

Abstract
CaBP4 is a calmodulin-like neuronal calcium-binding protein that is crucial for the development and/or maintenance of the cone and rod photoreceptor synapse. Previously, we showed that CaBP4 directly regulates Cav1 L-type Ca2+ channels, which are essential for normal photoreceptor synaptic transmission. Here, we show that the function of CaBP4 is regulated by phosphorylation. CaBP4 is phosphorylated by protein kinase C ζ (PKCζ) at serine 37 both in vitro and in the retina and colocalizes with PKCζ in photoreceptors. CaBP4 phosphorylation is greater in light-adapted than dark-adapted mouse retinas. In electrophysiological recordings of cells transfected with Cav1.3 and CaBP4, mutation of the serine 37 to alanine abolished the effect of CaBP4 in prolonging the Ca2+ current through Cav1.3 channel, whereas inactivating mutations in the CaBP4 Ca2+-binding sites strengthened Cav1.3 modulation. These findings demonstrate how light-stimulated changes in CaBP4 phosphorylation and Ca2+ binding may regulate presynaptic Ca2+ signals in photoreceptors.
Keywords: CaBP4, Ca2+-binding proteins, phosphorylation, protein kinase C ζ, Ca2+ channel, photoreceptors
Introduction
CaBP4 belongs to a subfamily of neuronal Ca2+-binding proteins (CaBPs) with high similarity to calmodulin (CaM) (Haeseleer et al., 2000, 2002, 2004; Haeseleer and Palczewski, 2002; Maeda et al., 2005). CaBPs bind to and yet differentially modulate a number of CaM targets, including voltage-gated Ca2+ channels (Lee et al., 2002; Zhou et al., 2004; Yang et al., 2006), transient receptor potential TRP channels (Kinoshita-Kawada et al., 2005), and inositol 1,4,5-trisphosphate (IP3) receptors (Yang et al., 2002; Haynes et al., 2004; Kasri et al., 2004). Through interactions with these and potentially other effectors, CaBPs may enhance the Ca2+ signaling potential in neurons and other excitable cells.
In the retina, CaBP4 is localized in the photoreceptor synaptic terminal and is required for normal neurotransmission to bipolar cells (Haeseleer et al., 2004; Maeda et al., 2005). Mice lacking CaBP4 (Cabp4−/−) exhibit synaptic defects and electroretinograms, indicating impaired cone and rod synaptic function. Multiple lines of evidence suggest that visual deficits in Cabp4−/− mice result from dysregulation of presynaptic Cav1 L-type Ca2+ channels that mediate transmitter release. First, CaBP4 interacts with the pore-forming α1 subunit of Cav1 L-type Ca2+ channels and modulates the activation properties of these channels in transfected human embryonic kidney HEK293T cells (Haeseleer et al., 2004; Yang et al., 2006). Second, defects in visual transmission and retina morphology in Cabp4−/− mice are similar to those in mice lacking α11.4 or the auxiliary Cav β2 subunit (Ball et al., 2002; Mansergh et al., 2005; Chang et al., 2006). Third, human mutations in genes encoding CaBP4 (Zeitz et al., 2006) and α11.4 (Bech-Hansen et al., 1998; Strom et al., 1998) are linked to incomplete congenital stationary night blindness (CSNB2). Thus, elucidating the factors controlling CaBP4 activity may yield insights into the underlying pathology of CSNB2.
Light-dependent phosphorylation regulates the physiological activity of several key players in the phototransduction cascade, including rhodopsin, opsin, phosducin, RGS-9 (regulator of G-protein signaling 9), centrins, β-subunit of the insulin receptor, abLIM (actin binding LIM protein family), peripherin, and phosphodiesterase (Kuhn and Dreyer, 1972; Lee et al., 1984; Boesze-Battaglia et al., 1997; Roof et al., 1997; Hayashi et al., 2000; Hu et al., 2001; Rajala et al., 2002; Trojan et al., 2003). Phosphorylation of Ca2+-binding proteins, including CaM, can affect their Ca2+-binding capability and interaction with target proteins (Benaim and Villalobo, 2002). Casein kinase 2 (CK2) phosphorylates several amino acid residues in CaM, including serine 101, which decreases the affinity of CaM for its substrates (Quadroni et al., 1998; Bildl et al., 2004; Kasri et al., 2004; Allen et al., 2007). Similar to CaM, an analogous residue (Serine 120) in CaBP1 is phosphorylated by casein kinase 2, which weakens the effect of CaBP1 in inhibiting Ca2+ release by the IP3 receptor (Kasri et al., 2004).
In this study, we investigated whether phosphorylation of CaBP4 might similarly regulate the activity of CaBP4. We provide evidence that CaBP4 is phosphorylated by protein kinase C ζ (PKCζ) in the retina and that phosphorylation and Ca2+ binding have opposite effects on CaBP4 modulation of Cav1 L-type Ca2+ channels. We also show that CaBP4 phosphorylation levels change with different light conditions, which may underlie light-dependent regulation of CaBP4 targets.
Materials and Methods
Animals.
C57BL/6J mice were originally obtained from The Jackson Laboratory (Bar Harbor, ME). C57BL/6J, Cabp4−/− (Haeseleer et al., 2004), Gnat1−/− (rod transducin α-subunit null; provided by Dr. Janis Lem, Tufts-New England Medical Center, Boston, MA) (Calvert et al., 2000) and Gnat1−/− Cabp4−/− (Maeda et al., 2005) mice were bred in the animal facility at the University of Washington. Their eyeballs were used as a source of retinal proteins and for immunohistochemistry. All the procedures for the maintenance and use of animals were approved by the Institutional Animal Care and Use Committee of the University of Washington.
Reagents.
Casein kinase II inhibitor I [4,5,6,7-tetrabromobenzotriazole (TBB)], H-89 (N-[2-(p-bromo-cinnamylamino)-ethyl]-5-isoquinoline-sulfon-amide 2HCl), Bisindolymaleimide I (Bis), 4-amino-5-(-4-chlorophenyl)-7-(t-butyl)pyrazolo [3,4-d] pyrimidine (PP2), Rottlerin, Gö6976 [12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo[2,3-a]pyrrolo[3,4-c] carbazole], Gö6983(2-[1-(3-dimethylaminopropyl)-5-methoxyindol-3-yl]-3-(1H-indol-3-yl)maleimide), protein kinase Cζ pseudosubstrate inhibitor, phorbol-12-myristate-13-acetate (PMA), 1,2-dioleoyl-sn-glycerol (DAG), and recombinant PKCζ were purchased from Calbiochem (La Jolla, CA). l-α-Phosphatidyl-l-serine (PS) was purchased from Sigma (St. Louis, MO). Recombinant PKCα and lipid activator (mix of PS and DAG) were obtained from Upstate Biotechnology (Lake Placid, NY). Doxycycline and hygromycinB were purchased from BD Biosciences (Palo Alto, CA). The 1 kb DNA ladder was purchased from Invitrogen (Carlsbad, CA).
Antibodies.
Commercially available antibodies were as follows: alkaline phosphatase-conjugated anti-mouse and anti-rabbit (Promega, Madison, WI); mouse anti-polyhistidine (6His) tag (EMD Biosciences Novagen, Madison, WI); mouse anti-glutathione S-transferase (GST) and anti-PKCα (Santa Cruz Biotechnology, Santa Cruz, CA); rabbit anti-PKCζ (Oxford Biomedical Research, Oxford, MI); cyanine 3 (Cy3) goat anti-rabbit (Jackson ImmunoResearch, West Grove, PA); and Alexa Fluor 488 goat anti-mouse (Invitrogen).
Development and characterization of rabbit anti-CaBP4 (UW145) was described by Haeseleer et al. (2004). Bacterially expressed CaBP4 was used to immunize BALB/c mice to obtain mouse anti-CaBP4 antisera.
The phospho-Ser37-specific antibody was targeted against a sequence in mouse CaBP4 (NH2-CPALTRRRpSKKESWHP-COOH). The corresponding peptide was obtained from Mimotopes (Clayton, Victoria, Australia) and contained an N-terminal cysteine residue for covalent linkage to keyhole limpet hemocyanin as a carrier protein for generating antibodies. The peptide was injected into a New Zealand white rabbit (Cocalico Biologicals, Reamstown, PA), and polyclonal antibodies were purified from the serum by affinity chromatography on a phospho-peptide–Sepharose column. After elution, the affinity-purified antibodies were depleted of the antibodies that react with the nonphosphorylated peptide by passing through a CaBP4–Sepharose column prepared with nonphosphorylated, bacterially expressed CaBP4.
Kinase assay.
Dark-adapted mouse retinas were dissected and sonicated in dim red light in 100 μl of 10 mm bis-Tris-propane (BTP), pH 8.0, containing a mixture of proteases inhibitor (Sigma). Phosphorylation experiments were performed in the dark using 5 μl of retinal extract and 5 μCi of [γ-32P]ATP in kinase buffer (10 mm BTP, pH 8.0, 1 mm MgCl2, 1 mm Na3VO4, 1 mm NaF, 1 mm dithiothreitol, and 500 μm ATP) for 10 min at 30°C. CaCl2 or EGTA (1 mm each) was added to the kinase assay when indicated. His-tagged or GST-tagged CaBP4 purified from bacteria (1 or 0.2 μg) was included in the reaction as indicated. The reaction was terminated with the addition of SDS sample buffer. The reaction product was subjected to SDS-PAGE followed by transfer to Immobilon membrane and Western blotting with the anti-CaBP4 antibody (UW145) or Ponceau staining. Alternatively, the gels were treated with Coomassie blue and dried. Incorporation of 32P in CaBP4 was detected by autoradiography. For characterization of the endogenous kinase, soluble proteins were isolated by centrifugation of the retinal extract for 15 min at 13,000 × g at 4°C. The pellet was washed with 10 mm BTP (100 μl) and centrifuged for 15 min at 13,000 × g at 4°C. The supernatants represented the soluble fraction. The pellet was resuspended in 10 mm BTP (100 μl) and constituted the membrane protein fraction. The following changes were made to the assay buffer when indicated: EDTA (10 mm) was added; MgCl2 (1 mm) was substituted with MnCl2 (1 mm); PMA (5 μm), PS (0.1 mg/ml), and/or DAG (0.01 mg/ml) were added. Protein kinase inhibitor concentrations used in the assay are described in the figures and were added to the reaction mix described above. Analysis of CaBP4 phosphorylation in HEK293 cells was performed as described above except that the retina extract was substituted with a lysate from HEK293 cells.
Kinase assay with recombinant PKC.
