

Abstract
A mild, convenient reaction sequence for the synthesis of Nazarov cyclization substrates is described. The [3+2] dipolar cycloaddition of a nitrone and an electron-deficient alkyne gives an isolable isoxazoline intermediate, which upon oxidation undergoes stereoselective extrusion of nitrosomethane to give aryl vinyl or divinyl ketones.
1. Introduction
One of the most important factors determining synthetic utility of Nazarov cyclization is the efficiency of divinyl ketone preparation. A number of specific methods have been developed for their synthesis, depending on the vinyl substitution pattern desired.i,ii,iii,iv,v For the preparation of substrates with electron-withdrawing groups such as those employed in Lewis acid-catalyzed Nazarov cyclizations of polarized substrates,vi Knoevenagel condensation has been the only option.vii Disadvantages of the procedure include: the requirement for a β-ketoester precursor, which is can be quite difficult to prepare efficiently; reaction conditions involving high temperatures; long reaction times; and either acidic or basic pH, all of which can destroy sensitive functionality. Because the condensation is thermodynamically controlled, mixtures of diastereomers are obtained. Finally, it can be difficult to prevent the divinyl ketone from undergoing premature Nazarov cyclization during acid-catalyzed Knoevenagel condensation.
An unusual reaction was reported by Padwa in 1987, in which Δ4-isoxazolines 1 underwent oxidative extrusion of nitrosomethane to give α-β-unsaturated ketones 3 (Scheme 1).viii
Scheme 1.

Padwa’s Oxidation of Δ4-Isoxazolines.
Upon treatment with m-CPBA, the ring nitrogen of isoxazoline 1 is thought to undergo oxidation to give intermediate 2, which cannot be isolated or even observed by NMR spectroscopy. Instead, immediate extrusion of nitrosomethane from putative N-oxide 2 occurs at low temperature to give 3 with surprisingly high stereoselectivity. The yields were good in the reported cases, although the scope was limited. This sequence provided us with a useful starting point to solve the problems associated with the preparation of Nazarov substrates.
This paper describes a mild and versatile [3+2] cycloaddition/nitrosomethane extrusion process for the synthesis of divinyl ketones for Nazarov cyclization. The reaction sequence is shown in Scheme 2: cycloaddition of a nitrone and an electron-deficient alkyne give a stable isoxazoline 5 (see Scheme 1). Oxidation leads to extrusion of nitrosomethane at low temperature to give a Nazarov precursor of type 6.
Scheme 2.
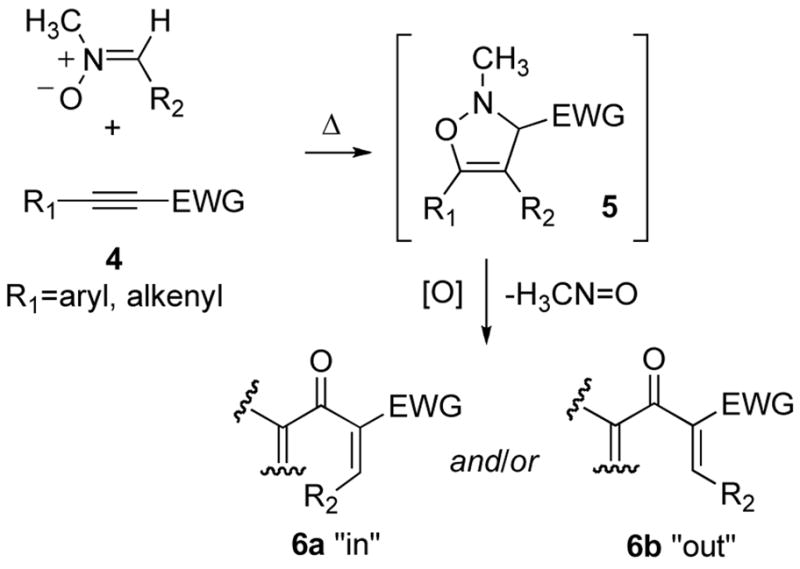
[3+2]/[O] sequence for synthesis of Nazarov substrates.
The reaction sequence has not only proven to be an efficient and stereoselective method for the synthesis of Nazarov cyclization precursors, but the unexpected stereoselectivities observed have also raised interesting questions about the factors controlling the “cheletropic” fragmentation of oxidized isoxazolines like 2. Cheletropic extrusions are a class of retro-cycloaddition reactions in which two sigma bonds flanking a single atom of a ring system are broken during a concerted bond reorganization.ix Two examples of cheletropic extrusions of (4n+2) systems that give stereochemistry consistent with Woodward-Hoffmann theory are shown in equations 1 and 2.
 |
(1) |
 |
(2) |
Extrusion can occur in either an inward or an outward direction,x and for unsymmetric substrates two diastereomeric products can be obtained. If one of these senses of rotation is favored over the other, the reaction is termed “torquoselective,” as defined by Houk. Typically, the sense of rotation is controlled by stereoelectronic effects rather than steric effects, and the stereochemical outcome of many torquoselective reactions, including Nazarov cyclizations, sigmatropic shifts, electrocyclic ring openings and closures, and the Cope rearrangement,xi,xii can be predicted using Houk’s theoretical model.xi An alternative theory, proposing a role for geminal bonds in the torquoselectivity of retro-electrocyclizations, has also been proposed.xif, xiii As will be discussed, the stereochemistry observed in the oxidation/extrusion sequences could not be rationalized using conventional methods, but computational studies led to new mechanistic proposals to explain the torquoselective extrusion.
2. Results
A majority of the acetylenic esters were prepared from the corresponding aldehydes via Corey-Fuchsxiv protocol, followed by acylation of the resulting alkyne (Table 1). Ester 4axv was made from commercially available piperonal 7a, in 89% overall yield (Entry 1). Pyranyl ester 4b was readily prepared from pyranyl aldehyde 7bxvi in 63% overall yield (Entry 2). Known methyl substituted cyclohexenyl aldehyde 7cxvii gave methylcyclohexenyl ester 4c in 78% yield over two steps (Entry 3). Cyclopentenyl ester 4d was prepared by the addition of lithiated ethyl propiolate to commercially available cyclopentanone 7d, followed by dehydration with POCl3 in pyridine, to give 58% yield over two steps (Entry 4). Cyclohexenyl ester 4e was obtained by acylation of enyne 7exviii with ethyl chloroformate in 95% yield (Entry 5).
Table 1.
Preparation of alkynoates
| entry | substrate | conditions | product | yield % |
|---|---|---|---|---|
| 1 |
 7a 7a
|
A |
 4a 4a
|
89 |
| 2 |
 7b 7b
|
A |
 4b 4b
|
63 |
| 3 |
 7c 7c
|
A |
 4c 4c
|
78 |
| 4 |
 7d 7d
|
B |
 4d 4d
|
58 |
| 5 |
 7e 7e
|
C |
 4e 4e
|
95 |
Reaction Conditions: A) 1. CBr4, PPh3, CH2Cl2 2. n-BuLi, ClC(O)OEt, THF, −78 °C B) LiCC-C(O)OEt, THF, −78 °C 2. POCl3, pyridine, 90 °C C) n-BuLi, ClC(O)OEt, THF, −78 °C
A series of substrates were prepared from the terminal alkyne 8, which was obtained from the methyl ketone by a 3-step 1-pot reaction sequence (Table 2).xix Deprotonation of alkyne 8 followed by addition of a range of electrophiles provided 9a-f in good overall yields. N-cyanobenzotriazole was selected as a cyanating reagent to prepare 9d. Sulfone 9e was synthesized by reacting alkyne 8 with sodium p-toluenesulfinate in the presence of ceric ammonium nitrate and sodium iodide, followed by base-induced elimination of H-I to recover the alkyne 9e.xx Haloalkyne 9f was easily synthesized by deprotonation of terminal alkyne 8 followed by addition of N-chlorosuccinimide. These alkynyl substrates could be prepared on multigram scale with no complications.
Table 2.
Synthesis of alkynes bearing different electron-withdrawing groups.
 | ||||
|---|---|---|---|---|
| Entry | Electrophile | Conditions | Product | Yield(%) |
| 1 |

|
A |
 9a 9a
|
98 |
| 2 |

|
A |
 9b 9b
|
77 |
| 3 |

|
A |
 9c 9c
|
95 |
| 4 |

|
A |
 9d 9d
|
74 |
| 5 |

|
B |
 9e 9e
|
83 |
| 6 |

|
A | -Cl 9f | 61 |
Reaction Conditions: a) i. LDA, THF, −78 °C; ii. ClP(O)(OEt)2, −78 to 23 °C; iii. LDA, THF, −78 to 23 °C A) n-BuLi, electrophile, THF, −78 °C B) i. CAN, NaI, p-tolSO2Na, CH3CN ii. K2CO3, acetone, Δ
Nitrones can be prepared in a variety of ways, most commonly by condensation of carbonyl compounds with N-alkyl hydroxylamines.xxi The condensations are typically run in the presence of either an acid scavenger or a water scavenger. We obtained the best results when both an acid scavenger and MgSO4 in refluxing methylene chloride were used (Table 3). In this way it was possible to prepare aryl, alkyl, and conjugated nitrones 10a–g (Entries 1–7). Additional nitrones bearing a different substituent on the nitrogen were prepared using t-butyl hydroxyl amine giving nitrones 10h–j (Entries 8–10).
Table 3.
Preparation of nitrones.
 | |||
|---|---|---|---|
| entry | R1 | R2 | yield (%) |
| 1 | phenyl | Me | 10a (84) |
| 2 | cyclohexyl | Me | 10b (89) |
| 3 | trans-cinnamyl | Me | 10c (82) |
| 4 | trans-α-methylcinnamyl | Me | 10d (89) |
| 5 | 2, 4, 6-trimethoxyphenyl | Me | 10e (83) |
| 6 | 3, 4, 5-trimethoxyphenyl | Me | 10f (89) |
| 7 | triethylphenyl | Me | 10g (25) |
| 8 | phenyl | t-Bu | 10h (69) |
| 9 | cyclohexyl | t-Bu | 10i (73) |
| 10 | trans-cinnamyl | t-Bu | 10j (75) |
With an array of alkynes and nitrones in hand, we applied the cyclization/oxidation reaction sequence to obtain dienones (Table 4). The [3+2] cycloaddition of alkynoates and nitrones proceeded without complication to provide isoxazolines, which were either isolated in good yield, or carried directly to the next step without purification. Oxidation was carried out using m-CPBA, generating trisubstituted olefins via extrusion of nitrosomethane at low temperature and in high yield. The resulting olefin geometry is denoted as “in” or “out” depending on the stereochemistry of the olefin substituent relative to the alkyl or aryl substituent that is installed from the alkyne starting material. Identification of stereoisomers was assigned by analysis of 3JC,H coupling constants.xxii It was found that for most substrates, the extrusion was highly stereoselective, favoring one olefin diastereoisomer. In most cases, the substituent R2 emerged cis to the ketone to give “in” stereoisomers in >15:1 selectivity (Entries 1, 7, 8). However, in a few cases the “out” stereoisomeric product, in which R2 is cis to the ester, was dominant (Entries 2, 4). When the ester substitution was changed from ethyl (Entry 5) to tert-butyl (Entry 6), the product mixture showed decreased selectivity for the “in” isomer.
Table 4.
[3+2]/[O] sequence for the synthesis of divinyl ketones.
 | ||||||
|---|---|---|---|---|---|---|
| entry | R1 | R2 | yield (%)[3+2] | major product from extrusion | yield (%)(a + b) | ratio(a: b) |
| 1 |

