB, Ottersen OP, Fujiyoshi Y, Engel A, Nielsen S. Aquaporin water channels--from atomic structure to clinical medicine. J Physiol. 2002;542:3–16. doi: 10.1113/jphysiol.2002.020818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agre P, Nielsen S, Ottersen OP. Towards a molecular understanding of water homeostasis in the brain. Neuroscience. 2004;129:849–850. doi: 10.1016/j.neuroscience.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Aoki K, Uchihara T, Tsuchiya K, Nakamura A, Ikeda K, Wakayama Y. Enhanced expression of aquaporin 4 in human brain with infarction. Acta Neuropathol. 2003;106:121–124. doi: 10.1007/s00401-003-0709-y. [DOI] [PubMed] [Google Scholar]
- 4.Badaut J, Hirt L, Granziera C, Bogousslavsky J, Magistretti PJ, Regli L. Astrocyte-specific expression of aquaporin-9 in mouse brain is increased after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:477–482. doi: 10.1097/00004647-200105000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Badaut J, Lasbennes F, Magistretti PJ, Regli L. Aquaporins in brain: distribution, physiology, and pathophysiology. J Cereb Blood Flow Metab. 2002;22:367–378. doi: 10.1097/00004647-200204000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Badaut J, Regli L. Distribution and possible roles of aquaporin 9 in the brain. Neuroscience. 2004;129:971–981. doi: 10.1016/j.neuroscience.2004.06.035. [DOI] [PubMed] [Google Scholar]
- 7.Bemeur C, Ste-Marie L, Desjardins P, Butterworth RF, Vachon L, Montgomery J, Hazell AS. Expression of superoxide dismutase in hyperglycemic focal cerebral ischemia in the rat. Neurochem Int. 2004;45:1167–1174. doi: 10.1016/j.neuint.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 8.Carmeliet P, Storkebaum E. Vascular and neuronal effects of VEGF in the nervous system: implications for neurological disorders. Semin Cell Dev Biol. 2002;13:39–53. doi: 10.1006/scdb.2001.0290. [DOI] [PubMed] [Google Scholar]
- 9.Chen C, Hu Q, Yan J, Lei J, Qin L, Shi X, Luan L, Yang L, Wang K, Han J, Nanda A, Zhou C. Multiple effects of 2ME2 and D609 on the cortical expression of HIF-1alpha and apoptotic genes in a middle cerebral artery occlusion-induced focal ischemia rat model. J Neurochem. 2007;102:1831–1841. doi: 10.1111/j.1471-4159.2007.04652.x. [DOI] [PubMed] [Google Scholar]
- 10.Danielisova V, Gottlieb M, Nemethova M, Burda J. Activities of endogenous antioxidant enzymes in the cerebrospinal fluid and the hippocampus after transient forebrain ischemia in rat. J Neurol Sci. 2007;253:61–65. doi: 10.1016/j.jns.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Ding Y, Young C, Li J, Luan X, McAllister JP, II, Clark J, Diaz FG. Reduced inflammatory mediator expression by pre-reperfusion infusion into ischemic territory: a real-time polymerase chain reaction analysis. Neurosci Lett. 2003;353:173–176. doi: 10.1016/j.neulet.2003.09.055. [DOI] [PubMed] [Google Scholar]
- 12.Gerard HC, Wang Z, Whittum H, El G, Goldbach M, Bardin T, Schumacher HR, Hudson AP. Cytokine and chemokine mRNA produced in synovial tissue chronically infected with Chlamydia trachomatis and C. pneumoniae. J Rheumatol. 2002;29:1827–1835. [PubMed] [Google Scholar]
- 13.Griesdale DE, Honey CR. Aquaporins and brain edema. Surg Neurol. 2004;61:418–421. doi: 10.1016/j.surneu.2003.10.047. [DOI] [PubMed] [Google Scholar]
- 14.Guo Q, Sayeed I, Baronne LM, Hoffman SW, Guennoun R, Stein DG. Progesterone administration modulates AQP4 expression and edema after traumatic brain injury in male rats. Exp Neurol. 2006;198:469–478. doi: 10.1016/j.expneurol.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 15.Harmey JH, Bouchier-Hayes D. Vascular endothelial growth factor (VEGF), a survival factor for tumour cells: implications for anti-angiogenic therapy. Bioessays. 2002;24:280–283. doi: 10.1002/bies.10043. [DOI] [PubMed] [Google Scholar]
- 16.Helton R, Cui J, Scheel JR, Ellison JA, Ames C, Gibson C, Blouw B, Ouyang L, Dragatsis I, Zeitlin S, Johnson RS, Lipton SA, Barlow C. Brain-specific knock-out of hypoxia-inducible factor-1alpha reduces rather than increases hypoxic-ischemic damage. J Neurosci. 2005;25:4099–4107. doi: 10.1523/JNEUROSCI.4555-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 18.Kaur C, Sivakumar V, Zhang Y, Ling EA. Hypoxia-induced astrocytic reaction and increased vascular permeability in the rat cerebellum. Glia. 2006;54:826–839. doi: 10.1002/glia.20420. [DOI] [PubMed] [Google Scholar]
- 19.Kleffner I, Bungeroth M, Schiffbauer H, Schabitz WR, Ringelstein EB, Kuhlenbaumer G. The role of aquaporin-4 polymorphisms in the development of brain edema after middle cerebral artery occlusion. Stroke. 2008;39:1333–1335. doi: 10.1161/STROKEAHA.107.500785. [DOI] [PubMed] [Google Scholar]