GST-tagged CaBP4 (10 μg) and GST–S37A (10 μg) mutant were phosphorylated in vitro with recombinant PKCα (rPKCα) (0.1 μg) in a 200 μl reaction containing 20 mm HEPES, pH 8.0, 0.03% Triton X-100, 0.1 mm ATP, 1 mm MgCl2, 1 mm CaCl2, 20 μl of lipid activator (0.1 mg/ml PS, 0.01 mg/ml DAG, final concentration) and 25 μCi of [γ-32P]ATP for 1 h at 30°C. The reactions were terminated by adding an equal volume of 2× sample buffer. For phosphorylation of the GST-tagged proteins (10 μg) by rPKCζ (0.2 μg), the reactions were performed in 200 μl containing 20 mm HEPES, pH 8.0, 0.03% Triton X-100, 0.1 mm ATP, 1 mm MgCl2, 1 mm EGTA, and 25 μCi of [γ-32P]ATP for 1 h at 30°C. The reactions were terminated by adding an equal volume of 2× sample buffer or on ice. Protein kinase inhibitor concentrations used in the assay are described in the figure legends and were added to the reaction mix. For affinity chromatography, purified His-tagged CaBP4 (300 μg) was coupled to cyanogen bromide (CNBr)-activated Sepharose, which was incubated with rPKCζ (3 μg) in a 1.5 ml kinase reaction as described above. The reactions were terminated by washing the CaBP4–Sepharose three times with PBS and 0.5% Triton X100.
Immunoprecipitation of radiolabeled native CaBP4.
CaBP4 was immunoprecipitated from the kinase assay reactions using anti-CaBP4 polyclonal antibodies. The phosphorylation experiments were performed as described above. The reactions were stopped by incubation on ice. For the immunoprecipitation experiments, the reactions were performed in light conditions unless specified as performed in the dark (dim red light). The samples were diluted with 2 vol of PBS containing 0.5% Triton-X100 and cleared by incubation with 40 μl of protein-G agarose for 4 h at 4°C. After centrifugation, 10 μg of affinity-purified anti-CaBP4 antibodies were added to the supernatant and incubated for 1 h at 4°C. Protein G agarose (40 μl) was added and incubated overnight at 4°C. The pellets were washed five times for 10 min with 1 ml of PBS and 0.5% Triton X-100. The immunoprecipitated proteins were eluted after boiling for 5 min in SDS-PAGE sample buffer and subject to SDS-PAGE followed by transfer on Immobilon membrane and Western blotting with anti-CaBP4 antibodies. The blot was then exposed to an x-ray film.
Generation of CaBP4 mutants.
The cloning of all deletion mutants was performed using PCR/subcloning. The deletion mutants of CaBP4 were generated by PCR with primers confined at the 5′ and 3′ end of the truncated segments and the resulting sequence subcloned into the pENTR-topo vector (Invitrogen). CaBP4–S37A was generated using appropriate mutagenic primers with the QuickChange Site-Directed mutagenesis kit (Stratagene, Cedar Creek, TX) according to the protocol of the manufacturer. To generate the mutant of CaBP4 that does not bind Ca2+ [CaBP4-EF-hand Ca2+-binding motif (EF)], the glutamic acid in position 12 of the EF1, EF3, and EF4 loops was mutated to a glutamine (Glu149Gln, Glu226Gln, Glu263Gln) using appropriate primers with the QuickChange Site-Directed mutagenesis kit (Stratagene). The sequence of all constructs was confirmed by DNA sequencing. The cDNA sequences encoding wild-type (WT) or mutated CaBP4 were then subcloned, using the Gateway Technology System (Invitrogen), into the Gateway expression vector pDest17 in fusion to a 6His tag and purified using Ni-NTA agarose (Qiagen, Valencia, CA) or into the pDest15 vector for fusion to a GST tag and purified on Glutathione resin (Promega) as recommended by the manufacturer.
The cDNA sequence encoding 6His-tagged wild-type or CaBP4–S37A was also subcloned into the pTRE-hyg vector (BD Biosciences) to generate an inducible stable cell line. T-Rex-293 cells (HEK293 cells stably expressing the tetracycline repressor) (Invitrogen) were transfected with the recombinant plasmids using Lipofectamine 2000 (Invitrogen). After doxycycline-induced CaBP4 expression, stable cell lines were selected for the highest protein levels from colonies isolated in the presence of hygromycin following the protocol of the manufacturer. The 6His-tagged proteins were purified from HEK293 cells using Ni-NTA agarose (Qiagen) following the protocol of the manufacturer.
Cloning of α11.4 and α11.3 cytoplasmic domains.
The mouse α11.4 C-terminal segment (CT1, amino acids 1445–1605) was amplified with primers FH586 (5′-CACCGATAACTTTGATTACCTAACCAGAG-3′) and FH585 (5′-CTTAAGTTACTTTTCTTTCCTTCTCCGGA-3′) that also includes a stop codon by PCR from mouse α11.4 plasmid (Haeseleer et al., 2004). The distal C-terminal downstream of the CT1 (fragment, CT2, was amplified with primers FH593 (5′-CACCGGGCTACTAGGAAGAGAGG-3′) and FH556 (5′-GGATCCTTAGAGGGCATGGACACAGGC-3) that also introduces a BamHI restriction site. The rat α11.3 CT1 (amino acids 1478–1644) was amplified with primers FH650 (5′-CACCTTGGACGAATTCAAAAGAATATGG-3′) and FH649 (5′-ctagagcatccgttcaagcatctg-3′) by PCR from rat brain α11.3 (Xu and Lipscombe, 2001). All PCR fragments were subcloned in pENTR/D-TOPO vector (Invitrogen) and sequenced by dideoxyterminator sequencing (ABI-Prism; PerkinElmer, Boston, MA) and then transferred into pDest17 for fusion to a 6His tag using the Gateway Technology System as described by the manufacturer (Invitrogen).
Cloning of human PKCζ and transfection in HEK293 cells.
Full-length PKCζ was amplified by PCR from human PKCζ cDNA (clone 3835020; Open Biosystems, Huntsville, AL) using primers FH690 (5′- CACCATGCCCAGCAGGACCGG-3′) and FH691 (5′-GCCTCACACCGACTCCTCG-3′). The PCR products were cloned into the pENTR/D-TOPO vector (Invitrogen) and sequenced by dideoxyterminator sequencing (ABI-Prism; PerkinElmer) and then transferred into the pcDNA3.1(−) vector. For the pull-down assay, T-Rex-293 stable cell line expressing CaBP4 or CaBP4–S37A were transfected with human PKCζ using Lipofectamine 2000 according to the instructions of the manufacturer (Invitrogen). Six hours after transfection, medium containing 1 μg/μl doxycycline was added to the cells, and the cells were induced for 48 h. The cells were then washed with PBS and collected for additional analysis.
Reverse transcription-PCR assay.
Total RNA (5 μg) from mouse retina was used for an initial reverse transcription step to prepare cDNA. The gene-specific primers used to amplify PKCα (5′-CCAACCGCTTCGCCCGCAAAG-3′ and 5′-GCTTGGCTTTCTCAAACTTCTGCC3-′) or PKCζ (5′-GCCTTCCGCCTGGTCTGTCAGG-3′ and 5′-TGCAGCCCCAGCCCCTGAGAG-3′) generated a fragment of ∼900 and 550 bp, respectively, after an initial PCR step at 94°C for 5 min and 35 cycles at 94°C for 30 s, 60°C for 30 s and 72°C for 1.5 min with YieldAce DNA polymerase (Stratagene). Amplification with glyceraldehyde-3-phosphate dehydrogenase (G3PDH)-specific primers (5′-GAAGGGCTAATGACCACAGTCCAT-3′ and 5′-TAGCCATATTCGTTGTCGATCCAGG-3′) was performed as a positive control.
Gel overlay assay.
Recombinant 6His-tagged purified proteins (2 μg) were separated on SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. After overnight saturation at 4°C in PBS, 0.1% Tween 20, and 3% nonfat milk, the membrane were incubated in PBS, 0.1% Tween 20, and 2% nonfat milk (blotting buffer) containing 2 μg/ml GST-tagged proteins for 1 h at room temperature. The blots were washed three times for 5 min with 10 ml of blot buffer and then incubated for 1 h with anti-GST antibody in blotting buffer at room temperature. After three washes of 5 min each, the blots were incubated with an anti-mouse antibody conjugated to alkaline phosphatase for 1 h at room temperature. The bound recombinant proteins were visualized by incubation with nitroblue-tetrazolium-chloride/5-bromo-4-chlor-indolyl-phosphate (Promega). The interactions were tested in the presence or absence of calcium. For the analysis in the absence of Ca2+, 5 mm EGTA was added to the blotting buffer.
Immunohistochemistry.
Mouse eyecups were fixed in 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4 (PB), for 4 h. After fixation, tissues were incubated with a sucrose series to 20% sucrose in PB and then embedded in 33% OCT compound (Miles, Elkhart, NY) diluted with 20% sucrose in PB. Eye tissues were cut in 12 μm sections. To block nonspecific labeling, retinal sections were incubated with 3% normal goat serum in PBST buffer (136 mm NaCl, 11.4 mm sodium phosphate, and 0.1% Triton X-100, pH 7.4) for 20 min at room temperature. Sections were incubated overnight at 4°C in a mix of diluted primary antibodies (1:400 for mouse anti-CaBP4; 1:250 for rabbit anti-PKCζ). A mixture of Cy3-conjugated goat anti-rabbit IgG and Alexa 488-conjugated goat anti-mouse IgG was reacted with sections for 1 h at room temperature. Then, sections were rinsed in PBST and mounted with Prolong antifade reagent (Invitrogen) to retard photobleaching. Sections were analyzed under a confocal microscope (LSM510; Zeiss, Thornwood, NY). Immunofluorescent images (0.8 μm thickness) were obtained with a Plan-Neofluar 40×/1.3 numerical aperture (Zeiss) objective lens.
CaBP4 affinity chromatography.
CaBP4 was coupled to CNBr-activated Sepharose (GE Healthcare, Piscataway, NJ) according to the protocol of the manufacturer. The CaBP4–Sepharose was either treated with rPKCζ or left untreated as described above (kinase assay with recombinant PKC). For binding in the presence of Ca2+, the CaBP4–Sepharose was equilibrated with 10 mm BTP, pH 8.0, 2 mm benzamidine, and 0.1 mm CaCl2. Purified His-tagged Cav1.4 CT1 was incubated with the Sepharose at 4°C for 2 h. The CaBP4–Sepharose was then washed with the equilibration buffer (40 times the volume of the column) followed by the same buffer containing 150 mm NaCl. The elution was performed with 3 mm EGTA followed by 0.1 m Gly, pH 2.5. Fractions were collected and aliquots were analyzed by Western blot with an anti-6His tag antibody. For the binding in the presence of EGTA, the CaBP4–Sepharose was equilibrated with 10 mm BTP, pH 8.0, 2 mm benzamidine, and 0.1 mm EGTA and washed with this buffer followed by the same buffer containing 150 mm NaCl. The elution was performed with 5 mm CaCl2 followed by 0.1 m Gly, pH 2.5.
Electrophysiological recordings.