|
Ph | 94 |
 11a 11a
|
94 | >15:1 |
| 2 |

|

|
67 |
 12b 12b
|
96 | 1:2.6 |
| 3 |

|

|
74 |
 13a 13a
|
83 | 6:1 |
| 4 |

|

|
67 |
 14b 14b
|
93 | 1:12 |
| 5 |

|
Ph | 61 |
 15a 15a
|
85 | 8:1 |
| 6 |

|
Ph | a |
 16a 16a
|
90 | 6:1 |
| 7 |

|
Ph | 64 |
 17a 17a
|
64 | >15:1 |
| 8 |

|
Ph | 53 |
 18a 18a
|
35 | >15:1 |
Reaction Conditions: i. 1 equiv alkyne, 2 equiv nitrone, 0.2 M toluene, 65 °C, 16 h; ii. 1.5 equiv m-CPBA, 0.9 M DCM, 0 °C, 1 h.
ester = t-Bu instead of Et, isoxazoline was not isolated.
We next investigated this reaction sequence using piperonal derived alkyne 4a and nitrones that contained aryl groups with varying steric bulk and electronics (Table 5). Using an unsubstituted phenyl ring on the nitrone, the major product after oxidative extrusion was the “in” stereoisomer in a ratio of >15:1 (Entry 1). When the alkynyl ester 4a was replaced with an analogous t-butyl ester, the same stereoisomer was favored, and the ratio dropped to 8:1 (Entry 2) as we had observed in Table 1 (Entries 5, 6). By replacing the phenyl substituent with 2, 4, 6-triethylphenyl, the product mixture flipped to favor the “out” stereoisomer by <1:15 (Entry 3), and changing to 2, 4, 6-trimethoxyphenyl lowered the ratio while still favoring the “out” isomer by 1:3 (Entry 4). Interestingly, when 2, 4, 6-trimethoxyphenyl substituent was changed to 3, 4, 5-trimethoxyphenyl, the product mixture again favored the “in” isomer >15:1 (Entry 5). Other less sterically bulky substituents such as cyclohexyl, trans-cinnamyl, and n-butyl all favored the “in” stereoisomeric product in a ratio of 12:1 or higher (Entries 6–8). C-aliphatic nitrones, ketonitrones and C-unsubstituted nitrones are difficult to prepare due to rapid dimerization behaviour of aliphatic/unsubstituted nitrones, and facile hydrolysis of ketonitrones. We therefore devised a one-pot method to trap these unstable nitrones by preparing them in situ with ynoate dipolarophiles, which successfully gave the corresponding β-ketoalkylidenes in high yields (Entries 8–10).
Table 5.
Scope of nitrone substitution in the [3+2]/[O] sequence.
 | |||||
|---|---|---|---|---|---|
| entry | R1, R2 | yield (%) [3+2] | major product from extrusion | yield (%) (a + b) | ratio (a: b) |
| 1 | Ph, H | 93 |
 19a 19a
|
85 | >15:1 |
| 2a | Ph, H | b |
 20a 20a
|
71 | 8:1 |
| 3 |
 , H , H |
b |
 21b 21b
|
89 | <1:15 |
| 4 |
 , H , H |
b |
 22b 22b
|
94 | 1:3 |
| 5 |
 , H , H |
b |
 23a 23a
|
85 | >15:1 |
| 6 |
 , H , H |
86 |
 24a 24a
|
93 | >15:1 |
| 7 |
 , H , H |
95 |
 25a 25a
|
92 | 12:1 |
| 8 | Bu, Hc | 81 |
 26a 26a
|
95 | 12:1 |
| 9 | H, Hc | 85 |
 27 27
|
91 | n/a |
| 10 | Me, Mec | 96 |
 28 28
|
96 | n/a |
Reaction Conditions: i. 1 equiv alkyne, 2 equiv nitrone, 0.2 M toluene, 65 °C, 16 h; ii. 1.5 equiv m-CPBA, 0.9 M DCM, 0 °C, 1 h.
ester = t-Bu instead of Et,
isoxazoline was not isolated, but subjected immediately to the oxidation procedure
nitrone prepared in situ from corresponding aldehyde.
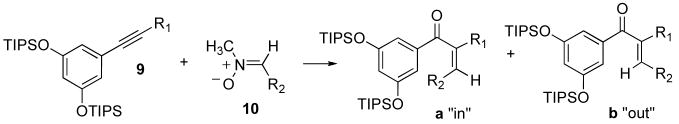
Alkynes bearing electron-withdrawing groups other than ester were also examined in the [3+2] cyclization/oxidative extrusion reaction sequence (Table 6).
Table 6.
[3+2]/[O] sequence applied to alkynes bearing varied electron-withdrawing groups.
 | |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Major product from extrusion | Yield a+b (%) | Ratio (a: b) |
| 1 |
 9a 9a
|

|
 29b 29b
|
65 | <1:19 |
| 2a |
 9a 9a
|

|
 30a 30a
|
67 | >19:1 |
| 3 |
 9b 9b
|

|
 31b 31b
|
63 | <1:19 |
| 4 |
 9c 9c
|

|
 32b 32b
|
57 | <1:19 |
| 5 |
 9d 9d
|

|
 33b 33b
|
57 | <1:19 |
| 6b |
 9e 9e
|

|
 34b 34b
|
43 | <1:19 |
| 7c | -Cl 9f |
|
 35b 35b
|
-- | -- |
Reaction Conditions: i. 1 equiv alkyne, 2 equiv nitrone, 0.2 M toluene, 80 °C, 16 h; ii. 2 equiv m-CPBA, 0.2 M CH2Cl2, 0 °C, 1 h.
N-methylnitrone prepared from trans-cinnamaldehyde was used.
reaction time = 5 days.
starting material was recovered unreacted, even with 1 equiv. Mg(OTf)2 or Zn(OTf)2
This reaction sequence worked well, giving β-ketoalkylidenes in moderate yield over two steps. Alkynyl ester 9a was reacted with two different nitrones (Entries 1–2), and the relative stereochemistry of the products reflected the substitution pattern of the nitrone utilized: when the nitrone contained a methyl substitution in the alpha position (Entry 1), the product favored the “out” stereoisomer 29b, however, when the nitrone replaced the methyl group with a hydrogen (Entry 2), the product distribution completely reversed to favor the “in” isomer 30a. Alkyne 9e was slow to undergo [3+2] cyclization (Entry 6); however, when the temperature was raised above 80 °C, the isoxazoline continued to react to give the β-ketoaziridine, which is a known transformation in the literature.xxiii Performing the [3+2] reaction at or below 80 °C circumvented this problem, though cycloaddition of alkynyl sulfone 9e took 5–8 days to go to completion. Haloalkyne 9f did not undergo [3+2] reaction and slowly dehalogenated to the terminal alkyne over extended reaction times. In an effort to bolster the reactivity of chloroalkyne 9f, Lewis acid promoters were added, as these have been used in [3+2] nitrone-alkene cycloadditions to improve regioselectivity.xxiv Unfortunately, catalytic and stoichiometric amounts of either Mg(OTf)2 or Zn(OTf)2 were unable to polarize the alkynyl chloride 9f toward reaction. A survey of the literature revealed surprisingly few examples of [3+2] cyclizations with either alkenyl or alkynyl halides, though the examples found were reported to cyclize in moderate yields.xxv
We also attempted the cycloaddition/oxidation sequence to prepare β-diketones (Table 7). Preliminary experiments were run with the N-methyl substituted isoxazolines (Entry 1–2). The results obtained were unexpectedly poor, giving only trace amounts of desired product and a large number of unidentified side products.
Table 7.
[3+2]/[O] sequence used to prepare β-diketone systems.
 | ||||||
|---|---|---|---|---|---|---|
| entry | R1 | R2 | yield [3+2] (%) | major product from extrusion | yield a+b (%) | ratio(a: b) |
| 1 | Me | Ph | 92 |
 36 36
|
-- | - |
| 2 | Me |
|
96 |
 37 37
|
12 | - |
| 3 | t-Bu | Ph | 50 |
 36b 36b
|
61 | <1:15 |
| 4 | t-Bu |

|
83 |
 37a 37a
|
66 | >15:1 |
| 5 | t-Bu |

|
64 |
 38a 38a
|
63 | 7:1 |
Reaction Conditions: i. 1 equiv alkyne, 1.5 equiv nitrone, 0.5 M toluene, 65 °C ii. 1.5 equiv m-CPBA, 0.9 M CH2Cl2, 0 °C.
A Hoffman-typexxvi elimination of the isoxazolino-N-oxide was considered as a decomposition pathway that could be responsible for the poor results (Scheme 3).
Scheme 3.

Potential competing Hoffman elimination pathway.
It was reasoned that an isoxazoline intermediate without α-hydrogens on the N-alkyl substituent would be unable to decompose via Hoffman elimination, so N-t-butyl substituted nitrones were tested in the sequence (Table 7, entries 3–5). We were pleased to observe smooth extrusion of the N-t-butyl isoxazolines, which gave β-diketones in good yields. Phenyl substitution on the nitrone gave the “out” isomer in a <1:15 ratio (Entry 3), and cyclohexyl completely reversed this selectivity to the “in” isomer by >15:1 (Entry 4). A trans-cinnamyl substituent on the nitrone gave a 7:1 ratio favoring the “in” isomer (Entry 5).
In addition to using the [3+2]/[O] sequence to the preparation of β-ketoesters and β-diketones, we attempted to apply these reactions to the synthesis of Baylis-Hillman-type adducts (Scheme 4). The Baylis-Hillman reactionxxvii is a popular method of preparing α-alkylidene-β-hydroxyketones, which are valuable building blocks for organic synthesis, especially in enantiopure form. However, while the Baylis-Hillman reaction is very effective for preparation of α-methylene-β-hydroxyketones (e.g. 43, R3 = H), reactivity often becomes a problem for more highly substituted analogs. Since an R3 substituent is easily introduced via a [3+2] cyclization, we sought to provide an alternative method for synthesis of α-alkylidene-β-hydroxyketones of type 43. It was envisioned that an isoxazoline of type 41 could be reduced to give the corresponding hydroxyisoxazoline 42, and subsequent oxidation/extrusion would provide the desired Baylis-Hillman adduct 43. Coupled with an enantioselective reduction of ketone 41,xxviii the strategy could provide chiral products with high enantiomeric excess.
Scheme 4.

Strategy for the preparation of Baylis-Hillman adducts.
To test this strategy, two complementary isoxazolines 44 and 47 were prepared via [3+2] cyclization. Isoxazoline 44 was reduced under Luchexxix conditions to yield the product alcohol 45 in 70 % yield, with a diastereomeric ratio of 1.3:1. Treatment of hydroxy isoxazoline 45 with m-CPBA under the standard oxidation conditions led to 73% yield of the desired β-hydroxy ketone isomers 46a and 46b in a ratio of 1:2 “in”:”out” relative to the ketone (eq 3). Reduction of isomeric isoxazoline 47 with DIBALxxx successfully gave alcohol 48 in 79% yield as a 3.6:1 mixture of diastereomers. Treatment of hydroxyl isoxazoline 48 with m-CPBA gave 73% yield of the desired β-hydroxyketone isomers 49a and 49b in a ratio of 1:1 (eq 4).xxxi So, it was possible to prepare the desired Baylis-Hillman adducts using this method, but mixtures of products were obtained. The reduction of isoxazoline ketones 44 and 47 using simple hydride reducing agents was not selective, which complicated analysis of selectivity in the oxidation/extrusion.
 |
(3 |
 |
(4) |
Fortunately, it was possible to separate the two diastereomers of hydroxyisoxazoline 45 by column chromatography and oxidize each diastereomer separately. Oxidation of the minor diastereomer gave a 7.8:1 mixture of products, and oxidation of the major diastereomer gave a 1:1 mixture of products. The complexity of the results obtained with isoxazolines 44 and 47 was discouraging, but our studies have shown that extrusion selectivity is highly substrate-dependent (vide supra), so other isoxazolines may be better candidates for this strategy.
3. Discussion
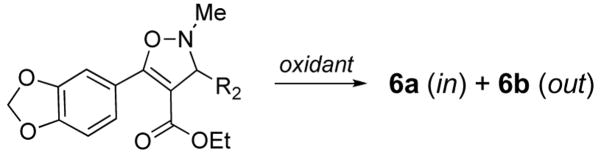
While the sequence represents a useful method for the synthesis of Nazarov cyclization precursors, we did not understand why the reaction was so stereoselective. For the intermediate N-oxides 2, the ring substituent R2 could rotate either inward (to give diastereomer 6a) or outward (to give diastereomer 6b) during the extrusion (Scheme 5). In the Padwa study, the reaction sequence always gave a single olefin isomer as product.viii Our experiments confirm the high selectivity of the sequence, and indicate that the olefin geometry of the product can change depending on the substitution pattern of the isoxazoline intermediate. The high torquoselectivities observed in both studies were rather surprising, especially since extrusion was selective for one olefin isomer (6a) in some cases and for the isomer with opposite olefin geometry (6b) in others.
Scheme 5.