- 20.Kreipke CW, Morgan R, Roberts G, Bagchi M, Rafols JA. Calponin phosphorylation in cerebral cortex microvessels mediates sustained vasoconstriction after brain trauma. Neurol Res. 2007;29:369–374. doi: 10.1179/016164107X204684. [DOI] [PubMed] [Google Scholar]
- 21.Li L, Xiong Y, Qu Y, Mao M, Mu W, Wang H, Mu D. The requirement of extracellular signal-related protein kinase pathway in the activation of hypoxia inducible factor 1 alpha in the developing rat brain after hypoxia-ischemia. Acta Neuropathol. 2008;115:297–303. doi: 10.1007/s00401-008-0339-5. [DOI] [PubMed] [Google Scholar]
- 22.Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, Willard MT, Zhong H, Simons JW, Giannakakou P. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3:363–375. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 23.Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- 24.Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J Neurosurg. 1994;80:291–300. doi: 10.3171/jns.1994.80.2.0291. [DOI] [PubMed] [Google Scholar]
- 25.miry-Moghaddam M, Ottersen OP. The molecular basis of water transport in the brain. Nat Rev Neurosci. 2003;4:991–1001. doi: 10.1038/nrn1252. [DOI] [PubMed] [Google Scholar]
- 26.Mu D, Jiang X, Sheldon RA, Fox CK, Hamrick SE, Vexler ZS, Ferriero DM. Regulation of hypoxia-inducible factor 1alpha and induction of vascular endothelial growth factor in a rat neonatal stroke model. Neurobiol Dis. 2003;14:524–534. doi: 10.1016/j.nbd.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 27.Nemethova M, Danielisova V, Gottlieb M, Burda J. Post-conditioning exacerbates the MnSOD immune-reactivity after experimental cerebral global ischemia and reperfusion in the rat brain hippocampus. Cell Biol Int. 2008;32:128–135. doi: 10.1016/j.cellbi.2007.08.023. [DOI] [PubMed] [Google Scholar]
- 28.Nielsen S, Nagelhus EA, miry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17:171–180. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nielsen S, Smith BL, Christensen EI, Agre P. Distribution of the aquaporin CHIP in secretory and resorptive epithelia and capillary endothelia. Proc Natl Acad Sci U S A. 1993;90:7275–7279. doi: 10.1073/pnas.90.15.7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papadopoulos MC, Krishna S, Verkman AS. Aquaporin water channels and brain edema. Mt Sinai J Med. 2002;69:242–248. [PubMed] [Google Scholar]
- 31.Papadopoulos MC, Verkman AS. Aquaporin-4 and brain edema. Pediatr Nephrol. 2007;22:778–784. doi: 10.1007/s00467-006-0411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rafols JA, Morgan R, Kallakuri S, Kreipke CW. Extent of nerve cell injury in Marmarou’s model compared to other brain trauma models. Neurol Res. 2007;29:348–355. doi: 10.1179/016164107X204657. [DOI] [PubMed] [Google Scholar]
- 33.Ribeiro MC, Hirt L, Bogousslavsky J, Regli L, Badaut J. Time course of aquaporin expression after transient focal cerebral ischemia in mice. J Neurosci Res. 2006;83:1231–1240. doi: 10.1002/jnr.20819. [DOI] [PubMed] [Google Scholar]
- 34.Rite I, Machado A, Cano J, Venero JL. Intracerebral VEGF injection highly upregulates AQP4 mRNA and protein in the perivascular space and glia limitans externa. Neurochem Int. 2008;52:897–903. doi: 10.1016/j.neuint.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 35.Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 36.Semenza GL. Expression of hypoxia-inducible factor 1: mechanisms and consequences. Biochem Pharmacol. 2000;59:47–53. doi: 10.1016/s0006-2952(99)00292-0. [DOI] [PubMed] [Google Scholar]
- 37.Semenza GL, Agani F, Feldser D, Iyer N, Kotch L, Laughner E, Yu A. Hypoxia, HIF-1, and the pathophysiology of common human diseases. Adv Exp Med Biol. 2000;475:123–130. doi: 10.1007/0-306-46825-5_12. [DOI] [PubMed] [Google Scholar]
- 38.Sorani MD, Zador Z, Hurowitz E, Yan D, Giacomini KM, Manley GT. Novel variants in human Aquaporin-4 reduce cellular water permeability. Hum Mol Genet. 2008;17:2379–2389. doi: 10.1093/hmg/ddn138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun MC, Honey CR, Berk C, Wong NL, Tsui JK. Regulation of aquaporin-4 in a traumatic brain injury model in rats. J Neurosurg. 2003;98:565–569. doi: 10.3171/jns.2003.98.3.0565. [DOI] [PubMed] [Google Scholar]
- 40.Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 2004;129:1021–1029. doi: 10.1016/j.neuroscience.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 41.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zador Z, Bloch O, Yao X, Manley GT. Aquaporins: role in cerebral edema and brain water balance. Prog Brain Res. 2007;161:185–194. doi: 10.1016/S0079-6123(06)61012-1. [DOI] [PubMed] [Google Scholar]