HEK293T cells were grown to 70–80% confluence and transfected using Gene Porter reagent (Gene Therapy Systems, San Diego, CA) according to the protocols of the manufacturer. Cells were transfected with ∼5 μg of total DNA [α11.3 (1.5 μg), β2A (0.8 μg), α2δ (0.8 μg) with or without CaBP4 (0.1 μg)] and green fluorescent protein expression plasmid (0.01 μg) for fluorescent detection of transfected cells. All electrophysiological data were acquired with EPC-9 patch-clamp amplifier driven by Pulse software (HEKA Elektronik, Lambrecht/Pfalz, Germany) and analyzed with Igor Pro software (WaveMetrics, Lake Oswego, OR). The time course of ICa decay was fit by Aslow[exp(−t/τslow)] + Afast[exp(−t/τfast)], where t is time, Aslow and Afast are the amplitudes of the slow and fast exponentials, respectively, at t = 0, and τslow and τfast are the time constants of the decay of the two processes. Extracellular recording solutions contained the following (in mm): 150 Tris, 1 MgCl2, and 10 BaCl2 or 10 CaCl2. Intracellular solutions consisted of the following (in mm): 140 N-methyl-d-glucamine, 10 HEPES, 2 MgCl2, 2 Mg-ATP, and 5 EGTA. The pH of intracellular and extracellular recording solutions was adjusted to 7.3 with methanesulfonic acid. Electrode resistances were typically 1–2 MΩ in the bath solution, and series resistance was ∼2–4 MΩ, compensated up to 80%. All averaged data are presented as the mean ± SEM. Statistical significance of differences between two groups was determined by ANOVA or Student's t test as indicated (SigmaStat; SPSS, Chicago, IL).
Results
Phosphorylation of CaBP4 by an endogenous protein kinase C
Given the importance of light-dependent protein phosphorylation in regulating signal transduction in photoreceptors, we tested whether CaBP4 is phosphorylated in mouse retina by immunoprecipitation with anti-CaBP4 antibody (UW145) from retinal extracts in the presence of [γ-32P]ATP. Because massive rhodopsin phosphorylation in light-adapted retina extract obscures detection of other phosphorylated proteins that nearly comigrate with rhodopsin, the phosphorylation assay was performed in the dark with extracts of dark-adapted retinas unless specified. Under these conditions, a 32P-labeled protein was immunoprecipitated, the identity of which was confirmed as CaBP4 by its presence in Cabp4+/+ (WT) but not Cabp4−/− [knock-out (KO)] retinal extracts (Fig. 1A). In addition, GST-tagged CaBP4 was phosphorylated by the endogenous kinase when incubated with retinal extract (Fig. 1B). Because we used retinal extract of Cabp4−/− mice in some assays, we also tested the kinase activity in Cabp4−/− extract. Similar levels of phosphorylated GST–CaBP4 were also detected using retinal extract of Cabp4−/− mice (Fig. 1B), indicating that the retinal kinase required for CaBP4 phosphorylation is not affected by loss of CaBP4. The endogenous kinase did not require Ca2+ in that CaBP4 phosphorylation was slightly higher in the absence than in the presence of Ca2+ (Fig. 1B).
Figure 1.

Phosphorylation of native and recombinant CaBP4 by PKCζ. A, Identification of native CaBP4 as a phosphorylated protein in retina. Retinal proteins were incubated in the presence of [γ-32P]ATP and subject to immunoprecipitation with anti-CaBP4 antibody. Autoradiograph (top) shows a labeled band in Cabp4+/+ (WT) but not CaBP4−/− (KO) retinal extract, which was also detected by immunoblotting with anti-CaBP4 antibody (bottom, top band). B, Ca2+-independent phosphorylation of recombinant CaBP4. GST–CaBP4 (1 μg) was incubated with a Cabp4+/+ (WT) or Cabp4−/− (KO) retina extract in the presence of [γ-32P]ATP and Ca2+ (1 mm) or EGTA (1 mm). Phosphorylation of GST–CaBP4 is stronger without Ca2+ in extracts of Cabp4−/− and Cabp4+/+ retina. Ponceau staining indicates equal levels of GST–CaBP4 between groups. C–H, Characterization of the CaBP4 kinase in retina as PKCζ. The same amount of bacterially expressed and purified 6His–CaBP4 (1 μg) was incubated with retinal extract in the presence of activators or inhibitors of protein kinases as indicated. Arrows indicate CaBP4, which was identified as the major protein of ∼43 kDa by Ponceau staining. Assays contained Ca2+ (1 mm) or EGTA (1 mm) as indicated. C, The CaBP4 kinase is soluble. Phosphorylation assays were conducted with retinal extracts separated into soluble (S) and membrane (M) fractions and in the presence or absence of Ca2+. D, Mg2+ is required for kinase activity. CaBP4 phosphorylation (lane Mg2+) was prevented by inclusion of 10 mm EDTA (lane EDTA) and inhibited when Mn2+ was substituted for Mg2+in the assay (lane Mn2+). E, The endogenous kinase is inhibited by PKC inhibitors. The kinase activity in the retina extract (lane −, left) was not affected by PP2 or TBB but was inhibited partially by H-89 and completely inhibited by Bis, each at 10 μm. F, CaBP4 phosphorylation is inhibited by a PKCζ-specific inhibitor. CaBP4 phosphorylation was not affected by Rottlerin, partially inhibited by Gö6976 and Gö6983, and strongly inhibited by a PKCζ-specific inhibitor. The kinase activity in the retina extract was tested in the presence of various concentrations of PKC inhibitors as indicated. G, H, Kinase activity is phospholipid independent. DAG (0.01 mg/ml), PS (0.1 mg/ml), and PMA (5 μm) did not affect CaBP4 phosphorylation in either the presence or absence of Ca2+. I, CaBP4 is phosphorylated by recombinant PKC isoforms in vitro. Phosphorylation of 6His–CaBP4 by recombinant PKCζ or PKCα was tested in the presence or absence of PKCζ inhibitor or Bis at the indicated concentrations.
We next undertook experiments to characterize the CaBP4 kinase in the retinal extract. First, we examined which subcellular fraction contained phosphorylated CaBP4. Greater levels of phosphorylated CaBP4 were detected in the soluble fraction than the membrane fraction, indicating that the endogenous kinase is primarily cytosolic rather than membrane associated (Fig. 1C). Because the activity of most protein kinases rely on Mg2+ (Hunter and Cooper, 1985; Edelman et al., 1987), we tested whether chelation of Mg2+ by EDTA prevented CaBP4 phosphorylation. Inclusion of EDTA prevented incorporation of 32P into CaBP4 and phosphorylation was only partially restored by Mn2+ (Fig. 1D). This result confirms the Mg2+ dependence of CaBP4 phosphorylation by the retinal kinase. We also tested the effects of various protein kinase inhibitors on CaBP4 phosphorylation in retinal extracts. Whereas inhibitors of the Src family of tyrosine kinases (PP2) and casein kinase II (TBB) had no effect, CaBP4 phosphorylation was strongly decreased by the PKC inhibitor Bis and partially reduced by H-89, which inhibits PKA but also PKC at higher concentrations (Fig. 1E,F). These results indicated that CaBP4 was phosphorylated by a member of the PKC family.
Because CaBP4 phosphorylation was not increased by Ca2+ (Fig. 1B), the retinal kinase was not likely to be a conventional PKC isoform (PKCα, PKCβ, and PKCγ), which would be activated by Ca2+ and phosphatidylserine and diacylglycerol. In addition, CaBP4 phosphorylation was strongly inhibited by Gö6983, an inhibitor of PKCα, PKCβ, and PKCδ that also inhibits PKCζ at higher concentrations (Fig. 1F) (Gschwendt et al., 1996) and was affected by the PKCα inhibitor Gö6976 only at high concentrations (100 μm) (Fig. 1F), at which this drug can also inhibit PKCζ (Martiny-Baron et al., 1993; Gschwendt et al., 1996). CaBP4 phosphorylation was also not altered by Rottlerin, an inhibitor of the novel PKC isoforms PKCδ and PKCθ (Fig. 1F) (Gschwendt et al., 1994). In contrast, a pseudosubstrate inhibitor of the atypical PKC isoform, PKCζ, nearly prevented CaBP4 phosphorylation (Fig. 1F). The involvement of PKCζ was further supported by the relative insensitivity of CaBP4 phosphorylation to phospholipid PKC activators, which do not influence the activity of PKCζ. In these experiments, nearly equivalent levels of phosphorylated CaBP4 were detected in the presence and absence of DAG, PS (Fig. 1G), and the DAG analog PMA (Fig. 1H). Finally, in a cell-free assay using GST-tagged CaBP4 and recombinant PKC isoforms, CaBP4 was efficiently phosphorylated by PKCζ, which was strongly inhibited by the pseudosubstrate PKCζ inhibitor (Fig. 1I). PKCα also promoted CaBP4 phosphorylation, which was blocked by Bis but was essentially insensitive to high concentrations of the PKCζ inhibitor (Fig. 1I), in contrast to the strong effects of the PKCζ inhibitor on CaBP4 phosphorylation in the retinal extract (Fig. 1F). Collectively, these results implicate PKCζ as the endogenous kinase that phosphorylates CaBP4 in the retina.
PKCζ is expressed in photoreceptors and colocalizes with CaBP4
If PKCζ is physiologically relevant as the CaBP4 kinase, it should be coexpressed and colocalized with CaBP4 in the retina. To test this, we first confirmed the expression of the PKCζ isoform in the retina by reverse transcription (RT)-PCR (Fig. 2A). As expected, PKCα was also amplified because it is primarily expressed in rod bipolar cells (Greferath et al., 1990). We confirmed the expression of PKCα and PKCζ in the retina by immunoblot. The specificity of the isoform-specific antibodies was first confirmed using recombinant proteins. The anti-PKCζ antibody recognized specifically PKCζ and not PKCα, whereas the anti-PKCα antibody strongly detected PKCα and only very weakly PKCζ (Fig. 2B). In Western blot analyses of retinal lysates, the anti-PKCα and anti-PKCζ antibodies detected proteins of ∼85 and ∼80 kDa, respectively, which were consistent in size with the corresponding recombinant proteins (Fig. 2B,C). These results confirmed that PKCζ is expressed at both the mRNA and protein level in the retina.
Figure 2.