N-Oxide Intermediates in the Oxidation/Extrusion Sequence.
The selectivities observed were not consistent with stereoelectronic predictions,xia and there were no obvious nonbonding interactions in the N-oxide intermediate that accounted for the product distribution. Neither is the product stereochemistry the result of thermodynamic equilibration, as product ratios were different from those obtained from synthesis via Knoevenagel condensation.xxxii Another possibility was that selectivity might arise from opposite directions of extrusion for the two diastereomeric N-oxide intermediates 2a and 2b, i.e. that one isomer undergoes selective inward extrusion, and the other selective outward extrusion to give the two product isomers 6a and 6b (Scheme 5).
Few literature cases of extrusions of this type could be found. One study on a system related to 2 has been reported, describing torquoselective extrusion of dimethylamine from N,N-dimethyl isoxazolinium salts, via an intermediate without a stereocenter at nitrogen.xxxiii Like our results, the stereoselectivity in these cases could not be rationalized easily. Since it was not possible to characterize the behavior of the N-oxides 2 directly, attempts were made to alter the ratio of N-oxides 2 generated in the oxidation step. Different oxidation reagents might have different selectivity for the two faces of the isoxazoline; in which case, we would expect to observe a different ratio of extrusion products 6a and 6b with different oxidants. Indeed, extrusion selectivity was slightly different when m-CPBA vs. oxaziridine 50 was employed in oxidation experiments with two different isoxazolines (Table 8).
Table 8.
Extrusion results using different oxidants.
 | ||
|---|---|---|
| R2 | oxidanta | ratio 6a/6b |
| 2,4,6-trimethoxyphenyl | m-CPBA | 1:2 |
| 2,4,6-trimethoxyphenyl | 50 | 1:10 |
| cinnamyl | m-CPBA | 12:1 |
| cinnamyl | 50 | 7:1 |
oxidant 50= 
Calculations on the oxidation/extrusion sequence were also performed in collaboration with the Houk group.xxxiv Oxidation by m-CPBA and oxaziridine models was calculated to favor a cis relationship between R2 and the N-methyl group (see 2b) in order to minimize repulsion between the oxidant and R2. These results are not consistent with the hypothesis that oxidation stereoselectivity dictates product stereochemistry. The change in the 6a/6b ratio may be explained by the fact that the activation barrier for oxidation by 50 was calculated to be significantly higher (by approximately 14 kcal/mol) than oxidation by m-CPBA. A closer interaction between 50 and the isoxazoline exists in this “later” transition state, which may alter the mode of decomposition of the resulting N-oxide. Thus, although experimental data suggests that the N-oxide stereochemistry affects torquoselectivity, because a single diastereomer may rotate “in” or “out,” depending on the oxidant and the isoxazoline substituents (see Scheme 7), a direct correlation between N-oxide stereochemistry and product stereochemistry cannot be made.
It was found that a diradical species was the most accessible reaction intermediate, consistent with a stepwise rather than a concerted extrusion process. Finally, the calculations predicted torquoselectivity favoring the “in” products for most substrates, consistent with the experimental findings. The selectivity arose from an extrusion pathway that maintained optimal charge separation in the diradical intermediate. Calculations were also performed on selected cases with “out” torquoselectivity, which appear to result from subtle nonbonded interactions in the diradical intermediate that can reverse the direction of rotation during extrusion.
4. Summary and Conclusions
In summary, a [3+2] cycloaddition/oxidation/extrusion sequence has been developed to synthesize aryl vinyl and divinyl ketones for Nazarov cyclization. The procedure is easy to conduct, and does not require either acidic or basic reaction conditions. Alkynyl esters, sulfones, phosphonates and amides all participate readily in the reaction. A range of substituents on the electron-deficient alkyne (i.e. R1) are tolerated, with best results obtained with R1=aryl. Depending on the substitution pattern, the reaction gave moderate to high selectivities of olefin isomers. The sequence was usually selective for the “in” extrusion product, although in some cases the “out” product dominated. It is important to note that both “in” and “out” isomers undergo efficient Nazarov cyclization to give the same cyclopentenone product,xxxii so this method will be valuable for the synthesis of any Nazarov cyclization substrates, even in the few cases when torquoselectivity is poor. Experimental and computational results show that the extrusion sequence is complicated and that torquoselectivity is not controlled by a single factor. There is strong computational evidence that the sequence proceeds through a stepwise pathway, and that stereoelectronic effects in the diradical intermediate have a strong influence on the direction of rotation during extrusion.xxxiv Investigations to further elucidate the mechanism are underway.
5. Experimental Section
5.1. General
Reagents were used as obtained from commercial supplier without further purification. Reaction solvents were purchased from Fisher and dispensed using the Glass Contour solvent purification system. ACS grade hexanes and ethyl acetate (EtOAc) are used for column chromatography. Thin layer chromatography (TLC) analysis was determined using precoated silica gel 60 F254 glass-supported plate (EMD product). Spots were visualized by UV light (254 nm), or by staining with potassium permanganate or p-anisaldehyde solutions followed by heating. Column chromatography separations were carried out on silica gel 60 (230–400 mesh) (EMD product).
1H NMR spectra were recorded on either a 400 MHz or a 500 MHz Avance spectrometer. 13C NMR spectra were recorded on either a 100 MHz or 125 MHz Avance spectrometer. Product E/Z stereochemistry was assigned based on carbon-hydrogen coupling constants obtained for the ester and(or) the ketone of the divinyl ketone, according to Kingsbury et al.xxxv Infrared spectra were recorded on a 8400s Shimadzu FTIR spectrometer using NaCl plates. High resolution mass spectra were done on a ThermoFinnigan MAT 95XL (ESI) at the Chemistry Instrumentation Center of the University of Buffalo.
5.2. Preparation of Electron-Deficient Alkynes
Alkynyl esters 4a–4d were prepared from commercially available aldehydes using the Corey-Fuchs protocol,xiv followed by carboalkoxylation of the resulting terminal alkyne with the appropriate chloroformate. A full description of the preparation of alkynes 9a–9f can be found in the supporting information accompanying reference xxxii.
2-(Benzo[1,3]dioxole)-propynoic acid ethyl ester (4a)
1H NMR (500 MHz, CDCl3): δ 7.10 (d, J = 8.0 Hz, 1H), 6.95 (s, 1H), 6.75 (d, J = 8.0 Hz, 1H), 5.97 (s, 2H), 1.5 (s, 9H). 13C NMR (100 MHz, CDCl3): δ 153.1, 149.7, 147.5, 128.5, 112.8, 112.3, 108.6, 101.6, 84.1, 83.2, 80.9, 65.8, 28.0, 27.3, 15.2. IR (neat) cm−1: 2213, 1706, 1105. HRMS (m/z): calcd for C14H14O4Na [M-Na+], 269.0771; found, 269.0777.
(5,6-Dihydro-4H-pyran-2-yl)-propynoic acid ethyl ester (4b)
1H NMR (500 MHz, CDCl3): δ 5.48 (m, 1H), 4.18 (q, J = 7.0 Hz, 2H), 3.99 (m, 2H), 2.09 (m, 2H), 1.80 (m, 2H), 1.24 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 153.3, 135.6, 114.9, 81.3, 78.5, 66.4, 61.8, 21.2, 20.9, 13.8. IR (neat) cm−1: 2976, 2936, 1712, 1463, 1444, 1244. HRMS (m/z): calcd for C10H12O3 [M+], 180.0781; found, 180.0786.
(2-Methyl-cyclohex-1-enyl)-propynoic acid ethyl ester (4c)
1H NMR (400 MHz, CDCl3): δ 4.11 (q, 2H J = 7.2 Hz), 2.05 (s, 2H), 1.98 (s, 2H), 1.82 (s, 3H), 1.5 (m, 4H), 1.19 (t, 3H J=7.2 Hz) 13C NMR (400 MHz, CDCl3): δ 154.1, 149.5, 111.9, 87.0, 82.9, 61.2, 31.3, 28.5, 22.4, 22.1, 21.8, 13.7. IR (neat) cm−1: 2981, 2933, 2862, 2825, 2208, 2192, 1745, 1704, 1629, 1442, 1423, 1365, 1336, 1282, 1259, 1215, 1178, 1143, 1095, 1024, 748. HRMS (m/z): calcd for C12H17O2 [M-H+], 193.1223; found, 193.1215.
(1-Hydroxy-cyclopentyl)-propynoic acid ethyl ester (4d)
1H NMR (400 MHz, CDCl3): δ 6.33 (m, 1H), 4.12 (q, J = 7.0 Hz, 2H), 2.45 (m, 4H), 1.88 (m, 2H), 1.24 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 153.8, 149.6, 121.8, 83.5, 81.7, 61.6, 35.4, 33.5, 23.0, 13.8. IR (neat) cm−1: 2935, 2202, 1704, 1446, 1365, 1265, 1207, 1141. HRMS (m/z): calcd for C10H13O2 [M-H+], 165.0910; found, 165.19189.
5.3. Preparation of Nitrones
Nitrones 10a–10d and 10h-j are known compounds and were prepared using the general procedure described below. Typically, the nitrones were not purified, but isolated in crude form and used directly in the [3+2] dipolar cycloaddition.
Representative procedure for preparation of nitrones
In a 250 mL round-bottom flask, MgSO4 (10.5 g), NaHCO3 (14.7 g, 0.2 mol), and N-methylhydroxylamine.HCl (7.6 g, 0.1 mol) were combined, and CH2Cl2 (75 mL) was added with the appropriate aldehyde (36.0 mmol). The solution was stirred for 6 h, then concentrated. The crude mixture was purified by column chromatography (2:1 hexanes:ethyl acetate) to give the nitrone.
Methanamine, N-((2,4,6-Trimethoxyphenyl)methylene)-N-oxide (10e)