Expression of PKCζ in the mouse retina. A, RT-PCR analysis of PKCζ and PKCα expression in the retina. PKCζ or PKCα cDNAs were amplified from total RNA from wild-type (Cabp4+/+) or CaBP4 knock-out (Cabp4−/−) mouse retina using isoform-specific primers or G3PDH-specific primers as positive controls. Standards, 1 kb DNA ladder. B, Specificity of the anti-PKC antibodies. Recombinant PKCα and PKCζ were subjected to Western blot analysis with commercial anti-PKC isoform-specific antibodies. C, Western blot of mouse retina extract with anti-PKCα and anti-PKCζ. A protein of ∼80 kDa was detected in a mouse retina extract with the anti-PKCζ (right), which was less intense than the ∼85 kDa protein detected by the anti- PKCα antibodies (left). D, Colocalization of PKCζ with CaBP4 in the mouse retina. Confocal micrographs of mouse retina sections double-immunostained for PKCζ and CaBP4. The image overlay of labeling for PKCζ (green, left, A′–C′) and CaBP4 (red, middle, A″–C″) is shown in the right panels (merge, A‴–C‴). A yellow color denotes colocalization of CaBP4 and PKCζ at the photoreceptor synapse and inner segment. Scale bar: A′–A‴, 20 μm. B′–B‴, Higher magnification of the photoreceptor layer; scale bar, 10 μm. C′–C‴, Retina treated with secondary antibody alone; scale bar, 20 μm. OS, Photoreceptor outer segment; IS, photoreceptor inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
Next, we determined whether PKCζ is colocalized with CaBP4 in the retina by confocal microscopy. In retina sections double-labeled with antibodies against PKCζ and CaBP4, PKCζ was localized to the photoreceptor inner segment, and weaker staining was also observed at the photoreceptor synapse in the outer plexiform layer (Fig. 2D, OPL). As reported previously, CaBP4 was localized to the photoreceptor synapse, and weak staining was also detected in the inner segment. Overlay of the two single-labeled images showed strong overlap of immunofluorescence for CaBP4 and PKCζ in the synaptic and inner segment layers (Fig. 2D), which is consistent with a role for PKCζ in regulating CaBP4.
Serine 37 is the major site of phosphorylation of mouse CaBP4 by PKCζ
To define potential sites in CaBP4 for PKCζ phosphorylation, we compared phosphorylation of truncated GST-tagged CaBP4 fragments in retinal extracts in the presence of [γ-32P]ATP (Fig. 3A,B). Whereas GST–CaBP4 fragments containing amino acids 1–34 and 60–271 showed no incorporation of 32P, the fragments containing amino acids 1–62 and 34–271 were efficiently phosphorylated (Fig. 3B), indicating phosphorylation site(s) between amino acids 34–62. Analysis of this region using the NetPhos 2.0 server (Technical University of Denmark) identified serine residues 37, 46, 50, and 51 as putative phosphorylation sites that are also are conserved or show conserved substitutions (S/T) in CaBP4 orthologs (Fig. 3A) and so may be physiologically important sites of phosphorylation. If so, then substitution of these serine residues with alanine should inhibit the incorporation of 32P into the GST–CaBP4 fragment containing amino acids 1–61. This result was obtained with fragments containing a serine 37 to alanine substitution (S37A), whereas serine to alanine substitution at residues 46, 50/51 did not affect levels of 32P incorporation compared with the wild-type CaBP4 fragment (Fig. 3C). When S37A was introduced into the full-length CaBP4, phosphorylation was almost completely abolished (Fig. 3D). The S37A mutation in both GST–CaBP4 (amino acids 1–61) and full-length CaBP4 also prevented phosphorylation by recombinant PKCζ (Fig. 3E), indicating that S37 is the major phosphorylation site for recombinant PKCζ. Whereas the S37A mutation also blocked phosphorylation of GST–CaBP4 (amino acids 1–61) by PKCα, 32P incorporation was still observed in the full-length CaBP4 containing S37A (Fig. 3E), suggesting that there is an additional site phosphorylated by PKCα in the cell-free assay.
Figure 3.

Identification of S37 as the major site of mouse CaBP4 phosphorylation. A, Scheme of mouse CaBP4 protein and CaBP4 partial segments fused to GST and tested for phosphorylation. The functional EF-hands are shown in black, whereas the nonfunctional EF-hand 2 is represented in gray. Alignment of amino acid residues 34 and 61 of mouse CaBP4 protein with the corresponding peptide sequence of rat, bovine, and human CaBP4. All amino acid numbers are for rodent peptides only. Large asterisk indicates potential phosphorylation sites conserved among species [serine 37, 46 (S/T conservative substitution), 50, and 51]. Those not conserved are shown with a small asterisk (serine 41, 55, 56, 57, and 60). B–F, Phosphorylation of GST-tagged CaBP4 truncated segments and serine to alanine CaBP4 mutants. GST-tagged CaBP4 and truncated segments (1 μg) were incubated with Cabp4−/− retina extract (B–D, F) or recombinant PKCζ or PKCα (E) in the presence of [γ-32P]ATP without Ca2+ unless indicated. Top panels, Autoradiograms of 32P-labeled GST–CaBP4 fusion proteins (arrow). Bottom panels, Coomassie blue or Ponceau staining shows levels of GST–CaBP4 fusion proteins used in the assay. The effects of serine to alanine substitutions on phosphorylation of the GST–CaBP4 fragment (amino acids 1–61; C, E) and the serine 37 to alanine (S37A) substitution in the full-length or partial (amino acids 1–61) GST–CaBP4 (D, E) were tested. In F, phosphorylation and Ca2+ shift of CaBP4, S37A mutant, and CaBP4–EF Ca2+-binding triple mutant are compared. S37A–CaBP4 and CaBP4–EF mutants
To test whether phosphorylation at S37 might affect Ca2+ binding, we compared the Ca2+-induced mobility shift of the S37A mutant and phosphorylated CaBP4 in SDS-PAGE gels. A similar Ca2+ shift was found for both CaBP4 and S37A mutant. No Ca2+ shift was observed for a mutant of CaBP4 unable to bind Ca2+ (CaBP4–EF; Ponceau-stained blot) (Fig. 3F), in which the glutamic acid in position 12 of the putative functional EF-hands 1, 3, and 4 was substituted with glutamine, which should disrupt Ca2+ coordination by these regions. This finding demonstrates that phosphorylation does not grossly influence the Ca2+ binding properties of CaBP4. In a reverse approach, we tested whether Ca2+ binding to CaBP4 might affect CaBP4 phosphorylation by comparing the phosphorylation of the wild-type CaBP4 with that of CaBP4–EF. The level of phosphorylation of CaBP4–EF was not different from that of wild-type CaBP4 in either the presence or absence of Ca2+ (Fig. 3F). These results show that Ca2+-dependent conformations of CaBP4 are not required for its phosphorylation at S37.
Role of CaBP4 phosphorylation in interactions with Cav1 channels
To evaluate the functional consequences of CaBP4 phosphorylation, we compared the ability of CaBP4 and S37A–CaBP4 to interact with and modulate Cav1 channels. We showed previously that CaBPs compete with CaM for binding to regions including the Pre-IQ and IQ-domains in the C-terminal domain of the Cav1 α1 subunit (Zhou et al., 2004; Tippens and Lee, 2007). Therefore, we tested whether the analogous region in the Cav1.4 α1 subunit, α11.4, associated with CaBP4 in a gel overlay assay. GST–CaBP4 bound to 6His-tagged fusion proteins containing the CaM-binding sequences (amino acids 1445–1605, CT1) but not those lacking these sites (amino acids 1600–1986, CT2) (Fig. 4A). The binding of CaBP4 to CT1 was stronger in the presence than in the absence of Ca2+ and was specific in that GST alone did not bind to this sequence. Binding of CaBP4 that was prephosphorylated by recombinant PKCζ (P-CaBP4) to CT1 was slightly stronger than unphosphorylated CaBP4 (Fig. 4A), which shows that phosphorylation of CaBP4 does not impair interactions with α11.4.
Figure 4.

Interaction of CaBP4 and S37A mutant with Cav1 channels. A, Gel overlay assay of recombinant CaBP4 with α11.4 cytoplasmic domains. His-tagged α11.4 C-terminal domain with (CT1) and without (CT2) CaM-binding sequences were separated on SDS-PAGE and transferred to PVDF membranes, which were incubated in the presence or absence of EGTA with GST or GST-tagged CaBP4, phosphorylated (P-CaBP4) or not (CaBP4) with rPKCζ. Bound proteins were detected with an anti-GST antibody. Ponceau staining (left) shows the relative amount of purified α11.4 proteins in the assay. B–E, Binding of α11.4 and α11.3 CT1 to CaBP4 by affinity chromatography. His-tagged α11.4 CT1 (B, C) or α11.3 CT1 (D, E) was incubated with Sepharose coupled with nonphosphorylated CaBP4 (CaBP4-Seph) or phosphorylated CaBP4 (P-CaBP4-Seph) in the presence of Ca2+ (B, D) or EGTA (C, E). After washes with buffer containing 150 mm NaCl, the proteins were eluted with 3 mm EGTA (B, D) or 5 mm CaCl2 (C, E), followed by 0.2 m glycine buffer, pH 2.1. The fractions were then analyzed by Western blot with anti-His tag antibody. Lane 1, Flow-through; lane 2, last wash; lanes 3–11, elution with Ca2+ or EGTA; lanes 12–17, elution with 0.1 m glycine.
We further investigated the binding of P-CaBP4 to α11.4 CT1 using affinity chromatography with CaBP4-coupled Sepharose that was pretreated or not with rPKCζ (P-CaBP4 and CaBP4, respectively) and incubated with 6His-tagged α11.4 CT1 (His-α11.4 CT1) in the presence of Ca2+. In this assay, Ca2+-dependent binding to CaBP4 is revealed by elution of His–α11.4 CT1 with a high concentration of EGTA. Western blot analysis revealed that His–α11.4 CT1 dissociated more slowly from P-CaBP4 than from CaBP4–Sepharose on EGTA elution (Fig. 4B), which suggests that phosphorylation of CaBP4 strengthens Ca2+-independent interactions with α11.4 CT1. Consistent with these results, when the protein incubation was performed in the presence of EGTA followed by Ca2+ elution, greater levels of His–α11.4 CT1 was eluted from P-CaBP4–Sepharose than CaBP4–Sepharose (Fig. 4C). Interestingly, binding of the 6His-tagged Cav1.3 α1 subunit (His–α11.3 CT1) was generally weaker to P-CaBP4 than to CaBP4 both with and without Ca2+ (Fig. 4D,E). These results demonstrate that phosphorylation of CaBP4 may differentially affect the strength and Ca2+ dependence of its interactions with targets.
To further test this, we compared the ability of CaBP4 and S37A–CaBP4 to modulate Cav1 channels in electrophysiological recordings of transfected HEK293T cells. Although we initially analyzed how phosphorylated CaBP4 influenced Cav1.4 channels, these experiments were hindered by the low expression levels of Cav1.4 particularly in combination with CaBP4 and S37A–CaBP4. Therefore, we shifted our focus to Cav1.3 channels because CaBP4 interacted efficiently with these channels in our biochemical assays (Fig. 4E,F).
As shown previously (Yang et al., 2006), Cav1.3 channels undergo considerably faster decay (inactivation) when Ca2+ rather than Ba2+ is used as the charge carrier (Fig. 5A,B). This Ca2+-dependent inactivation is because of to CaM and is opposed by CaBP4 binding to α11.3 (Yang et al., 2006). In our experiments, inactivation of Ca2+ and Ba2+ currents (ICa, IBa) was measured as Ires/Ipk, which was the current amplitude at the end of a 1 s depolarizing pulse normalized to the peak current amplitude. We found CaBP4 to have a significant effect in suppressing inactivation of ICa (∼70%; p < 0.05), which was not observed with S37A–CaBP4 (Fig. 5A). Neither CaBP4 nor S37A–CaBP4 influenced inactivation of IBa (Fig. 5B), indicating a selective effect of CaBP4 on Ca2+-dependent inactivation. CaBP4 but not S37A–CaBP4 significantly inhibited inactivation of ICa for various test voltages (−20 to 0 mV) (Fig. 5C). Kinetic analyses revealed that CaBP4 suppressed inactivation of ICa primarily by increasing the time constant for slow (τslow) but not fast (τfast) inactivation (Fig. 5D). In contrast, τslow and τfast were not different in cells transfected with Cav1.3 alone and those cotransfected with S37A–CaBP4 (Fig. 5C).
Figure 5.

CaBP4 phosphorylation is required for modulation of Cav1.3. A, B, ICa and IBa were evoked by 1 s pulses from −90 to −10 mV (ICa) or −20 mV (IBa) in HEK293T cells transfected with Cav1.3 (α11.3, β2A, α2δ) alone or cotransfected with CaBP4 or CaBP4-S37A. Ires/Ipk represents amplitude of the current at the end of the pulse normalized to the peak current amplitude. Numbers of cells are indicated in parentheses. *p < 0.05. Representative current traces are shown above. Vertical calibration bars: A, 0.2 nA; B, 0.4 nA. C, For cells transfected as in A, Ires/Ipk was measured for ICa during 1 s pulses from −90 mV to various voltages and plotted against test voltage. Asterisks indicate voltages at which values for with CaBP4 significantly differ from Cav1.3 alone (p < 0.05). Points represent mean ± SE of 15–21 cells. D, Fast and slow time constants for inactivation (τfast, τslow) obtained from double-exponential fits of ICa evoked as in A for cells transfected with Cav1.3 alone (n = 8), with CaBP4 (n = 9), or with S37A (n = 9). *p < 0.05. E, Western blot of lysates from cells transfected as in A probed with anti-CaBP4 antibodies.
The ineffectiveness of S37A–CaBP4 suggests the importance of CaBP4 phosphorylation for modulating inactivation of Cav1.3 ICa. Alternatively, lower expression levels of S37A–CaBP4 compared with CaBP4 could also account for our results. However, Western blots with CaBP4 antibodies showed strong levels of both S37A–CaBP4 and CaBP4 in lysates of HEK293T cells transfected as for electrophysiological experiments (Fig. 5E). To confirm that CaBP4 in transfected cells was subject to phosphorylation at S37 as we found in the retina (Fig. 3C–F), we compared 32P incorporation into GST–CaBP4 and GST–S37A–CaBP4 during incubation with cell lysates. In these experiments, GST–CaBP4 but not S37A–CaBP4 was efficiently phosphorylated, which was inhibited by Bis (Fig. 6A).
Figure 6.
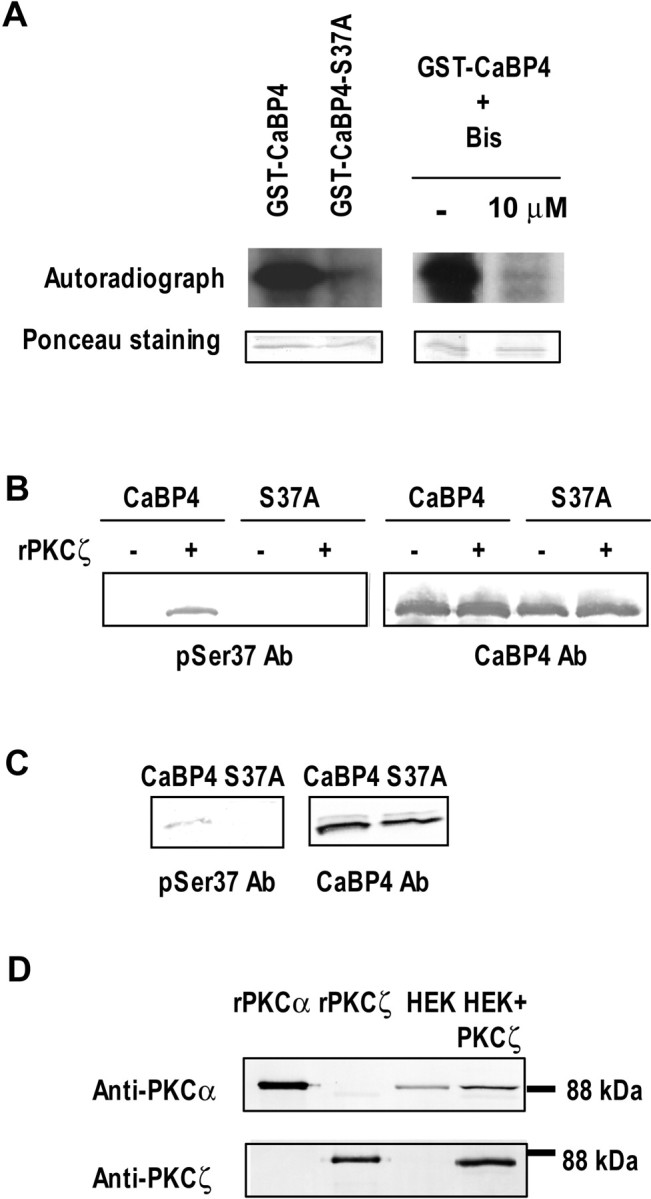
CaBP4 is phosphorylated at S37 in transfected HEK293 cells. A, CaBP4 is phosphorylated by the endogenous PKC in HEK293 cells. Autoradiograph and Ponceau staining of recombinant GST-tagged CaBP4 (1 μg) or GST-tagged S37A mutant (1 μg) incubated with an equivalent amount of cell lysate from HEK293 cells in the presence of [γ-32P]ATP (left). The kinase activity in the cell lysate was tested with GST-tagged CaBP4 in the presence or absence of 10 μm Bisindolymaleimide (right). B, C, Detection of phosphorylated CaBP4 by a phospho-specific antibody. In B, anti-phospho-Ser37–CaBP4 antibody detected GST-tagged CaBP4 (0.2 μg) but not GST-tagged S37A mutant (0.2 μg) only when phosphorylated by recombinant PKCζ (left). Labeling with anti-CaBP4 (UW145, right) confirms equal levels of GST–CaBP4 proteins in each lane. In C, 6His-tagged CaBP4 or 6His-tagged S37A mutant were purified from stably transfected HEK293 cells and analyzed by Western blotting with anti-phospho-Ser37–CaBP4 antibody (left) or with anti-CaBP4 as a positive control (right). CaBP4 but not S37A mutant immunoreacted with anti-phospho-Ser37–CaBP4. D, PKCα is expressed in HEK293 cells. Equivalent amount of total proteins from lysates of HEK293 cells transfected or not with PKCζ were analyzed for PKCα and PKCζ expression by Western blot (lanes 3, 4). rPKCα or rPKCζ (10 ng each) were also loaded as positive controls (lanes 1, 2).
To further confirm the importance of serine 37 for CaBP4 phosphorylation in transfected cells and in the retina, we developed and characterized an antibody that specifically recognizes the sequence including phosphorylated S37 (anti-phospho-S37). In Western blots, anti-phospho-S37 recognized recombinant CaBP4 that was phosphorylated by recombinant PKCζ and not unphosphorylated CaBP4 (Fig. 6B). Moreover, anti-phospho-S37 did not detect S37A–CaBP4 incubated with or without PKCζ in the cell-free assay (Fig. 6B), which further validated the specificity of this antibody for the phospho-S37. The anti-phospho-S37 antibodies detected a band in HEK293T cells transfected with CaBP4 but not S37A–CaBP4 (Fig. 6C). The identity of this band as CaBP4 was confirmed by Western blotting with CaBP4 antibodies that recognize both wild-type and mutant forms. To determine which PKC isoform in HEK293T cells may mediate phosphorylation of CaBP4, we performed Western blots with antibodies against PKC antibodies. These experiments showed that PKCα but not PKCζ was expressed endogenously in HEK293T cells (Fig. 6D), which suggested that, unlike in the retina, phosphorylation of S37 in CaBP4 in these cells is performed by PKCα.
Our findings demonstrate that phosphorylation of CaBP4 is required to maintain functional interactions with α11.3, somewhat at odds with our biochemical data indicating weaker binding of P-CaBP4 to α11.3 (Fig. 4D,E). One mechanism to reconcile these results would be if low-affinity interactions of P-CaBP4 with α11.3 were important for the net effect of suppressing ICa inactivation. Because we also found that P-CaBP4 and CaBP4 binding to α1CT1 was weaker in the absence of Ca2+ (Fig. 4), one possibility is that CaBP4 modulates the channel more strongly when it is not associated with Ca2+. To test this, we analyzed the effects of the mutant of CaBP4 that can no longer bind Ca2+ (CaBP4–EF) but was still efficiently phosphorylated by the retinal extract (Fig. 3F). As predicted, CaBP4–EF robustly inhibited ICa inactivation (∼113%) (Fig. 7A) across a larger range of test voltages than CaBP4 (−30 to +10 mV) (Fig. 7B). Moreover, unlike CaBP4, CaBP4–EF inhibited both fast and slow time constants for inactivation (Fig. 7C), which suggests that Ca2+ binding to CaBP4 may have additional effects on ICa inactivation compared with CaBP4 phosphorylation. Interestingly, CaBP4–EF also significantly increased inactivation of IBa (Fig. 7A), suggesting additional mechanistic differences in the roles of Ca2+ and phosphorylation in CaBP4 modulation of Cav1.3 function.
Figure 7.