Prepared as above. 1H NMR (400 MHz, CDCl3): δ 7.32 (s, 1H), 6.13 (s, 2H), 2.88 (m, 12H). 13C NMR (100 MHz, CDCl3): δ 163.0, 159.8, 130.2, 101.4, 90.7, 55.8, 55.4, 53.4. IR (neat) cm−1: 3048, 3014, 2934, 2838, 1606, 1575, 1499, 1456, 1407, 1398. HRMS: calcd for C12H15ON2Na [M-Na+], 226.1077; found, 226.1077.
Methanamine, N-((3,4,5-Trimethoxyphenyl)methylene)-N-oxide (10f)
Prepared as above. 1H NMR (500 MHz, CDCl3): δ 7.53 (s, 2H), 3.81 (m, 12H). 13C NMR (100 MHz, CDCl3): δ 152.7, 139.7, 134.8, 125.9, 105.7, 60.7, 56.0, 54.1. IR (neat) cm−1: 3116, 3015, 2964, 2935, 2831, 1583, 1568, 1497, 1471, 1454, 1435, 1412, 1400, 1338, 1306, 1232, 1167. HRMS: calcd for C11H16O4N [M-H+], 226.1074; found, 226.1072.
Methanamine, N-((2,4,6-Trimethylphenyl)methylene)-N-oxide (10g)
Prepared as above. 1H NMR (400 MHz, CDCl3): δ 7.56 (s, 1H), 6.93 (s, 2H), 3.83 (s, 3H), 2.5 (m, 6H), 1.19 (m, 9H). 13C NMR (100 MHz, CDCl3): δ 145.8, 143.4, 135.4, 125.4, 124.8, 87.4, 53.0, 44.1, 28.8, 26.7, 15.4, 15.1. HRMS: calcd for C14H22ON [M-H+], 220.1694; found, 220.1678.
5.4. General Procedures for the [3+2] Cycloaddition/Cheletropic Extrusion Sequence
General Procedure A for the Preparation of Isoxazolines ([3+2] Cycloaddition)
Alkyne (0.56 mmol) and nitrone (1.23 mmol) were covered with toluene (0.5 mL) at rt under argon. The resulting suspension was heated to 65 °C and stirred until completion as indicated by TLC (about 18 h). The reaction mixture was loaded directly onto a silica gel column and eluted with ethyl acetate/hexane to give the isoxazoline product.
General Procedure B for the Preparation of Isoxazolines ([3+2] Cycloaddition)
Alkyne (0.48 mmol), aldehyde (0.72 mmol), N-methylhydroxylamine hydrochloride (0.64 mmol) and diisopropylethyl amine (0.64 mmol) were all combined at rt, covered with ethanol (0.30 mL) and heated to 50 °C for 12 h. The resulting solution was then heated to 80 °C for another 12 h. The solution was cooled to rt, diluted with Et2O (2 mL), washed with sat NH4Cl (2 × 1 mL), brine (1 mL), dried over MgSO4 and concentrated. Purification by column chromatography (ethyl acetate/hexane) gave the desired isoxazoline.
General Procedure C (Oxidation of Isoxazoline Intermediate)
m-CPBA (0.20 mmol) was added to a solution of isoxazoline (0.14 mmol) in DCM (0.16 mL) at 0°C. The reaction was stirred at 0 °C until complete by TLC (about 5 min). The reaction was diluted with Et2O (1 mL), washed with 1M NaOH (1 mL), and the organic layer was dried over MgSO4 and concentrated. Column chromatography (ethyl acetate/hexane) gave the desired product.
General Procedure D (One-pot [3+2] Cycloaddition/Oxidation)
In a dry 100 mL round-bottom flask was weighed the nitrone (8.0 mmol), alkyne (7.1 mmol), and toluene (35 mL), and the solution was heated at 80 °C for 16 h. The solution was concentrated, and CH2Cl2 (35 mL) was added, cooled to −20 °C, and m-CPBA (11.6 mmol) was added and stirred for 30 min. The reaction was quenched with saturated Na2SO3 (50 mL), the layers were separated and the aqueous phase was extracted with CH2Cl2 (2 × 50 mL), the organic phases were combined and washed with brine (50 mL). The organic phase was dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (ethyl acetate/hexane).
5.5. Preparation of Aryl Vinyl and Divinyl Ketones
2-(Cyclohex-1-enecarbonyl)-3-phenyl-acrylic acid ethyl ester (11a)
The intermediate isoxazoline (5-Cyclohex-1-enyl-2-methyl-3-phenyl-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure A (88 % yield): 1H NMR (400 MHz, CDCl3): δ 7.28–7.37 (m, 5H), 6.5 (m, 1H), 4.93 (s, 1H), 2.92 (s, 3H), 2.22–2.5 (m, 4H), 1.66–1.73 (m, 4H), 1.14 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 164.0, 141.6, 135.8, 128.2, 127.5, 127.0, 126.2, 109.3, 102.3, 76.3, 59.5, 46.8, 31.4, 26.0, 25.5, 22.1, 21.4, 13.9. IR (neat) cm−1: 2817, 2269, 1703, 1618, 1501, 1475, 1392, 1315, 1225, 1068, 911. HRMS (m/z): calcd for C19H23NO3 [M+], 313.1672; found, 313.1668.
General Procedure C provided the title compound (11a) from the intermediate isoxazoline (94 % yield). 1H NMR (400 MHz, CDCl3): δ 7.79 (s, 1H), 7.32 (m, 5H), 6.82 (m, 1H), 4.27 (q, J = 7.2 Hz, 2H), 2.35 (m, 2H), 2.13 (m, 2H), 1.68–1.55 (m, 4H), 1.28 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 196.5, 165.1, 144.6, 141.3, 139.2, 133.2, 131.7, 129.9, 129.8, 128.5, 61.2, 26.1, 22.6, 21.6, 21.3, 14.0 IR (neat) cm−1: 3050, 3020, 2974, 2924, 2860, 1662, 1654, 1633, 1570, 1497, 1448, 1250, 1190. HRMS (m/z): calcd for C18H20O3 [M+], 284.1407; found 284.1411. JC-H: 7.8 Hz.
2-(Cyclohex-1-enecarbonyl)-3-(2,4,6-trimethoxy-phenyl)-acrylic acid ethyl ester (12b)
The intermediate isoxazoline (2-Methyl-5-(2-methyl-cyclohex-1-enyl)-3-(2,4,6-trimethoxy-phenyl)-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure A (67 % yield): 1H NMR (400 MHz, CDCl3): δ 6.31 (s, 1H), 6.08 (s, 2H), 5.65 (s, 1H), 3.93 (m, 2H), 3.76 (m, 9 H), 2.87 (s, 3H), 2.36–2.17 (m, 4H), 1.67 (m, 4H), 1.03 (t, J = 7 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 164.44, 164.42, 160.5, 159.6, 133.6, 127.0, 110.5, 99.8, 90.8, 67.1, 58.9, 55.7, 55.0, 47.9, 26.1, 25.3, 22.2, 21.5, 13.8. IR (neat) cm−1: 2996, 2971, 2942, 2928, 2852, 2840, 1672, 1649, 1625, 1606, 1588. HRMS (m/z): calcd for C22H30NO6 [M-H+], 404.2068; found 404.2074.
General Procedure C provided the title compound (12b) from the intermediate isoxazoline (96 % combined yield with 12a). 1H NMR (500 MHz, CDCl3): δ 7.79 (s, 1H), 7.53 (dd, Jab = 8.0 Hz, Jac = 1.5 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.59 (s, 2H), 6.03 (s, 2H), 4.25 (q, J = 7.0 Hz, 2H), 3.79 (s, 3H), 3.66 (s, 6H), 1.21 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 195.1, 167.0, 163.2, 159.4, 141.1, 138.5, 134.8, 129.3, 105.5, 90.2, 90.1, 65.8, 35.4, 35.3, 35.0, 26.0, 23.0, 22.1, 21.8, 14.2. IR (neat) cm−1: 2928, 2850, 2841, 2832, 1706, 1646, 1629, 1607, 1584, 1508. HRMS (m/z): C21H27O6 [M-H+], 375.1738; found, 375.1711. JC-H: 9.3 Hz.
2-(Cyclohex-1-enecarbonyl)-5-(phenyl-penta-2,4-dienoic acid ethyl ester ((E)-13a)
The intermediate isoxazoline (5-Cyclohex-1-enyl-2-methyl-3-styryl-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure A (74% yield). 1H NMR (400 MHz, CDCl3): δ 7.40–7.20 (m, 5H), 6.66 (d, J = 16 Hz, 1H), 6.48 (m, 1H), 6.28 (dd, Jab = 16 Hz, Jac = 7 Hz, 1H), 4.60 (d, J = 7 Hz, 1H), 4.19–4.11 (m, 2H), 2.85 (s, 3H), 2.42 (m, 1H), 2.21 (m, 3H), 1.72–1.65 (m, 4H), 1.26 (t, J = 7 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ164.1, 136.8, 135.9, 130.6, 128.3, 127.6, 127.4, 126.4, 126.3, 100.6, 74.0, 59.6, 45.9, 26.0, 25.5, 22.1, 21.4, 14.1. IR (neat) cm−1: 2913, 2842, 1784, 1614. HRMS (m/z): calcd for C21H25NO3Na [M-Na+], 362.1727; found 362.1722.
General Procedure C provided the title compound (13a) from the intermediate isoxazoline: 1H NMR (500 MHz, CDCl3): δ 7.53–7.31 (m, 6H), 6.99 (d, J = 15.0 Hz, 1H), 6.77–6.69 (m, 2H), 4.24 (q, J = 7.0 Hz, 2H), 2.39 (m, 2H), 2.24 (m, 2H), 1.73–1.63 (m, 4H), 1.27 (q, J = 7.0 Hz, 3H) 13C NMR (100 MHz, CDCl3): δ 195.0, 165.1, 144.8, 142.5, 142.0, 140.2, 135.6, 132.0, 129.3, 128.6, 127.6, 127.4, 123.2, 109.5, 60.8, 26.2, 22.6, 21.7, 21.4, 14.0. IR (neat) cm−1: 3050, 3020, 2974, 2924, 2860, 2842, 2753, 1684, 1674, 1603, 1542, 1505, 1448, 1250, 1190. HRMS: calcd for C21H25O3 [M-H+], 325.1785; found 325.1787. (Z)-11f d 7.82 (m, 1H), 1.53 (m, 2H), 7.35 (m, 4H), 6.98–6.94 (m, 2H), 6.69 (m, 1H), 4.28 (q, J = 7.0 Hz, 2H), 2.33 (m, 2H), 2.24 (m, 2H), 1.67 (m, 4H), 1.30 (t, J = 7.0 Hz, 3H). JC-H: 7.8 Hz.
2-(2-Methyl-cyclohex-1-enecarbonyl)-3-(2,4,6-trimethoxy-phenyl)-acrylic acid ethyl ester (14b)
The intermediate isoxazoline (2-Methyl-5-(2-methyl-cyclohex-1-enyl)-3-(2,4,6-trimethoxy-phenyl)-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure A (49 % yield): 1H NMR (400 MHz, CDCl3): δ 6.10 (s, 2H), 5.70 (s, 1H), 3.78–4.00 (m, 2H), 3.78 (s, 9H), 2.94 (s, 3H), 2.30–2.68 (m, 2H), 2.05 (m, 2H), 1.73 (s, 3H), 1.66 (m, 4H), 1.06 (t, J = 7 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 164.5, 164.1, 160.5, 159.6, 137.2, 120.3, 109.3, 90.9, 66.9, 58.8, 55.6, 55.1, 31.2, 27.8, 22.4, 22.3, 20.5, 14.0. IR (neat) cm−1: 2998, 2971, 2940, 2926, 2852, 2840, 1672, 1606, 1588. HRMS (m/z): calcd for C23H32NO6 [M-H+], 418.2224, found 418.2236.
General Procedure C provided the title compound (14b) from the intermediate isoxazoline (96 % combined yield with 14a). 1H NMR (500 MHz, CDCl3): δ 7.70 (s, 1H), 6.07 (s, 2H), 4.18 (q, J = 7.5 Hz, 2H), 3.84 (s, 3H), 3.78 (s, 6H), 2.24 (m, 2H), 2.03 (m, 2H), 1.70 (s, 3H), 1.65 (m, 4H), 1.23 (t, J = 7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 199.4, 167.4, 163.6, 159.9, 136.9, 134.7, 133.4, 132.9, 105.7, 90.3, 60.3, 55.45, 55.43, 31.3, 27.4, 22.6, 22.3, 21.1, 14.1. IR (neat) cm−1: 2926, 2852, 2841, 2830, 1707, 1646, 1629, 1607, 1584, 1508. HRMS (m/z): calcd for C22H29O6 [M-H+], 389.1935; found 389.1944. JC-H: 12.4 Hz.
2-(2-Methyl-cyclohex-1-enecarbonyl)-3-phenyl-acrylic acid ethyl ester (15a)
The intermediate isoxazoline (2-Methyl-5-(2-methyl-cyclohex-1-enyl)-3-phenyl-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure A (61% yield): 1H NMR (400 MHz, CDCl3): δ 7.39 (d, J = 7.2 Hz, 2H), 7.33 (t, J = 6.8 Hz, 1H), 7.27 (m, 1H), 4.09–3.98 (m, 2H), 2.96 (s, 3H), 2.23 (m, 2H), 2.06 (m, 2H), 1.72 (s, 3H), 1.68 (m, 4H), 1.14 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 164.7, 163.7, 142.2, 138.1, 182.2, 127.5, 127.0, 119.6, 103.9, 75.8, 59.4, 47.2, 31.2, 27.7, 22.3, 22.2, 20.6, 13.9. IR (neat) cm−1: 2977, 2927, 2858, 2360, 2337, 1693, 1627, 1442, 1373, 1330, 1276, 1234, 1172, 1076. HRMS (m/z): calcd for C20H26NO3 [M-H+], 328.1907; found, 328.1915.