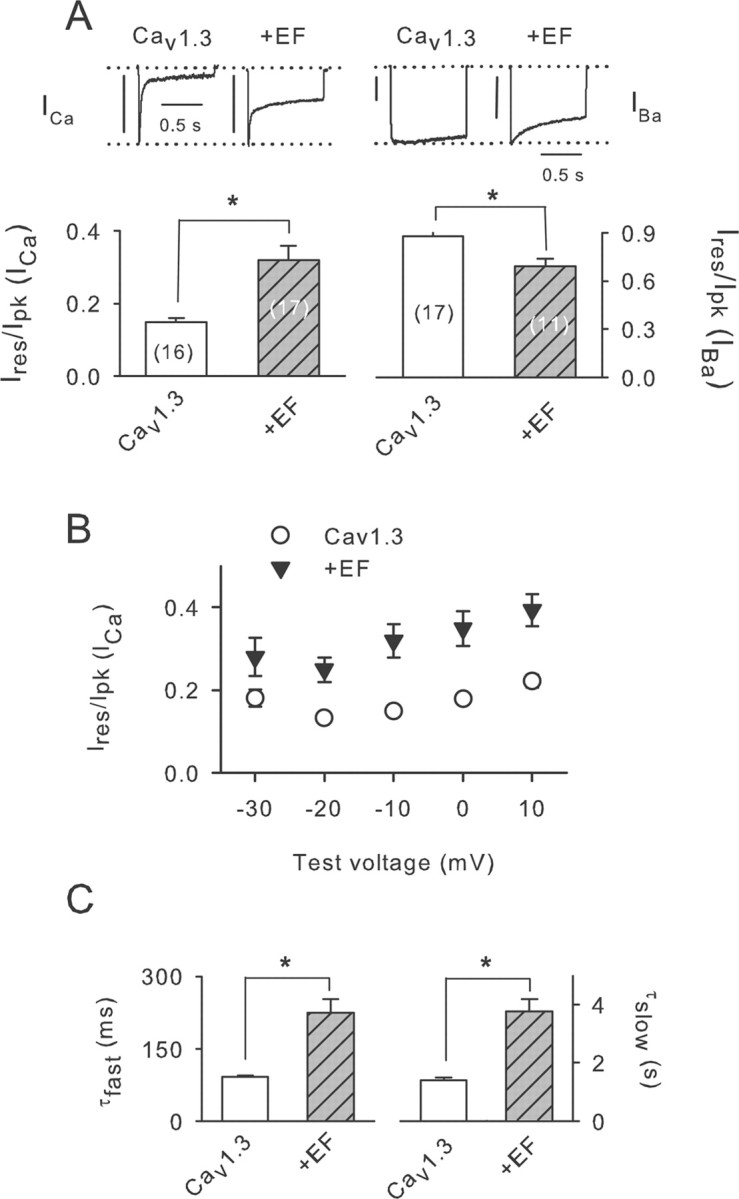
Modulation of Cav1.3 does not require Ca2+ binding to CaBP4. A, Ires/Ipk was determined for ICa and IBa as in Figure 5, A and B, in cells transfected with Cav1.3 alone or cotransfected with CaBP4–EF (+EF). Numbers of cells are indicated in parentheses. Representative current traces are shown above. Vertical calibration bars, 0.3 nA. B, Ires/Ipk plotted against test voltage as in Figure 5C. Points represent mean ± SE of 12–21 cells. C, τfast and τslow for cells transfected with Cav1.3 with or without EF were determined as in Figure 5D for cells transfected with Cav1.3 alone (n = 8) or with EF (n = 9). *p < 0.01.
Light-dependent phosphorylation of CaBP4 in mouse retina
Our results illustrate that phosphorylation of CaBP4 can significantly alter its interactions with target molecules. Light-dependent alterations in CaBP4 phosphorylation could therefore significantly impact the consequence of the light signal in photoreceptors. To test whether phosphorylation of CaBP4 changes with the light conditions, we compared the level of CaBP4 phosphorylation in dark-adapted or light-adapted mouse retina. In Western blots, the anti-phospho-S37 antibody detected a protein consistent in size with CaBP4 (∼35 kDa) more strongly in light-adapted than dark-adapted retina (Fig. 8A). This result was not attributable to alterations in CaBP4 expression because an equal level of CaBP4 was detected in light- and dark-adapted retina with anti-CaBP4 antibodies. Neither of the antibodies detected the 34 kDa band in light-adapted retina of Cabp4−/− mice, which verified the identity of this protein as CaBP4. Light-dependent phosphorylation of CaBP4 was also observed by immunoprecipitation from dark-adapted or light-adapted retinal extracts preincubated with [γ-32P]ATP. Anti-CaBP4 antibodies immunoprecipitated a 32P-labeled band corresponding to the phosphorylated CaBP4 much more strongly in light-adapted than dark-adapted conditions (Fig. 8B), whereas no signal was detected in parallel experiments with retinal extracts of Cabp4−/− mice. Light-dependent changes in CaBP4 phosphorylation required activation of the phototransduction cascade in rods, because no changes in the levels of phosphorylated CaBP4 were detected between light-adapted and dark-adapted retina from mice lacking transducin (Gnat1−/−) (Fig. 8C,D), the G-protein that couples rhodopsin to the electrical photoresponse (Calvert et al., 2000).
Figure 8.

CaBP4 phosphorylation in the retina increases in response to light. A, Western blot analysis of native CaBP4 in wild-type mouse retina using the phospho-S37 antibody. Phosphorylation of CaBP4 was analyzed by Western blot with the phospho-Ser37-specific antibody in three groups (1–3; 2 retinas per group) of dark-adapted (WT, Dark) or light-adapted (WT, Light) mouse retina (top). Bottom shows equivalent levels of CaBP4 detected by the anti-CaBP4 antibody. Lysate of light-adapted retina from Cabp4−/− mice (Cabp4−/−, Light; right) was used as a negative control. B, Western blot and autoradiograph of immunoprecipitated 32P-labeled phosphorylated CaBP4 from dark-adapted and light-adapted mouse retinas. Retinal proteins from dark-adapted (WT, Dark) or from light-adapted Cabp4+/+ (WT, Light) retinas were incubated with [γ-32P]ATP under dim red light or in light, respectively, and subject to immunoprecipitation with anti-CaBP4 antibody. Incorporation of 32P in CaBP4 is indicated in autoradiograph (top). Western blot with anti-CaBP4 antibody (bottom) shows levels of immunoprecipitated CaBP4. Reactions with retina from light-adapted Cabp4−/− (Cabp4−/−, Light) mice was performed in parallel as a negative control. C, D, Absence of light-dependent CaBP4 phosphorylation in Gnat1−/− mouse retina. C shows similar experiment as in A except that retina was from Gnat1−/− mouse. Retina from mice lacking both CaBP4 and Gnat 1 (Cabp4−/−/Gnat1−/−) was used as a negative control. D shows similar experiment as in B except with retina from WT, Gnat1−/−, and Cabp4−/−/Gnat1−/− mice.
Together, our results indicate that CaBP4 phosphorylation is favored in the light, which may significantly influence the role of this protein in regulating photoreceptor synaptic transmission.
Discussion
CaM-like CaBPs have emerged as essential regulators of Ca2+ signaling in neurons (Lee et al., 2002; Haeseleer et al., 2004; Zhou et al., 2004, 2005). Our study provides the first evidence that CaBP4, one CaBP family member, is subject to phosphorylation by PKC and that phosphorylation can modulate interactions of CaBP4 with Cav1.3 Ca2+ channels. Light-stimulated phosphorylation of CaBP4 in the retina suggests a mechanism by which the activity of CaBP4 may be upregulated or downregulated with respect to photoreceptor light signal transmission.
CaBP4 is phosphorylated by PKCζ
PKCs are serine/threonine kinases that are classified according to their dependence on Ca2+, phospholipid, and DAG (Mellor and Parker, 1998). The classical PKCs (α, βI, βii, and γ) are Ca2+ and DAG dependent, whereas the novel PKCs (δ, ε, η, and θ) require DAG but not Ca2+. Atypical PKCs (ζ, δ, ε, η, and θ) are insensitive to both Ca2+ and DAG. Our conclusion that PKCζ phosphorylates CaBP4 in the retina is based on our findings that CaBP4 phosphorylation does not require Ca2+ or DAG and is inhibited by the specific pseudosubstrate inhibitor for PKCζ and Bisindolymaleimide I, a common PKC inhibitor (Fig. 1). This conclusion is further supported by the expression of PKCζ in the retina and colocalization with CaBP4 in inner segments and at the photoreceptor synapse in the outer plexiform layer (Fig. 2D). This result is consistent with other studies that have described expression of PKCζ in the retina and in photoreceptors (Fujisawa et al., 1992; Ghalayini et al., 1994). Our results also indicate that CaBP4 can be phosphorylated by PKCα in vitro and in transfected HEK293T cells (Figs. 3E, 6). However, in the retina, PKCα is primarily expressed in bipolar cells and not at the photoreceptors synapse. Although biochemical evidence of PKCα expression in rod outer segments have been reported, PKCα is unlikely to participate in the regulation of CaBP4 in vivo because CaBP4 is not localized in photoreceptor outer segments (Williams et al., 1997; Sokal et al., 2003). Although our results indicate S37 to be the major site targeted by PKCζ, CaBP4 contains additional consensus sites for phosphorylation by various protein kinases. One of these sites, threonine 223, is analogous to serine 120 of CaBP1 and serine 101 of CaM, which are phosphorylated by CK2 (Nakajo et al., 1988; Kasri et al., 2004). Although CaBP4 can be phosphorylated by recombinant CK2 in vitro (data not shown), CaBP4 phosphorylation in the retinal extract was not affected by the CK2 inhibitor (Fig. 1E) or by mutation of threonine 223 to alanine in CaBP4 (data not shown). Our findings that the S37A mutation in CaBP4 completely eliminates phosphorylation of CaBP4, by both recombinant and native PKC (Figs. 3, 6), demonstrates that S37 is the primary site of phosphorylation in CaBP4 and that PKCζ is the physiologically relevant kinase for CaBP4 in the retina.
Multiple lines of evidence indicate that CaBP4 is phosphorylated under conditions when intracellular Ca2+ is minimal. First, CaBP4 phosphorylation is favored in the presence of EGTA, which chelates Ca2+, and under light-adapted conditions when Ca2+ concentrations are low in photoreceptors compared with in darkness (Rieke and Schwartz, 1996). Second, the CaBP4 EF-hand mutant unable to bind Ca2+ was phosphorylated as efficiently as the wild-type CaBP4. This result also shows that Ca2+-induced conformational change does not mediate the decreased phosphorylation of CaBP4 in the presence of Ca2+. More likely, dephosphorylation of CaBP4 in the dark may be mediated by a Ca2+-dependent phosphatase (Kutuzov and Bennett, 1996; Sherman et al., 1997) and its phosphorylation promoted by light-dependent activation of PKCζ.
Functional significance of CaBP4 phosphorylation
Phosphorylation of S37 did not affect binding of Ca2+ to CaBP4 (Fig. 3E) and therefore is unlikely to influence Ca2+-dependent activities of CaBP4. However, CaBPs, including CaBP4, associate with Cav voltage-gated Ca2+ channels independent of Ca2+ (Lee et al., 2002; Haeseleer et al., 2004; Zhou et al., 2004, 2005; Cui et al., 2006). CaBP1 and CaBP4 compete with CaM in binding to the C-terminal IQ motif in the Cav1.2 and Cav1.3 α1 subunit (Zhou et al., 2004, 2005; Cui et al., 2006; Yang et al., 2006). CaM binding to this site mediates Ca2+-dependent inactivation of Cav1 channels (Peterson et al., 1999; Zuhlke et al., 1999). CaBP1 binding to this site, as well as to a modulatory site in the N-terminal domain of the Cav1.2 α1 subunit (α11.2), opposes the actions of CaM and causes prolonged activation of Cav1.2 Ca2+ currents (Zhou et al., 2004, 2005). CaBP4 also associates with the IQ domain of α11.3 but causes a more modest suppression of Ca2+-dependent inactivation than CaBP1 (Cui et al., 2006; Yang et al., 2006). Whereas CaBP4 also interacts with the C-terminal domain of α11.4 (Haeseleer et al., 2004), Cav1.4 channels alone show no Ca2+-dependent inactivation attributable to an autoinhibitory module that inhibits this process located in the distal C-terminal domain of α11.4 (Singh et al., 2006; Wahl-Schott et al., 2006). However, the presence of this autoinhibitory module does not interfere with CaBP4 binding to or modulation of Cav1.4 (Haeseleer et al., 2004).
Perhaps because of distinct sequence elements in their C-terminal domains, α11.4 and α11.3 interacted differentially with phosphorylated CaBP4 (Fig. 4). CaBP4 phosphorylation strengthened Ca2+-independent interactions with α11.4 (Fig. 4B,C) but slightly weakened binding to α11.3 both with and without Ca2+ (Fig. 4D,E). For Cav1.3, phosphorylation of S37 in CaBP4 may facilitate low-affinity interactions that transduce inhibitory modulation of Cav1.3 inactivation. Phosphorylation-induced conformational changes would be prevented by the S37A mutation in CaBP4, as shown by the negative effects on Cav1.3 inactivation (Fig. 5). The importance of low-affinity interactions of CaBP4 with α11.3 for channel modulation is further supported by the stronger effects of the CaBP4 EF-hand mutant (CaBP4–EF) on suppressing Cav1.3 inactivation (Fig. 7). Because CaBP4 binding to α11.3 was weaker in the absence of Ca2+ (Fig. 4), it is likely that CaBP4–EF maintains the low-affinity interaction required for channel modulation (Fig. 7). Light-dependent decreases in the photoreceptor Ca2+ concentration may increase CaBP4 phosphorylation. Physiologically, we would thus predict that phosphorylation of CaBP4 may have an opposite effect on CaBP4 activity than Ca2+ binding, which is consistent with our biochemical and electrophysiological analyses.
Light-dependent protein phosphorylation is a common regulatory mechanism in photoreceptors (Kuhn and Dreyer, 1972; Lee et al., 1984; BoeszeBattaglia et al., 1997; Roof et al., 1997; Hayashi et al., 2000; Hu et al., 2001; Rajala et al., 2002; Trojan et al., 2003). Because CaBP4 phosphorylation is increased under light conditions, CaBP4 may therefore adjust Cav1.3 and Cav1.4 Ca2+ signals as a function of light intensity. Cav1.3 channels are expressed primarily in cones (Morgans et al., 1998; Morgans, 1999; Barnes and Kelly, 2002), which show faster rates of neurotransmitter release compared with rods in darkness (Choi et al., 2005; Sheng et al., 2007). This difference in transmission may result from a higher intraterminal Ca2+ concentration in cones compared with rods in the dark (Sheng et al., 2007). We have shown previously that CaBP4 is required for normal cone to bipolar cell transmission (Maeda et al., 2005). For Cav1.3, light-dependent phosphorylation of CaBP4 may maintain activation of the presynaptic Ca2+ current at various light intensities, thus ensuring high release rates even at more hyperpolarized voltages. Cav1.4 channels are expressed primarily in rods (Morgans, 2001; Morgans et al., 2001; Barnes and Kelly, 2002), which are biochemically specialized for responses to dim illumination. Phosphorylation of CaBP4 during a light stimulus may help regulate Cav1.4 Ca2+ signals, thus increasing the signal-to-noise ratio and reliability of transmission at the rod synapse.
Although the upstream signaling events that ultimately lead to CaBP4 phosphorylation by PKCζ remain to be addressed, the activation of the rod phototransduction cascade is required because light-dependent phosphorylation of CaBP4 was not observed in retina from Gnat1−/− mice (Fig. 8C,D), which lack functional rods (Calvert et al., 2000). Although CaBP4 phosphorylation might still occur in cones of Gnat1−/− mice, cones constitute only 3% of the photoreceptors in mouse retina (Carter-Dawson and Lavail, 1979) such that this small fraction of phosphorylated CaBP4 may not have been not detected in our assay.
Although we have identified Cav1 channels as important targets for CaBP4 modulation, we cannot exclude that CaBP4 also interacts with and/or regulates other synaptic proteins. Phosphorylation of CaBP4 may differentially regulate the function of a variety of effectors, helping to shape presynaptic Ca2+ signals in photoreceptors and their neurotransmitter release properties in response to light.
Footnotes
This work was supported by National Institutes of Health Grants EY014561 (F.H.) and NS044922 (A.L.). We thank Izabela Sokal for her advice with the phosphorylation assay. We thank Dr. Diane Lipscombe for the cDNA encoding Cav1.3 α subunit.
References
- Allen D, Fakler B, Maylie J, Adelman JP. Organization and regulation of small conductance Ca2+-activated K+ channel multiprotein complexes. J Neurosci. 2007;27:2369–2376. doi: 10.1523/JNEUROSCI.3565-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball SL, Powers PA, Shin HS, Morgans CW, Peachey NS, Gregg RG. Role of the beta(2) subunit of voltage-dependent calcium channels in the retinal outer plexiform layer. Invest Ophthalmol Vis Sci. 2002;43:1595–1603. [PubMed] [Google Scholar]
- Barnes S, Kelly MEM. Calcium channels at the photoreceptor synapse. In: Baehr W, Palczewski K, editors. Photoreceptors and calcium. Georgetown, TX: Landes Bioscience; 2002. pp. 465–476. [Google Scholar]
- Bech-Hansen NT, Naylor MJ, Maybaum TA, Pearce WG, Koop B, Fishman GA, Mets M, Musarella MA, Boycott KM. Loss-of-function mutations in a calcium-channel alpha(1)-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19:264–267. doi: 10.1038/947. [DOI] [PubMed] [Google Scholar]
- Benaim G, Villalobo A. Phosphorylation of calmodulin—functional implications. Eur J Biochem. 2002;269:3619–3631. doi: 10.1046/j.1432-1033.2002.03038.x. [DOI] [PubMed] [Google Scholar]
- Bildl W, Strassmaier T, Thurm H, Andersen J, Eble S, Oliver D, Knipper M, Mann M, Schulte U, Adelman JP, Fakler B. Protein kinase CK2 is coassembled with small conductance Ca2+-activated K+ channels and regulates channel gating. Neuron. 2004;43:847–858. doi: 10.1016/j.neuron.2004.08.033. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Kong FS, Lamba OP, Stefano FP, Williams DS. Purification and light-dependent phosphorylation of a candidate fusion protein, the photoreceptor cell peripherin/rds. Biochemistry. 1997;36:6835–6846. doi: 10.1021/bi9627370. [DOI] [PubMed] [Google Scholar]
- Calvert PD, Krasnoperova NV, Lyubarsky AL, Isayama T, Nicolo M, Kosaras B, Wong G, Gannon KS, Margolskee RF, Sidman RL, Pugh EN, Makino CL, Lem J. Phototransduction in transgenic mice after targeted deletion of the rod transducin alpha-subunit. Proc Natl Acad Sci USA. 2000;97:13913–13918. doi: 10.1073/pnas.250478897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-Dawson LD, Lavail MM. Rods and cones in the mouse retina. 1. Structural-analysis using light and electron-microscopy. J Comp Neurol. 1979;188:245–262. doi: 10.1002/cne.901880204. [DOI] [PubMed] [Google Scholar]
- Chang B, Heckenlively JR, Bayley PR, Brecha NC, Davisson MT, Hawes NL, Hirano AA, Hurd RE, Ikeda A, Johnson BA, McCall MA, Morgans CW, Nusinowitz S, Peachey NS, Rice DS, Vessey KA, Gregg RG. The nob2 mouse, a null mutation in Cacnalf: anatomical and functional abnormalities in the outer retina and their consequences on ganglion cell visual responses. Vis Neurosci. 2006;23:11–24. doi: 10.1017/S095252380623102X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Borghuis B, Rea R, Levitan ES, Sterling P, Kramer RH. Encoding light intensity by the cone photoreceptor synapse. Neuron. 2005;48:555–562. doi: 10.1016/j.neuron.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Cui G, Haeseleer F, Lee A. Ca2+-binding protein 4 is expressed in cochlear hair cells and modulates Cav1.3 L-type Ca2+ channels. Soc Neurosci Abstr. 2006;32:131–9. [Google Scholar]
- Edelman AM, Blumenthal DK, Krebs EG. Protein serine threonine kinases. Annu Rev Biochem. 1987;56:567–613. doi: 10.1146/annurev.bi.56.070187.003031. [DOI] [PubMed] [Google Scholar]
- Fujisawa N, Ogita K, Saito N, Nishizuka Y. Expression of protein-kinase-C subspecies in rat retina. FEBS Lett. 1992;309:409–412. doi: 10.1016/0014-5793(92)80818-2. [DOI] [PubMed] [Google Scholar]
- Ghalayini AJ, Koutz CA, Wetsel WC, Hannun YA, Anderson RE. Immunolocalization of Pkc-zeta in rat photoreceptor Inner segments. Curr Eye Res. 1994;13:145–150. doi: 10.3109/02713689409042409. [DOI] [PubMed] [Google Scholar]
- Greferath U, Grunert U, Wassle H. Rod bipolar cells in the mammalian retina show protein kinase-C-like immunoreactivity. J Comp Neurol. 1990;301:433–442. doi: 10.1002/cne.903010308. [DOI] [PubMed] [Google Scholar]
- Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F. Rottlerin, a novel protein-kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–98. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C mu by various inhibitors. Differentiation from protein kinanse C isoenzymes. FEBS Lett. 1996;392:77–80. doi: 10.1016/0014-5793(96)00785-5. [DOI] [PubMed] [Google Scholar]
- Haeseleer F, Palczewski K. Calmodulin and Ca2+-binding proteins (CaBPs): variations on a theme. In: Baehr W, Palczewski K, editors. Photoreceptors and calcium. Georgetown, TX: Landes Bioscience; 2002. [DOI] [PubMed] [Google Scholar]
- Haeseleer F, Sokal I, Verlinde C, Erdjument-Bromage H, Tempst P, Pronin AN, Benovic JL, Fariss RN, Palczewski K. Five members of a novel Ca2+-binding protein (CABP) subfamily with similarity to calmodulin. J Biol Chem. 2000;275:1247–1260. doi: 10.1074/jbc.275.2.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeseleer F, Imanishi Y, Sokal I, Filipek S, Palczewski K. Calcium-binding proteins: intracellular sensors from the calmodulin superfamily. Biochem Biophys Res Commun. 2002;290:615–623. doi: 10.1006/bbrc.2001.6228. [DOI] [PubMed] [Google Scholar]