General Procedure C provided the title compound (15a) from the intermediate isoxazoline (85 % combined yield with 15b) and characterized as a 2:1 mixture. 1H NMR (500 MHz, CDCl3): δ 7.68 (s, 1H), 7.47 (m, 2.5H), 7.33 (m, 5H), 4.35 (m, 2.82H), 2.23 (m, 0.9H), 2.18 (m, 2H), 2.10 (m, 3H), 2.02 (s, 3H), 1.70 (m, 2.5H), 1.48 (m, 4.5H), 1.26 (m, 4.6H). 13C NMR (100 MHz, CDCl3): δ 197.3, 194.6, 165.3, 149.5, 142.7, 140.4, 135.6, 134.7, 133.5, 133.2, 131.87, 131.83, 131.5, 130.6, 129.9, 129.7, 129.6, 128.8, 128.5, 61.5, 61.3, 34.5, 31.1, 27.3, 26.3, 22.48, 22.46, 22.3, 22.1, 22.0, 21.1, 14.2, 13.9. IR (neat): 3051, 3022, 2974, 2924, 2862, 1662, 1654, 1636, 1570, 1497, 1448, 1253, 1192. HRMS (m/z): calcd for C19H22O3 [M+], 298.1563; found 298.1567.
2-(Cyclopent-1-enecarbonyl)-3-phenyl-acrylic acid ethyl ester(17a)
The intermediate isoxazoline (2-Methyl-5-(2-methyl-cyclohex-1-enyl)-3-phenyl-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure A (89 % yield): 1H NMR (400 MHz, CDCl3): δ 7.26–7.38 (m, 5H), 6.92 (m, 1H), 4.98 (s, 1H), 4.04 (m, 2H), 2.92 (s, 3H), 2.52–2.81 (m, 4H), 1.98 (m, 2H), 1.13 (t, J = 7 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 163.9, 158.7, 141.7, 141.5, 130.8, 128.2, 127.5, 127.1, 103.3, 59.6, 46.8, 33.5, 23.1, 13.9. IR (neat) cm−1: 3093, 3035, 2958, 2939, 2906, 2873, 2852, 1703, 1681, 1610, 1581, 1456, 1392, 1371, 1323, 1298, 1276, 1261, 1232, 1195, 1172, 1130, 1074, 950. HRMS (m/z): calcd for C18H21NO3 [M+], 299.1516, found 299.1510.
General Procedure C provided the title compound (17a) from the intermediate isoxazoline (64 % yield). 1H NMR (500 MHz, CDCl3): δ 7.77 (s, 1H), 7.35 (m, 5H), 6.60 (s, 1H), 4.26 (q, J = 7.2 Hz, 2H), 2.65 (m, 2H), 2.43 (m, 2H), 1.99 (m, 2H), 1.33 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 193.5, 165.1, 148.3, 145.4, 141.2, 133.2, 132.3, 130.1, 129.9, 128.7, 61.4, 34.0, 30.2, 22.9, 14.1. IR (neat) cm−1: 2974, 2955, 1714, 1703, 1650, 1609, 1448. HRMS (m/z): calcd for C17H18O3 [M+], 269.1172; found 269.1175. JC-H: 7.8 Hz.
2-(5,6-Dihydro-4H-pyran-2-carbonyl)-3-phenyl-acrylic acid ethyl ester ((E)-18a)
The intermediate isoxazoline (5-(5,6-Dihydro-4H-pyran-2-yl)-2-methyl-3-phenyl-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure A (53% yield). 1H NMR (400 MHz, CDCl3): δ 7.38–7.26 (m, 5H), 5.71 (t, J = 4 Hz, 1H), 4.97 (s, 1H), 4.14–4.03 (m, 4H), 2.93 (s, 3H), 2.22 (m, 2H), 1.92 (m, 2H), 1.14 (t, J = 7 Hz, 3H) 13C NMR (100 MHz, CDCl3): δ 163.4, 156.5, 142.2, 140.8, 128.2, 127.7, 127.2, 109.0, 104.5, 76.6, 66.4, 59.8, 46.6, 21.7, 20.4, 13.9. IR (neat) cm−1: 3028, 2927, 2874, 2840, 1700, 1685, 1666, 1607, 1453, 1391, 1387, 1371. HRMS: calcd for C18H21O4N [M+], 315.1465; found 315.1466.
General Procedure C provided the title compound (18a) from the intermediate isoxazoline. 1H NMR (500 MHz, CDCl3): δ 7.81 (s, 1H), 7.39–7.33 (m, 5H), 6.00 (t, J = 4.5 Hz, 1H), 4.28 (t, J = 7.0 Hz, 2H), 4.07 (m, 2H), 2.14 (m, 2H), 1.82 (m, 2H), 1.29 (t, J = 7.0 Hz, 3H) 13C NMR (100 MHz, CDCl3): δ 190.5, 164.9, 151.2, 142.9, 133.0, 130.3, 130.2, 128.7, 116.0, 66.5, 61.5, 21.3, 21.0, 14.1. IR (neat) cm−1: 3070, 3000, 2945, 2892, 2860, 1675, 1614, 1601, 1570, 1497, 1448, 1250, 1190. JC-H: 7.3 Hz.
2-(Benzo[1,3]dioxole-5-carbonyl)-3-phenyl-acrylic acid ethyl ester (19a)
The intermediate isoxazoline (5-Benzo[1,3]dioxol-5-yl-2-methyl-3-phenyl-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure A (85 % yield). 1H NMR (500 MHz, CDCl3): δ 7.50 (d, J = 8 Hz, 1H), 7.43 (m, 3H), 7.37 (t, J = 7 Hz, 2H), 7.31 (m, 1H), 6.84 (d, J = 8 Hz, 1H), 6.05 (s, 2H), 5.07 (s, 1H), 4.11 (q, J = 7 Hz, 2H), 3.01 (s, 3H), 1.14 (t, J = 7 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 164.1, 161.1, 150.0, 147.2, 141.6, 128.4, 127.8, 127.2, 125.1, 120.7, 110.2, 107.8, 102.6, 101.5, 76.8, 60.0, 53.4, 14.0. IR (neat) cm−1: 3101, 3015, 29, 81, 28, 97, 1701, 1692, 1633, 1604, 1488, 1446, 1326, 1255, 1232, 1074, 1039, 813. HRMS (m/z): calcd for C20H20NO5 [M-H+], 354.1336; found, 354.1341.
General Procedure C provided the title compound (19a) from the intermediate isoxazoline (94 % yield). 1H NMR (400 MHz, CDCl3): δ 7.91 (s, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.48 (s, 1H), 7.37–7.22 (m, 5H), 6.78 (d, J = 8.0 Hz, 1H), 6.02 (s, 1H), 4.25 (q, J = 7.2 Hz, 2H), 1.21 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 193.6, 165.0, 152.6, 148.4, 142.1, 132.8, 131.3, 130.3, 130.1, 128.7, 126.4, 108.1, 102.0, 61.5, 14.1 IR (neat) cm−1: 2980, 2903, 1712, 1658, 1600, 1502, 1486, 1438, 1244. HRMS (m/z): calcd for C19H16O5 [M-H+], 325.1071; found 325.1076. JC-H: 7.8 Hz.
2-(Benzo[1,3]dioxole-5-carbonyl)-3-(2,4,6-triethyl-phenyl)-acrylic acid ethyl ester (21b)
The intermediate isoxazoline (5-Benzo[1,3]dioxol-5-yl-2-methyl-3-(2,4,6-triethyl-phenyl)-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure A (38 % yield). 1H NMR (500 MHz, CDCl3): δ 7.39 (d, J = 8.5 Hz, 1H), 7.33 (s, 1H), 6.88 (m, 2H), 6.02 (s, 2H), 5.75 (s, 1H), 3.95 (m, 2H), 3.05 (s, 3H), 2.98–2.84 (m, 4H), 2.60 (q, J = 7.5 Hz, 2H), 1.23 (m, 9H), 0.95 (t, J = 7 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 163.9, 160.7, 149.6, 147.1, 143.5, 131.7, 126.7, 124.3, 121.1, 109.8, 107.8, 103.2, 101.4, 73.4, 59.6, 47.8, 28.4, 16.3, 15.3, 13.7. IR (neat) cm−1: HRMS (m/z): calcd for C26H32NO5 [M-H+], 438.2275; found 438.2268.
General Procedure C provided the title compound (21b) from the intermediate isoxazoline (89 % yield). 1H NMR (400 MHz, CDCl3): δ 7.52 (s, 1H), 7.5 (d, J = 8.0 Hz, 1H), 7.41 (s, 1H), 6.9 (s, 2H), 6.88 (d, J = 8.0 Hz, 1H), 6.06 (s, 2H), 3.98 (q, J = 7.0 Hz, 2H), 2.60 (m, 6H), 1.26–1.17 (m, 9H), 0.89 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 191.0, 164.9, 151.9, 148.2, 144.8, 140.7, 137.5, 131.6, 130.2, 125.6, 125.0, 108.8, 107.8, 101.9, 60.8, 28.6, 26.9, 15.5, 14.8, 13.4. IR (neat) cm−1: 2963, 2930, 2871, 1728, 1603, 1483, 1439, 1371. HRMS (m/z): calcd for C25H29O5 [M-H+], 409.2010; found 409.2013. JC-H: 12.4 Hz.
2-(Benzo[1,3]dioxole-5-carbonyl)-3-(2,4,6-trimethoxy-phenyl)-acrylic acid ethyl ester (22b)
The intermediate isoxazoline (5-Benzo[1,3]dioxol-5-yl-2-methyl-3-(2,4,6-trimethoxy-phenyl)-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure A (72 % yield). 1H NMR (400 MHz, CDCl3); δ 7.41 (d, J = 8.4 Hz, 1H), 7.39 (s, 1H), 6.83 (d, J = 8 Hz,1H), 6.10 (s, 2H), 5.97 (s, 2H), 5.75 (s, 1H), 3.96 (m, 2H), 3.76 (m, 9H), 2.96 (s, 3H), 1.02 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3); δ 164.3, 161.2, 160.7, 159.6, 149.3, 146.9, 124.2, 121.6, 109.8, 107.6, 101.2, 100.2, 90.9, 67.6, 60.2, 59.1, 55.8, 55.1, 13.8. IR (neat) cm−1: 2999, 2976, 2949, 2932, 2874, 1690, 1636, 1591, 1505, 1487. HRMS (m/z): calcd for C23H26NO8 [M-H+], 444.1653; found, 444.1655.
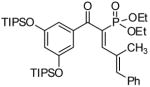
General Procedure C provided the title compound (22b) from the intermediate isoxazoline (94 % combined yield with 22a). 1H NMR (500 MHz, CDCl3); δ 8.09 (s, 1H), 7.47 (d, J = 8.0 Hz, 1H), 7.42 (s, 1H), 6.78 (d, J = 8.0 Hz, 1H), 6.01 (s, 2H), 5.96 (s, 2H), 4.19 (q, J = 7.2 Hz, 2H), 3.77 (s, 3H), 3.51 (s, 6H), 1.56 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 192.0, 176.6, 166.7, 163.4, 159.6, 150.9, 147.7, 135.5, 132.8, 128.7, 125.1, 108.3, 107.5, 105.1, 101.6, 90.2, 60.8, 55.3, 54.7, 14.1. IR (neat) cm−1: 2975, 2938, 2901, 2839, 1703, 1661, 1598, 1573, 1436. HRMS (m/z): calcd for C22H22O8Na [M-Na+], 415.1387;found 415.1393. JC-H: 8.5 Hz.
2-(Benzo[1,3]dioxole-5-carbonyl)-3-(3,4,5-trimethoxy-phenyl)-acrylic acid ethyl ester (23a)
The intermediate isoxazoline (5-Benzo[1,3]dioxol-5-yl-2-methyl-3-(3,4,5-trimethoxy-phenyl)-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure A (70 % yield). 1H NMR 400 MHz (CDCl3); δ 7.46 (d, J = 8 Hz, 1H), 7.44 (s, 1H), 6.83 (d, J = 8 Hz, 1H), 6.64 (s, 2H), 5.98 (s, 2H), 4.99 (s, 1H), 4.09 (q, J = 7.2 Hz, 2H), 3.86 (s, 6H), 3.84 (s, 3H), 2.98 (s, 3H), 1.14 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3); δ 163.9, 161.0, 153.2, 150.0, 147.2, 137.5, 137.1, 125.0, 120.6, 110.1, 107.8, 104.0, 102.4, 101.5, 60.6, 59.9, 56.0, 46.8, 20.9, 14.1. IR (neat) cm−1: 2968, 2942, 2926, 2901, 2829, 1765, 1688, 1635, 1592, 1504, 1486, 1442, 1421. HRMS (m/z): calcd for C23H26NO8 [M-H+], 444.1653; found 444.1654.
General Procedure C provided the title compound (23a) from the intermediate isoxazoline (85 % yield). 1H NMR (500 MHz, CDCl3); δ 7.79 (s, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.45 (s, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.59 (s, 2H), 6.04 (s, 2H), 4.24 (q, J = 7.2 Hz 2H), 3.80 (s, 3H), 3.68 (s, 6H), 1.22 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3); δ 193.8, 165.0, 152.9, 152.6, 148.4, 142.0, 139.9, 131.2, 130.3, 128.1, 126.2, 108.2, 108.0, 107.6, 102.0, 61.4, 60.8, 55.8, 14.0. IR (neat) cm−1: 2937, 2903, 2838, 1712, 1657, 1601, 1579, 1503, 1486, 1439, 1244. HRMS (m/z): calcd for C22H23O8 [M-H+], 415.1387; found 415.1387. JC-H: 8.3 Hz.
2-(Benzo[1,3]dioxole-5-carbonyl)-3-cyclohexyl-acrylic acid ethyl ester (24a)