- Haeseleer F, Imanishi Y, Maeda T, Possin DE, Maeda A, Lee A, Rieke F, Palczewski K. Essential role of Ca2+-binding protein 4, a Ca(v) 1.4 channel regulator, in photoreceptor synaptic function. Nat Neurosci. 2004;7:1079–1087. doi: 10.1038/nn1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi F, Matsuura I, Kachi S, Maeda T, Yamamoto M, Fujii Y, Liu H, Yamazaki M, Usukura J, Yamazaki A. Phosphorylation by cyclin-dependent protein kinase 5 of the regulatory subunit of retinal cGMP phosphodiesterase II. Its role in the turnoff of phosphodiesterase in vivo. J Biol Chem. 2000;275:32958–32965. doi: 10.1074/jbc.M000703200. [DOI] [PubMed] [Google Scholar]
- Haynes LP, Tepikin AV, Burgoyne RD. Calcium-binding protein 1 is an inhibitor of agonist-evoked, inositol 1,4,5-trisphosphate-mediated calcium signaling. J Biol Chem. 2004;279:547–555. doi: 10.1074/jbc.M309617200. [DOI] [PubMed] [Google Scholar]
- Hu G, Jang GF, Cowan CW, Wensel TG, Palczewski K. Phosphorylation of RGS9–1 by an endogenous protein kinase in rod outer segments. J Biol Chem. 2001;276:22287–22295. doi: 10.1074/jbc.M011539200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T, Cooper JA. Protein-tyrosine kinases. Annu Rev Biochem. 1985;54:897–930. doi: 10.1146/annurev.bi.54.070185.004341. [DOI] [PubMed] [Google Scholar]
- Kasri NN, Holmes AM, Bultynck G, Parys JB, Bootman MD, Rietdorf K, Missiaen L, McDonald F, De Smedt H, Conway SJ, Holmes AB, Berridge MJ, Roderick HL. Regulation of InsP(3) receptor activity by neuronal Ca2+-binding proteins. EMBO J. 2004;23:312–321. doi: 10.1038/sj.emboj.7600037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita-Kawada M, Tang JS, Xiao R, Kaneko S, Foskett JK, Zhu MX. Inhibition of TRPC5 channels by Ca2+-binding protein 1 in Xenopus oocytes. Pflügers Arch. 2005;450:345–354. doi: 10.1007/s00424-005-1419-1. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Dreyer WJ. Light dependent phosphorylation of rhodopsin by Atp. FEBS Lett. 1972;20:1–6. doi: 10.1016/0014-5793(72)80002-4. [DOI] [PubMed] [Google Scholar]
- Kutuzov MA, Bennett N. Calcium-activated opsin phosphatase activity in retinal rod outer segments. Eur J Biochem. 1996;238:613–622. doi: 10.1111/j.1432-1033.1996.0613w.x. [DOI] [PubMed] [Google Scholar]
- Lee A, Westenbroek RE, Haeseleer F, Palczewski K, Scheuer T, Catterall WA. Differential modulation of Ca(v) 2.1 channels by calmodulin and Ca2+-binding protein 1. Nat Neurosci. 2002;5:210–217. doi: 10.1038/nn805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RH, Brown BM, Lolley RN. Light-induced dephosphorylation of a 33k protein in rod outer segments of rat retina. Biochemistry. 1984;23:1972–1977. doi: 10.1021/bi00304a014. [DOI] [PubMed] [Google Scholar]
- Maeda T, Lem J, Palczewski K, Haeseleer F. A critical role of CaBP4 in the cone synapse. Invest Ophthalmol Vis Sci. 2005;46:4320–4327. doi: 10.1167/iovs.05-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansergh F, Orton NC, Lalonde MR, Stell WK, Tremblay F, Barnes S, Rancourt DE, Bech-Hansen NT. Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina. Hum Mol Genet. 2005;14:3035–3046. doi: 10.1093/hmg/ddi336. [DOI] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective-inhibition of protein-kinase-C isozymes by the indolocarbazole Go 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J. 1998;332:281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgans CW. Calcium channel heterogeneity among cone photoreceptors in the tree shrew retina. Eur J Neurosci. 1999;11:2989–2993. doi: 10.1046/j.1460-9568.1999.00719.x. [DOI] [PubMed] [Google Scholar]
- Morgans CW. Localization of the alpha(1F) calcium channel subunit in the rat retina. Invest Ophthalmol Vis Sci. 2001;42:2414–2418. [PubMed] [Google Scholar]
- Morgans CW, El Far O, Berntson A, Wassle H, Taylor WR. Calcium extrusion from mammalian photoreceptor terminals. J Neurosci. 1998;18:2467–2474. doi: 10.1523/JNEUROSCI.18-07-02467.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgans CW, Gaughwin P, Maleszka R. Expression of the alpha(1F) calcium channel subunit by photoreceptors in the rat retina. Mol Vis. 2001;7:202–209. [PubMed] [Google Scholar]
- Nakajo S, Masuda Y, Nakaya K, Nakamura Y. Determination of the phosphorylation sites of calmodulin catalyzed by casein kinase-2. J Biochem. 1988;104:946–951. doi: 10.1093/oxfordjournals.jbchem.a122588. [DOI] [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of 1-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- Quadroni M, L'Hostis EL, Corti C, Myagkikh I, Durussel I, Cox J, James P, Carafoli E. Phosphorylation of calmodulin alters its potency as an activator of target enzymes. Biochemistry. 1998;37:6523–6532. doi: 10.1021/bi972930+. [DOI] [PubMed] [Google Scholar]
- Rajala RVS, McClellan ME, Ash JD, Anderson RE. In vivo regulation of phosphoinositide 3-kinase in retina through light-induced tyrosine phosphorylation of the insulin receptor beta-subunit. J Biol Chem. 2002;277:43319–43326. doi: 10.1074/jbc.M206355200. [DOI] [PubMed] [Google Scholar]
- Rieke F, Schwartz EA. Asynchronous transmitter release: control of exocytosis and endocytosis at the salamander rod synapse. J Physiol (Lond) 1996;493:1–8. doi: 10.1113/jphysiol.1996.sp021360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roof DJ, Hayes A, Adamian M, Chishti AH, Li TS. Molecular characterization of abLIM, a novel actin-binding and double zinc finger protein. J Cell Biol. 1997;138:575–588. doi: 10.1083/jcb.138.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Z, Choi SY, Dharia A, Li J, Sterling P, Kramer RH. Synaptic Ca2+ in darkness is lower in rods than cones, causing slower tonic release of vesicles. J Neurosci. 2007;27:5033–5042. doi: 10.1523/JNEUROSCI.5386-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman PM, Sun H, Macke JP, Williams J, Smallwood PM, Nathans J. Identification and characterization of a conserved family of protein serine/threonine phosphatases homologous to Drosophila retinal degeneration C (rdgC) Proc Natl Acad Sci USA. 1997;94:11639–11644. doi: 10.1073/pnas.94.21.11639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Hamedinger D, Hoda JC, Gebhart M, Koschak A, Romanin C, Striessnig J. C-terminal modulator controls Ca2+-dependent gating of Ca(v)1.4 L-type Ca2+ channels. Nat Neurosci. 2006;9:1108–1116. doi: 10.1038/nn1751. [DOI] [PubMed] [Google Scholar]
- Sokal I, Hu G, Liang Y, Mao ML, Wensel TG, Palczewski K. Identification of protein kinase C isozymes responsible for the phosphorylation of photoreceptor-specific RGS9–1 at Ser(475) J Biol Chem. 2003;278:8316–8325. doi: 10.1074/jbc.M211782200. [DOI] [PubMed] [Google Scholar]
- Strom TM, Nyakatura G, Apfelstedt-Sylla E, Hellebrand H, Lorenz B, Weber BHF, Wutz K, Gutwillinger N, Ruther K, Drescher B, Sauer C, Zrenner E, Meitinger T, Rosenthal A, Meindl A. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19:260–263. doi: 10.1038/940. [DOI] [PubMed] [Google Scholar]
- Tippens AL, Lee A. Caldendrin, a neuron-specific modulator of Ca(v) 1.2 (L-type) Ca2+ channels. J Biol Chem. 2007;282:8464–8473. doi: 10.1074/jbc.M611384200. [DOI] [PubMed] [Google Scholar]
- Trojan P, Giessl A, Pulvermuller A, Hofmann KP, Wolfrum U. Light-dependent phosphorylation of Centrins in vertebrate retina. Eur J Cell Biol. 2003;82:76. [Google Scholar]
- Wahl-Schott C, Baumann L, Cuny H, Eckert C, Griessmeier K, Biel M. Switching off calcium-dependent inactivation in L-type calcium channels by an autoinhibitory domain. Proc Natl Acad Sci USA. 2006;103:15657–15662. doi: 10.1073/pnas.0604621103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DS, Liu XR, Schlamp CL, Ondek B, Jaken S, Newton AC. Characterization of protein kinase C in photoreceptor outer segments. J Neurochem. 1997;69:1693–1702. doi: 10.1046/j.1471-4159.1997.69041693.x. [DOI] [PubMed] [Google Scholar]
- Xu WF, Lipscombe D. Neuronal Cav 1.3 α1 L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci. 2001;21:5944–5951. doi: 10.1523/JNEUROSCI.21-16-05944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, McBride S, Mak DOD, Vardi N, Palczewski K, Haeseleer F, Foskett JK. Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca2+ release channels. Proc Natl Acad Sci USA. 2002;99:7711–7716. doi: 10.1073/pnas.102006299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang PS, Alseikhan BA, Hiel H, Grant L, Mori MX, Yang WJ, Fuchs PA, Yue DT. Switching of Ca2+-dependent inactivation of CaV1.3 channels by calcium binding proteins of auditory hair cells. J Neurosci. 2006;26:10677–10689. doi: 10.1523/JNEUROSCI.3236-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitz C, Kloeckener-Gruissem B, Forster U, Kohl S, Magyar I, Wissinger B, Matyas G, Borruat FX, Schorderet DF, Zrenner E, Munier FL, Berger W. Mutations in CABP4, the gene encoding the Ca2+-binding protein 4, cause autosomal recessive night blindness. Am J Hum Genet. 2006;79:657–667. doi: 10.1086/508067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Kim SA, Kirk EA, Tippens AL, Sun H, Haeseleer F, Lee A. Ca2+-binding protein-1 facilitates and forms a postsynaptic complex with Cav1.2 (L-Type) Ca2+ channels. J Neurosci. 2004;24:4698–4708. doi: 10.1523/JNEUROSCI.5523-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Yu K, McCoy KL, Lee A. Molecular mechanism for divergent regulation of Ca(v) 1.2 Ca2+ channels by calmodulin and Ca2+-binding protein-1. J Biol Chem. 2005;280:29612–29619. doi: 10.1074/jbc.M504167200. [DOI] [PubMed] [Google Scholar]
- Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]