The intermediate isoxazoline (5-Benzo[1,3]dioxol-5-yl-3-cyclohexyl-2-methyl-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure A (86% yield). 1H NMR (400 MHz, CDCl3): δ 7.39 (d, J = 8 Hz, 1H), 7.33 (s, 1H), 6.83 (d, J = 8 Hz, 1H), 6.00 (s, 2H), 4.17 (q, J = 7 Hz, 2H), 3.87 (d, J = 6.6 Hz, 1H), 2.81 (s, 3H), 1.17–1.34 (m, 13H). 13C NMR (125 MHz, CDCl3): δ 164.6, 161.2, 125.0, 121.2, 110.2, 107.7, 101.4, 77.8, 59.8, 47.6, 42.0, 29.7, 26.7, 26.5, 26.4, 26.2, 14.2. IR (neat) cm−1: 2970, 2956, 2849, 1769, 1693, 1606, 1589, 1503, 1490, 1448, 1357, 1344, 1241, 1067, 1082, 947. HRMS (m/z): calcd for C20H26NO5 [M-H+], 360.1805; found, 360.1811.
General Procedure C provided the title compound (24a) from the intermediate isoxazoline (93 % yield). 1H NMR (400 MHz, CDCl3): δ 7.43 (m, 2H), 6.94 (d, J = 10.8 Hz, 1H), 6.85 (d, J = 8.0 Hz, 1H), 6.06 (s, 2H), 4.16 (q, J = 7.2 Hz, 2H), 2.17 (m, 1H), 1.64 (m, 5H), 1.14 (m, 9H). 13C NMR (100 MHz, CDCl3): δ 192.5, 164.8, 152.3, 152.1, 148.2, 131.9, 131.7, 126.2, 108.1, 107.9, 101.9, 61.0, 38.5, 31.5, 25.4, 24.9, 13.9. IR (neat) cm−1: 2924, 2850, 1715, 1663, 1602, 1503, 1486, 1439. HRMS (m/z): calcd for C19H22O5Na [M-Na+], 353.1359, found 353.1359. JC-H: 8.6 Hz.
2-(Benzo[1,3]dioxole-5-carbonyl)-5-phenyl-penta-2,4-dienoic acid ethyl ester (25a)
The intermediate isoxazoline (5-Benzo[1,3]dioxol-5-yl-2-methyl-3-styryl-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure A (96 % yield). 1H NMR (500 MHz, CDCl3): δ 7.49 (d, J = 8 Hz, 1H), 7.42 (m, 3H), 7.30 (m, 2H), 7.23 (m, 1H), 6.86 (d, J = 8.5 Hz, 1H), 6.72 (d, J = 16 Hz, 1H), 6.36 (dd, Jab = 16 Hz, Jac = 6.5 Hz, 1H), 5.99 (s, 2H), 4.75 (d, J = 6.5 Hz, 1H), 4.20 (m, 2H), 2.95 (s, 3H), 1.27 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 164.1, 161.2, 150.0, 147.2, 136.8, 131.0, 128.5, 127.6, 127.4, 126.6, 125.0, 120.8, 110.1, 107.8, 101.5, 101.0, 74.5, 60.1, 46.1, 14.2. IR (neat) cm−1: 3031, 2981, 2964, 2904, 1764, 1701, 1632, 1613, 1600, 1503. HRMS (m/z): calcd for C22H22NO5 [M-H+], 308.1492; found 380.1499.
General Procedure C provided the title compound (25a) from the intermediate isoxazoline (92 % combined yield with 25b.) 1H NMR (400 MHz, CDCl3): δ 7.68 (d, J = 11.6 Hz, 1H), 7.49–7.30 (m, 7H), 7.05 (d, J = 15.4 Hz, 1H), 6.86 (d, J = 8.5 Hz, 1H), 6.78 (dd, Jab = 15.4 Hz, Jac = 8.5 Hz, 1H), 6.08 (s, 2H), 4.23 (q, J = 7.2 Hz, 2H), 1.21 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 192.5, 165.1, 152.4, 148.3, 143.5, 143.2, 135.5, 131.9, 131.3, 129.6, 128.7, 127.6, 126.6, 122.9, 108.3, 108.1, 102.0, 61.2, 14.1. IR (neat) cm−1: 2979, 2909, 1715, 1648, 1612, 1600, 1590, 1502, 1487, 1439, 1248. HRMS (m/z): calcd for C21H19O5 [M-H+], 351.1227; found 351.1236. JC-H: 8.1 Hz.
2-(Benzo[1,3]dioxole-5-carbonyl)-hept-2-enoic acid ethyl ester (26a)
The intermediate isoxazoline (5-Benzo[1,3]dioxol-5-yl-3-butyl-2-methyl-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure B (72 % yield). 1H NMR (400, MHz CDCl3): δ 7.42 (d, J = 8 Hz, 1H), 7.36 (s, 1H), 6.84 (d, J = 8.4 Hz, 1H), 6.01 (s, 2H), 4.18 (m, 2H), 3.98 (m, 1H), 2.84 (s, 3H), 1.32–1.77 (m, 6H), 1.27 (t, J = 7.2 Hz, 3H), 0.82 (t, J = 7 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 164.4, 160.9, 149.7, 147.0, 124.8, 121.1, 110.0, 107.7, 101.9, 101.4, 73.2, 59.8, 46.9, 34.4, 27.6, 22.5, 14.1, 13.9. IR (neat) cm−1: 2979, 2902, 1697, 1627, 1600, 1504, 1488, 1448, 1332, 1251, 1226, 1130, 1108, 1039, 968, 933, 813, 761. HRMS (m/z): calcd for C18H24NO5 [M-H+], 334.1653; found, 334.1655.
General Procedure C provided the title compound (26a) from the intermediate isoxazoline (95 % combined yield with 26b). 1H NMR (500 MHz, CDCl3): δ 7.44–7.41 (m, 2H), 7.11 (t, J = 8.0 Hz, 1H), 6.82 (d, J = 8.0 Hz, 1H), 6.05 (s, 2H), 4.17 (q, J = 7.2 Hz, 2H), 2.10 (q, J = 7.5, 2H), 1.43–1.20 (m, 4H), 1.18 (t, J = 7.5 Hz, 3H), 0.82 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 192.6, 164.5, 152.3, 148.3, 147.9, 133.6, 131.8, 126.3, 108.2, 108.0, 101.9, 61.0, 30.4, 29.3, 22.3, 14.0, 13.7. IR (neat) cm−1: 2957, 2930, 2905, 2861, 1711, 1663, 1602, 1503, 1487, 1439, 1366. HRMS (m/z): calcd for C17H21O5 [M-H+], 305. 1384; found 305.1388. JC-H: 7.3 Hz.
2-(Benzo[1,3]dioxole-5-carbonyl)-acrylic acid ethyl ester (27)
The intermediate N-methyl isoxazoline (5-Benzo[1,3]dioxol-5-yl-2-methyl-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure B (85% yield). 1H NMR (500 MHz, CDCl3): δ 7.45 (d, J = 8 Hz, 1H), 7.38 (s, 1H), 6.84 (d, J = 8 Hz, 1H), 6.00 (s, 2H), 4.58 (m, 1H), 4.21 (q, J = 7 Hz, 2H), 3.96 (m, 1H), 2.87 (s, 3H), 1.28 (t, J = 7 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 164.0, 161.1, 149.7, 147.1, 124.6, 120.7, 109.8, 107.7, 101.4, 98.0, 62.5, 59.9, 47.5, 14.2. IR (neat) cm−1: 3085, 2979, 2960, 2902, 2875, 2781. HRMS: calcd for C14H16NO5 [M-H+], 278.1023; found, 278.1031.
Alternatively, the intermediate N-t-butyl isoxazoline (5-Benzo[1,3] dioxol-5-yl-2,3,3-trimethyl-2,3-dihydro-isoxazole-4-carboxylic acid ethyl ester) was prepared according to General Procedure B (79% yield). 1H NMR (500 MHz, CDCl3): δ 7.20 ((dd, Jab = 8 Hz, Jab = 1 Hz, 1H), 7.12 (d, J = 1 Hz, 1H), 5.99 (s, 2H), 4.16 (q, J = 7 Hz, 2H), 2.77 (s, 3H), 1.42 (s, 6H), 1.20 (t, J = 7Hz, 3H) 13C NMR (125 MHz, CDCl3): δ 164.5, 161.7, 149.5, 147.0, 124.3, 121.9, 109.8, 107.7, 107.4, 101.4, 69.9, 59.6, 53.4, 38.4, 14.05. IR (neat) cm−1: 2975, 2929, 2902, 1689, 1604, 1504, 1488, 1446, 1371. HRMS: calcd for C16H20NO5 [M-H+], 306.1336; found, 306.1344.
General Procedure C provided the title compound (27) from either the N-methyl or N-t-butyl intermediate isoxazoline. 1H NMR (400 MHz, CDCl3): δ 7.45 (m, 2H), 6.87 (d, J = 8.0 Hz, 1H), 6.66 (s, 1H), 6.08 (s, 2H), 6.00 (s, 1H), 4.28 (q, J = 7.0 Hz, 2H), 1.25 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 191.3, 164.3, 152.3, 148.2, 141.3, 131.0, 130.4, 126.6, 108.6, 107.8, 101.9, 61.4, 13.9. IR (neat) cm−1: 2981, 2897, 1718, 1661, 1603, 1502, 1487, 1439, 1392, 1367, 1354, 1316, 1248, 1219, 1092. HRMS: calcd for C13H13O5 [M-H+], 249.0758; found 249.0753.
2-(Benzo[1,3]dioxole-5-carbonyl)-3-methyl-but-2-enoic acid ethyl ester (28)
The intermediate isoxazoline (5-Benzo[1,3] dioxol-5-yl-2,3,3-trimethyl-2,3-dihydro-isoxazole-4-carbxylic acid ethyl ester) was prepared by method B (0.248 g, 79%). 1H NMR (500 MHz, CDCl3): δ 7.20 ((dd, Jab = 8 Hz, Jab = 1 Hz, 1H), 7.12 (d, J = 1 Hz, 1H), 5.99 (s, 2H), 4.16 (q, J = 7Hz, 2H), 2.77 (s, 3H), 1.42 (s, 6H), 1.20 (t, J = 7 Hz, 3H) 13C NMR (125 MHz, CDCl3): δ 164.5, 161.7, 149.5, 147.0, 124.3, 121.9, 109.8, 107.7, 107.4, 101.4, 69.9, 59.6, 53.4, 38.4, 14.05. IR (neat) cm−1: 2975, 2929, 2902, 1689, 1604, 1504, 1488, 1446, 1371. HRMS: calcd for C16H20NO5 [M-H+], 306.1336; found, 306.1344.
General Procedure C provided the title compound (28) from the intermediate isoxazoline. 1H NMR (400 MHz, CDCl3): δ 7.47 (dd, Jab = 8.0 Hz, Jac = 1.2 Hz, 1H), 7.42 (d, J = 1.2 Hz, 1H), 6.84 (d, J = 8.0 Hz, 2H), 6.06 (s, 2H), 4.11 (d, J = 7.0 Hz, 2H), 2.30 (s, 3H), 1.79 (s, 3H), 1.09 (t, J = 8.0 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 193.7, 164.7, 153.9, 152.1, 148.3, 132.0, 129.3, 125.9, 108.2, 108.0, 101.9, 60.4, 24.1, 22.1, 13.9. IR (neat) cm−1: 2979, 2899, 1714, 1661, 1601, 1503, 1485, 1436, 1366, 1354, 1281, 1223. HRMS: calcd for C15H17O5 [M-H+], 277.1071; found, 277.1070.
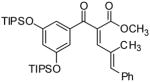
α-[(2E)-2-Methyl-3-phenyl-2-propen-1-ylidene]-β-oxo-3,5-bis[[tris(1-methylethyl)silyl]oxy]-, (αZ) benzenepropanoic acid methyl ester (29b)
Alkynyl ester 9a was subjected to General Procedure D using the nitrone 10d to yield 3.0 g (65%) of 29b. 1H NMR (CDCl3, 400 MHz): δ 7.66 (d, J = 0.8 Hz, 1H), 7.34 (m, 5H), 7.07 (d, J = 2.4 Hz, 2H), 6.98 (s, 1H), 6.65 (t, J = 2.0 Hz, 1H), 3.69 (s, 3H), 1.79 (s, 3H), 1.24 (m, 6H), 1.12 (d, J = 9.0 Hz, 36H); 13C NMR (CDCl3, 100 MHz): δ 195.0, 165.8, 157.3, 147.8, 141.9, 138.9, 136.2, 133.3, 129.7, 129.2, 128.3, 128.0, 117.3, 113.7, 52.3, 17.8, 12.5; IR (neat, cm−1): 3024, 2945, 2892, 1724, 1714, 1679, 1672, 1588, 1443, 1334, 1249, 1198; HRMS (EI) calculated for C38H58O5Si2 650.3817, found 650.3817. 3JC-H=4.2 Hz for Cketone-H, 3JC-H=7.5 Hz for Cester-H.
β-Oxo-α-[(2E)-3-phenyl-2-propen-1-ylidene]-3,5-bis[[tris(1-methyl ethyl)silyl]oxy]-, (αE) benzenepropanoic acid methyl ester (30a)
Alkynyl ester 9a (0.8 g, 1.5 mmol) was subjected to General Procedure D using the nitrone 10c to yield 320 mg (67%) of 30a. 1H NMR (500 MHz, CDCl3): ™7.69 (d, J = 12.0 Hz, 1 H), 7.34 (m, 5 H), 7.05 (d, J = 2.5 Hz, 2 H), 7.02 (d, J = 16.0 Hz, 1 H), 6.70 (dd, J = 2.5 Hz, 3.0 Hz, 1 H), 6.66 (t, J = 2.4 Hz, 1 H), 3.71 (s, 3 H), 1.26 (m, 6 H), 1.10 (d, J = 8.5 Hz, 18 H); 13C NMR (100 MHz, CDCl3): δ 194.0, 176.6, 165.4, 157.4, 143.6, 143.4, 138.7, 135.5, 131.0, 129.6, 128.7, 127.6, 123.0, 117.5, 113.9, 52.2, 17.8, 12.6; IR (neat, cm−1): 2944, 2866, 1719, 1672, 1584, 1462, 1438, 1279, 1171; HRMS (EI) calculated for C37H56O5Si2Na: 659.3558, found 659.3542. 3JC-H = 9.0 Hz for Cketone-H, 3JC-H = 6.6 Hz for Cester-H.
N,N-Dimethyl-α-[(2E)-2-methyl-3-phenyl-2-propen-1-ylidene]-β-oxo-3,5-bis[[tris(1-methylethyl)silyl]oxy]-, (αZ) benzenepropanamide (31b)
Alkynyl amide 9b (1.6 g, 2.8 mmol) was subjected to General Procedure D using the nitrone 10d to obtain 1.2 g (63%) of 31b. 1H NMR (CDCl3, 400 MHz): δ 7.34 (m, 5H), 7.02 (s, 1H), 6.88 (d, J = 2.4 Hz, 2H), 6.79 (s, 1H), 6.62 (t, J = 2.0 Hz, 1H), 3.08 (s, 3H), 3.01 (s, 3H), 2.08 (s, 3H), 1.24 (m, 6H), 1.10 (d, J = 9.0 Hz, 36H); 13C NMR (CDCl3, 100 MHz): δ 194.2, 168.0, 156.9, 147.9, 141.5, 139.4, 136.3, 134.4, 133.7, 129.5, 128.3, 128.1, 115.5, 113.9, 38.2, 34.6, 17.8, 14.6, 12.6; IR (neat, cm−1): 2945, 2867, 1643, 1585, 1437, 1393, 1336, 1239, 1072; HRMS (EI) calculated for C39H61NO4Si2Na 686.4031, found 686.4056. 3JC-H = 2.5 Hz for Cketone-H, 3JC-H = 10.5 Hz for Cester-H.
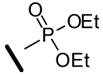
P-[(1Z,3E)-1-[3,5-Bis[[tris(1-methylethyl)silyl]oxy]benzoyl]-3-methyl-4-phenyl-1,3-butadien-1-yl]-, phosphonic acid diethyl ester (32b)
Alkynyl phosphate 9c (2.7 g, 4.5 mmol) was subjected to General Procedure D using the nitrone 10d to obtain 1.9 g (57%) of 32b. 1H NMR (CDCl3, 400 MHz): δ 7.47 (d, J = 26.0 Hz, 1H), 7.27 (m, 5H), 7.11 (d, J = 2.0 Hz, 2H), 6.87 (s, 1H), 6.64 (t, J = 2.0 Hz, 1H), 4.13 (quint., J = 7.2 Hz, 4H), 1.75 (s, 3H), 1.24 (m, 12H), 1.12 (d, J = 9.0 Hz, 36H); 13C NMR (CDCl3, 100 MHz): δ 195.0, 157.2, 155.9, 151.2, 140.4, 139.0, 136.2, 133.8, 133.6, 129.3, 128.2, 127.9, 117.3, 114.3, 114.0, 62.6, 17.8, 16.1, 12.5; IR (neat, cm−1): 2868, 2728, 2359, 1666, 1587, 1445, 1334, 1173, 1027; HRMS (EI) calculated for C40H65O6PSi2Na 751.3950, found 751.3964. 3JC-H = 7.8 Hz for Cketone-H.
α-[(2E)-2-Methyl-3-phenyl-2-propen-1-ylidene]-β-oxo-3,5-bis[[tris(1-methylethyl)silyl]oxy]-, (αE) benzenepropanenitrile (33b)
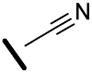
Alkynyl nitrile 9d (2.4 g, 5.1 mmol) was subjected to General Procedure D using the nitrone 10d to obtain 1.8 g (57%) of 33b. 1H NMR (CDCl3, 400 MHz): δ 7.68 (s, 1H), 7.41 (m, 5H), 7.09 (s, 1H), 6.90 (d, J = 2.0 Hz, 2H), 6.65 (t, J = 2.0 Hz, 1H), 2.50 (s, 3H), 1.24 (m, 6H), 1.10 (d, J = 9.0 Hz, 36H); 13C NMR (CDCl3, 100 MHz): δ 189.6, 160.3, 157.2, 147.5, 137.7, 135.4, 133.8, 130.0, 129.3, 128.6, 116.5, 113.7, 109.5, 17.9, 15.6, 12.6; IR (neat, cm−1): 2943, 2866, 2210, 1555, 1581, 1435, 1172, 1030; HRMS (EI) calculated for C37H55NO3Si2 617.3715, found 617.3743. 3JC-H = 6.0 Hz for Cketone-H, 3JC-H = 13.4 Hz for Cester-H.
1-[3,5-Bis[[tris(1-methylethyl)silyl]oxy]phenyl]-4-methyl-2-[(4-methylphenyl)sulfonyl]-5-phenyl-, (2Z,4E) 2,4-pentadien-1-one (34b)
Alkynyl sulfone 9e (739 mg, 1.2 mmol) was subjected to General Procedure D using the nitrone 10d to obtain 191 mg (43%) of 34b. 1H NMR (CDCl3, 400 MHz): δ 7.74 (d, J = 8.0 Hz, 2H), 7.69 (d, J = 0.5 Hz, 1H), 7.30 (m, 5H), 7.17 (d, J = 8.0 Hz, 2H), 7.03 (d, J = 2.0 Hz, 2H), 6.92 (s, 1H), 6.64 (t, J = 2.0 Hz, 1H), 2.42 (s, 3H), 1.68 (s, J = 1.0 Hz, 3H), 1.24 (m, 6H), 1.10 (d, J = 9.0 Hz, 36H); 13C NMR (CDCl3, 100 MHz): δ 191.6, 176.5, 157.3, 145.8, 144.4, 142.5, 139.0, 138.2, 137.3, 135.7, 131.9, 130.2, 129.6, 129.0, 128.3, 128.1, 117.8, 114.2, 21.6, 17.4, 16.2, 12.5; IR (neat, cm−1): 2948, 2729, 1746, 1666, 1453, 1244, 1087; HRMS (EI) calculated for C43H62O5SSi2Na 769.3749, found 769.3740. 3JC-H = 8.8 Hz for Cketone-H.
2-Benzylidene-1-phenyl-heptane-1,3-dione (36b)
The intermediate N-methyl isoxazoline (5-Butyl-2-methyl-3-phenyl-2,3-dihydro-isoxazol-4-yl)-phenyl-mathanone) was prepared according to General Procedure A. 1H NMR (400 MHz, CDCl3): δ 7.55 (m, 2H), 7.53–7.23 (m, 8H), 5.27 (s, 1H), 2.96 (s, 3H), 2.22 (t, J = 7 Hz, 2H), 1.55 (m, 2H), 1.22 (m, 2H), 0.80 (t, J = 7 Hz, 3H) 13C NMR (100 MHz, CDCl3): δ191.9, 166.0, 141.0, 140.5, 140.2, 131.4, 128.4, 128.2, 127.7, 127.4, 127.2, 114.1, 77.1, 47.1, 28.9, 26.5, 22.2, 13.5. IR (neat) cm−1: 2958, 2931, 1612, 1577, 1450, 1361, 1230, 1157. HRMS: calcd for C21H23O2N [M+]; 321.1723, found 321.1717.
The intermediate N-t-butyl isoxazoline (5-Butyl-2-tert-butyl-3-phenyl-2,3-dihydro-isoxazol-4-yl)-phenyl-mathanone) was prepared according to General Procedure A. 1H NMR (500 MHz, CDCl3): δ 7.44–7.39 (m, 5H), 7.35 (m, 2H), 2.26 (m, 2H), 7.20 (m, 1H), 5.64 (s, 1H), 5.29 (s, 1H), 2.15 (t, J = 8 Hz, 2H), 1.53 (m, 2H), 1.27 (m, 3H), 1.18 (s, 9H), 1.88 (t, J = 8 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 191.7, 166.0, 143.5, 140.4, 131.2, 128.28, 128.21, 127.6, 127.5, 127.1, 115.5, 68.9, 61.2, 29.1, 26.5, 24.9, 22.4, 13.5. IR (neat) cm−1: 2974, 2958, 2928, 2873, 2857, 1633, 1595, 1574, 1494, 1457, 1447, 1371, 1363. HRMS: calcd for C24H30O2N [M-H+], 364.2271; found, 364.2260.
General Procedure C provided the title compound (36b) from the N-t-butyl intermediate isoxazoline (61% yield). 1H NMR (500 MHz, CDCl3): δ 7.92 (m, 2H), 7.80 (m, 1.6H), 7.61–7.21 (m, 15H), 2.66 (t, J = 7.0 Hz, 2H), 2.60 (t, J = 7.0 Hz, 0.7H), 1.64 (m, 4.2H), 1.34 (m, 4.5H), 0.90 (m, 5H). 13C NMR (100 MHz, CDCl3): δ 198.2, 198.0, 141.8, 140.2, 139.4, 136.1, 134.5, 134.0, 133.0, 132.6, 130.4, 130.3, 130.2, 129.8, 129.5, 129.1, 128.95, 128.91, 128.7, 128.5, 127.9, 43.9, 39.3, 26.1, 25.3, 22.2, 22.0, 13.85, 13.81. IR (neat) cm−1: 3060, 3029, 2919, 1706, 1700, 1640, 1609, 1598, 1449, 1320. HRMS: calcd for C20H21O2 [M-H+]; 293.1562, found 293.1569.
2-Cyclohexylmethylene-1-phenyl-heptane-1,3-dione (37a)
The intermediate N-methyl isoxazoline ((5-Butyl-3-cyclohexenyl-2-methyl-2,3-dihydro-isoxazol-4-yl)-phenyl-mathanone) was prepared according to General Procedure A (96% yield). 1H NMR (400 MHz, CDCl3): δ 7.62 (m, 2H), 7.51 (m, 1H), 7.4 3(m, 2H), 4.06 (d, J = 4 Hz, 1H), 2.79 (s, 3H), 2.07 (m, 2H), 1.71–1.61 (m, 8H), 1.40 (m, 2H), 1.23–1.10 (m, 7H), 0.74 (t, J = 7 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 192.5, 166.1, 140.4, 131.2, 128.1, 127.7, 110.8, 77.7, 48.3, 41.5, 29.4, 28.8, 27.1, 26.2, 26.1, 25.9, 22.0, 13.3. IR (neat) cm−1: 2923, 2850, 1608, 1573, 1446, 1357, 1242. HRMS: calcd for C21H30O2N [M-H+]; 328.2271, found 328.2278.
The intermediate N-t-butyl isoxazoline ((5-Butyl-2-tert-butyl-3-cyclohexyl-2,3-dihydro-isoxazol-4-yl)-phenyl-mathanone) was prepared according to General Procedure A (83% yield). 1H NMR (500 MHz, CDCl3): δ 7.57 (m, 2H), 7.48 (m, 1H), 7.41 (m, 2H), 4.45 (d, J = 3.5 Hz, 1H), 2.03 (m, 2H), 1.72–1.38 (m, 9H), 1.25–1.12 (m, 18H, 0.72 (t, J = 7.5 Hz, 3H) 13C NMR (125 MHz, CDCl3): δ 192.0, 167.9, 140.8, 131.1, 128.2, 113.1, 68.4, 60.6, 43.4, 29.8, 29.0, 27.8, 26.53, 26.51, 26.4, 25.1, 22.5, 13.4. IR (neat) cm−1: 2983, 2959, 2917, 2849, 1630, 1595, 1577, 1460, 1444, 1376, 1364, 1298, 1234, 1204, 1172. HRMS: calcd for C24H36NO2 [M-H+], 370.2741; found, 370.2756.
General Procedure C provided the title compound (37a) from the N-t-butyl intermediate isoxazoline (66% yield). 1H NMR (400 MHz, CDCl3): δ 7.76 (dd, Jab = 7.0 Hz, Jac = 5.0 Hz, 2H), 7.58 (m, 1H), 7.46 (m, 2H), 6.25 (d, J = 10.0 Hz, 1H), 2.61 (t, J = 7.0 Hz, 1H), 1.80–1.56 (m, 8H), 1.34–1.11 (m, 7H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 202.9, 195.4, 154.5, 140.6, 137.4, 132.7, 129.5, 128.5, 43.4, 38.6, 32.1, 25.7, 25.6, 25.1, 22.1, 13.8. IR (neat) cm−1: 3060, 3029, 2932, 2856, 1709, 1700, 1657, 1652, 1613, 1450. HRMS: calcd for C20H26O2 [M-H+]; 298.1927, found 298.1939.
1-Phenyl-2-(3-phenyl-allylidene)-heptane-1,3-dione (38)
The intermediate isoxazoline (5-Butyl-2-tert-butyl-3-styryl-2,3-dihydro-isoxazol-4-yl)-phenyl-mathanone) was prepared according to General Procedure A (64% yield). 1H NMR (500 MHz, CDCl3): δ 7.57 (m, 2H), 7.49 (m, 1H), 7.42 (m, 2H), 7.36, (M, 2H), 7.26 (m, 2H), 7.20 (m, 1H), 6.56 (d, J = 16 Hz, 1H), 6.34 (dd, Jab = 16 Hz, Jac = 6.5 Hz, 1H), 5.36 (d, J = 6.5 Hz, 1H), 2.14 (m, 2H), 1.52–1.44 (m, 2H), 1.22 (m, 11H), 0.79 (t, J = 6.5 Hz, 3H) 13C NMR (125 MHz, CDCl3): δ 191.8, 166.9, 140.6, 136.9, 131.3, 130.5, 129.6, 129.5, 128.8, 128.5, 128.39, 128.32, 128.0, 127.7, 127.3, 126.6, 113.2, 67.0, 60.9, 29.0, 26.6, 25.0, 22.4, 13.5. IR (neat) cm−1: 3024, 2959, 2931, 2870, 1597, 1574, 1446, 1359, 1228, 1209, 1191. HRMS: calcd for C26H32NO2 [M-H+], 390.2428; found, 390.2424.
General Procedure C provided the title compounds (38a and 38b) as a mixture of isomers (7: 1) from the intermediate isoxazoline (63% yield). 1H NMR (500 MHz, CDCl3): δ 7.81 (d, J = 7.0 Hz, 0.49H), 7.62 (d, J = 7.0 Hz, 2H), 7.59–7.26 (m, 15H), 7.01 (m, 2.43H), 6.69 (m, 0.22H), 2.64 (m, 2.63H), 1.63 (m, 3.18H), 1.32 (m, 4.02H), 0.89 (m, 4.37H). 13C NMR (125 MHz, CDCl3): δ 203.3, 198.1, 196.9, 195.5, 145.7, 145.5, 144.0, 141.2, 139.5, 139.0, 137.7, 137.1, 135.7, 135.5, 134.5, 133.9, 1328, 129.9, 129.7, 129.4, 129.3, 128.9, 128.8, 127.9, 127.6, 123.8, 123.3, 77.2, 43.1, 39.4, 30.3, 26.2, 26.1, 22.3, 22.2, 13.8. IR (neat) cm−1: 3060, 3029, 2932, 2856, 1709, 1700, 1657, 1652, 1613, 1575, 1449. HRMS: calcd for C22H22O2 [M+], 318.1614; found, 318.1612.
5.6. Assignment of olefin geometry for β-ketoalkylidene compounds
Product geometries were assigned based on carbon-hydrogen coupling constants obtained for the ester and/or the ketone of the divinyl ketone. A detailed study by Kingsbury gives tabulated splitting constants for a wide variety of differentially substituted olefins.xxxv
As shown, J13C-H ester (trans) couplings are on the order of ~12–13 Hz (I) and J13C-H ketone (cis) couplings are on the order of ~6–7 Hz (I). J13C-H ester (cis) couplings are on the order of ~8–9 Hz and J13C-H ketone (cis) couplings are on the order of ~10–11 Hz. Because these coupling constants can be easily distinguished, either the J13C-H ester or the J13C-H ketone can be measured to assign olefin geometry, and in ambiguous cases both J13C-H ketone and J13C-H ester can be measured to clarify the hydrogen-carbonyl relationship.

The above data (along with data previously tabulated within our group)vib assignments of product olefin geometry can be done with a high degree of confidence. All J13C-H were obtained using J13C-H ester with the exception of 15a. In that particular case J13C-H ketone was used.
5.7. Preparation of Baylis-Hillman Adducts
1-(2-tert-Butyl-3,5-diphenyl-2,3-dihydro-isoxazole-4-yl)-pentan-1-one (44)
Prepared by General Procedure A. 1H NMR (400 MHz, CDCl3): δ 7.66–7.26 (m, 10H), 5.61 (s, 1H), 2.11 (t, J = 7.2 Hz, 2H), 1.35 (m, 2H), 1.28 (s, 9H), 0.99 (m, 2H), 0.67 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 195.7, 162.8, 143.4, 131.0, 129.3, 128.5, 128.48, 128.42, 127.5, 127.3, 116.6, 68.8, 61.5, 40.2, 26.6, 25.0, 22.0, 13.6. IR (neat) cm−1: 3060, 3029, 2956, 2930, 2871, 1671, 1622, 1593, 1490, 1452, 1445, 1363, 1234, 1205, 1137. HRMS: calcd for C24H30NO2 [M-H+], 364.2271; found, 364.2265.
1-(2-tert-Butyl-3,5-diphenyl-2,3-dihydro-isoxazole-4-yl)-pentan-1-ol (45)
NaBH4 (0.010 g, 0.28 mmol) was added slowly to a stirred solution of isoxazoline 44 (0.100 g, 0.28 mmol) in MeOH (1 mL) at 0 °C. This was stirred for about 1 h, when it was quenched with H2O (2 mL). This was extracted with Et2O (3 × 5 mL), dried over MgSO4 and concentrated to give pure products as a 2:1 mixture of diastereomers. 1H NMR (500 MHz, CDCl3): δ 7.68 (m, 1.4H), 7.55 (m, 1H), 7.47–7.26 (m, 11.4H), 5.37 (s, 0.3H), 5.23 (0.7H), 4.56 (t, J = 6.5 Hz, 0.3H), 4.41 (t, J = 6.5 Hz, 0.7H), 1.67–1.60 (m, 2.1H), 1.35–1.18 (m, 9H), 0.89 (t, J = 6.5 Hz, 3H), 0.78 (t, J = 6.5 Hz, 1.1H). 13C NMR (100 MHz, CDCl3): δ 151.0, 150.1, 145.0, 144.7, 131.0, 129.6, 129.4, 129.3, 129.2, 129.1, 128.8, 128.58, 128.53, 128.4, 128.1, 128.0, 127.8, 127.6, 127.5, 127.4, 127.2, 127.0, 126.8, 126.7, 112.8, 111.6, 68.9 (2), 67.9 (2), 61.2, 61.0, 35.9, 30.3, 28.3, 26.1, 25.1, 22.6, 22.3, 22.2, 14.0. IR (neat) cm−1: 3513, 3052, 3025, 2945, 2928, 2853, 1666, 1629, 1593, 1494, 1447, 1445, 1370, 1234, 1210, 1137 HRMS: calcd for C18H28NO2 [M-H+], 290.2115; found, 290.2120.
2-Benzylidene-3-hydroxy-1-phenyl-heptan-1-one (46)xxxvi
Oxidized according to General Procedure C. 1H NMR (400 MHz, CDCl3): δ 7.83–7.02 (m, 11H), 4.80 (m, 1H), 3.89 (m, 1H), 1.94 (m, 1H), 1.72 (m, 1H), 1.48 (m, 1H), 1.34 (m, 3H), 0.877 (m, 3H). 13C NMR (100 MHz, CDCl3): δ 201.1, 142.7, 141.3, 137.9, 134.4, 132.7, 129.8, 129.2, 128.9, 128.6, 128.3, 70.0, 36.1, 28.4, 22.5, 14.0. IR (neat) cm−1: 3413, 2956, 1650, 1595, 1447, 1377, 1229, 1070, 1001, 950.
(5-Butyl-2-tert-butyl-3-phenyl-2,3-dihydro-isoxazol-4-yl)-phenyl-mathanone (47)
Prepared according to General Procedure A. 1H NMR (500 MHz, CDCl3): δ 7.44–7.39 (m, 5H), 7.35 (m, 2H), 2.26 (m, 2H), 7.20 (m, 1H), 5.64 (s, 1H), 5.29 (s, 1H), 2.15 (t, J = 8.0 Hz, 2H), 1.53 (m, 2H), 1.27 (m, 3H), 1.18 (s, 9H), 1.88 (t, J = 8.0 Hz, 3H.) 13C NMR (100 MHz, CDCl3): δ 191.7, 166.0, 143.5, 140.4, 131.2, 128.28, 128.21, 127.6, 127.5, 127.1, 115.5, 68.9, 61.2, 29.1, 26.5, 24.9, 22.4, 13.5. IR (neat) cm−1: 2974, 2958, 2928, 2873, 2857, 1633, 1595, 1574, 1494, 1457, 1447, 1371, 1363. HRMS: calcd for C24H30O2N [M-H+], 364.2271; found, 364.2260.
(5-Butyl-2-tert-butyl-3-phenyl-2,3-dihydro-isoxazol-4-yl)-phenyl-methanol (48)
DIBAL (1.0 M/Toluene, 0.21 mL) was added slowly to a stirred solution of isoxazoline 10f (0.050 g, 0.14 mmol) in dichloromethane (0.28 mL) at 0 °C. This was stirred for about 10 min, quenched with MeOH (0.30 mL) and warmed to rt. The solid was filtered off, and the filtrate was concentrated. Column chromatography provided alcohol 47 (0.035 g, 73%) as a 3:1 mixture of diastereomers: 1H NMR (500 MHz, CDCl3): δ 7.36–7.14 (m, 13 H), 5.16 (m, 0.3 H), 4.86 (m, 1H), 4.62 (s, 0.3H), 2.39 (m, 3H), 1.64–1.37 (m, 9.2 H), 1.07 (s, 9H), 0.96 (m, 9.2H). 13C NMR (100 MHz, CDCl3): δ 153.1, 151.9, 143.8, 142.1, 142.0, 128.8, 128.3, 128.1, 128.0, 127.65, 127.60, 127.2, 127.08, 127.01, 126.0, 125.5, 110.1, 109.6, 70.0, 69.9, 69.2, 68.1, 60.59, 60.53, 29.7, 29.6, 24.9, 24.7, 24.3, 22.79, 22.73, 13.8. IR (neat) cm−1: 3346, 2962, 2957, 2928, 2877, 2863, 1587, 1571, 1488, 1457, 1444, 1378, 1362. HRMS: calcd for C24H32NO2 [M-H+], 366.2479; found, 366.2473.
1H and 13C spectra for all compounds can be found in the supplementary information.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- i.a) Thibonnet J, Vu VA, Berillon L, Knochel P. Tetrahedron. 2002;58:4787. [Google Scholar]; b) Acuna C, Zapata A. Synth Commun. 1988;18:1133. [Google Scholar]
- ii.a) Marino JP, Lindermann RJ. J Org Chem. 1981;46:3696. [Google Scholar]; b) Marino JP, Lindermann RJ. J Org Chem. 1983;48:4621. [Google Scholar]; c) Kerr DJ, Metje C, Flynn BL. J Chem Soc, Chem Commun. 2003:1380. [PubMed] [Google Scholar]
- iii.Jones TK, Denmark SE. Helv Chim Acta. 1983;66:2377. [Google Scholar]
- iv.Mazzola RD, Giese S, Benson CL, West FG. J Org Chem. 2004;69:220. doi: 10.1021/jo034788q. [DOI] [PubMed] [Google Scholar]
- v.Occhiato EG, Prandi C, Ferrali A, Guarna A, Deagostino A, Venturello P. J Org Chem. 2002;67:7144. doi: 10.1021/jo025930a. [DOI] [PubMed] [Google Scholar]
- vi.He W, Sun X, Frontier AJ. J Am Chem Soc. 2003;125:14278–14279. doi: 10.1021/ja037910b.addition/correction J Am Chem Soc. 2004;126:10493.Malona JA, Colbourne JM, Frontier AJ. Org Lett. 2006;8:5661. doi: 10.1021/ol062403v.
- vii.a) Tietze LF, Beifuss U. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 2. Pergamon Press; Oxford: 1991. p. 341. [Google Scholar]; b) Tanikaga R, Konya N, Hamamura K, Kaji A. Bull Chem Soc Jpn. 1988;61:3211. [Google Scholar]; c) Desimoni G, Faita G, Ricci M, Righetti PP. Tetrahedron. 1998;54:9581. [Google Scholar]
- viii.Padwa A, Kline DN, Perumattam J. Tetrahedron Lett. 1987;28:913.Padwa A, Chiacchio U, Kline DN, Perumattam J. J Org Chem. 1988;53:2238.c) According to the authors, only one product isomer was isolated, and the geometry drawn for the product would have arisen from “outward” disrotation. The method used to assign the olefin geometry was not described.
- ix.a) Woodward RB, Hoffmann R. The Conservation of Orbital Symmetry. Verlag Chemie; Weinheim: 1970. [Google Scholar]; b) Woodward RB, Hoffmann R. Angew Chem Int Edit. 1969;8:781–932. [Google Scholar]
- x.a) Mock WL. J Am Chem Soc. 1966;88:2857. [Google Scholar]; b) McGregor SD, Lemal DM. J Am Chem Soc. 1966;88:2858. [Google Scholar]; c) Lemal DM, McGregor SD. J Am Chem Soc. 1966;88:1335. [Google Scholar]; d) Mock WL. J Am Chem Soc. 1975;97:3666. [Google Scholar]
- xi.Rondan NG, Houk KN. J Am Chem Soc. 1985;107:2099.Doulbier WR, Jr, Koroniak H, Houk KN, Sheu C. Acc Chem Res. 1996;29:471.Lee PS, Zhang XY, Houk KN. J Am Chem Soc. 2003;125:5072. doi: 10.1021/ja0287635. and references therein.Evanseck JD, Thomas BE, Spellmeyer DC, Houk KN. J Org Chem. 1995;60:7134.Luo L, Bartberger MD, Dolbier WJ. J Am Chem Soc. 1997;119:12366.For another study proposing a role for geminal bonds in the torquoselectivity of retroelectrocyclizations, see Yasui M, Naruse Y, Inagaki S. J Org Chem. 2004;69:7246. doi: 10.1021/jo049081y. and references therein.
- xii.For recent theoretical investigations of torquoselective Nazarov-type cyclizations, see: Dieker J, Froehlich R, Wuerthwein EU. Eur J Org Chem. 2006;23:5339.Cavalli A, Masetti M, Recanatini M, Prandi C, Guarna A, Occhiato EG. Chem Eur J. 2006;12:2836. doi: 10.1002/chem.200501391.Harmata M, Schreiner PR, Lee DR, Kirchhoefer PL. J Amer Chem Soc. 2004;126:10954. doi: 10.1021/ja048942h.For a recent theoretical investigation of a torquoselective sigmatropic shift, see: Faza ON, Lopez CS, de Lera AR. J Org Chem. 2007;72:2617. doi: 10.1021/jo070050n.For recent theoretical investigations of torquoselective electrocyclic ring openings and closures, see: Murakami M, Hasegawa M, Igawa H. J Org Chem. 2004;69:587. doi: 10.1021/jo035433+.Walker MJ, Hietbrink BN, Thomas BE, IV, Nakamura K, Kallel EA, Houk KN. J Org Chem. 2001;66:6669. doi: 10.1021/jo010466f.Ikeda H, Kato T, Inagaki S. Chem Lett. 2001;3:270.Murakami M, Miyamoto Y, Ito Y. Angew Chem Int Edit. 2001;40:189.Luo L, Bartberger MD, Dolbier WR., Jr J Am Chem Soc. 1997;119:12366.For a recent theoretical investigation of a torquoselective Cope Rearrangement, see: Zhao YL, Suhrada CP, Jung ME, Houk KN. J Am Chem Soc. 2006;128:11106. doi: 10.1021/ja060913e.
- xiii.Naruse Y, Hayashi Y, Inagaki S. Tetrahedron Lett. 2003;44:8509. [Google Scholar]
- xiv.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;13:3769. [Google Scholar]
- xv.Sai H. Synthesis. 1995:582. [Google Scholar]
- xvi.Bolin DG, Paquette LA, Schulze MM. J Org Chem. 1994;59:2043. [Google Scholar]
- xvii.Harding KE, Ligon RC. Synth Commun. 1974;4:297. [Google Scholar]
- xviii.Chaidez MA, Inman WD, Paulson DR, Sanchez AKJ. J Org Chem. 1989;54:4872. [Google Scholar]
- xix.Negishi E-i, King AO, Klima WL, Patterson W, Silveira A., Jr J Org Chem. 1980;45:2526. [Google Scholar]
- xx.Nair V, Augustine A, Suja TD. Synthesis. 2002;15:2259. [Google Scholar]
- xxi.Hamer J, Macaluso A. Chem Rev. 1964;64:473. [Google Scholar]
- xxii.Kingsbury CA, Draney D, Sopchik A, Rissler W, Durham D. J Org Chem. 1976;41:3863. [Google Scholar]
- xxiii.a) Baldwin JE, Pudussery RG, Qureshi AK, Sklarz B. J Am Chem Soc. 1968;90:5325. [Google Scholar]; b) Adachi I, Harada K, Kano H. Tetrahedron Lett. 1969;55:4875. [Google Scholar]; c) Ishikawa T, Kudoh T, Yoshida J, Yasuhara A, Manabe S, Saito S. Org Lett. 2002;4:1907. doi: 10.1021/ol025906j. [DOI] [PubMed] [Google Scholar]
- xxiv.a) Merino P, Tejero T, Laguna M, Cerrada E, Moreno A, Lopez JA. Org Biomol Chem. 2003;1:2336. doi: 10.1039/b304112c. [DOI] [PubMed] [Google Scholar]; b) Kanemasa S, Ueno N, Shirahase M. Tetrahedron Lett. 2002;43:657. and references therein. [Google Scholar]
- xxv.a) Stang PJ, Murch P. Tetrahedron Lett. 1997;38:8793. [Google Scholar]; b) Hems WP, Tan CH, Stork T, Feeder N, Holmes AB. Tetrahedron Lett. 1999;40:1393. [Google Scholar]; c) Chermier MOJ, Moussalli N, Chanet-Ray J, Chou S. J Chem Research (S) 1999:566. [Google Scholar]
- xxvi.(a) Hoffman AW. Ann. 1851;78:253. [Google Scholar]; (b) Hoffman AW. Ann. 1851;79:11. [Google Scholar]; (c) Hoffmann AW. Ber. 1881;14:659. [Google Scholar]; (d) Cope AC, Trumbull ER. Org React. 1960;11:317. [Google Scholar]
- xxvii.Basavaiah D, Rao AJ, Satyanarayana T. Chem Rev. 2003;103:811. doi: 10.1021/cr010043d. [DOI] [PubMed] [Google Scholar]
- xxviii.Singh VK. Synthesis. 1992:607. [Google Scholar]
- xxix.Luche JL. J Am Chem Soc. 1978;100:2226. [Google Scholar]
- xxx.Masamune S, Seidner RT, Wilson KE. J Chem Soc, Chem Commun. 1970:213. [Google Scholar]
- xxxi.For experimental data for compounds 49, see Trost BM, Shuichi O. J Am Chem Soc. 2001;123:1230. doi: 10.1021/ja003629a.
- xxxii.He W, Herrick IR, Atesin TA, Caruana PA, Kellenberger CA, Frontier AJ. J Am Chem Soc. 2008;130:1003. doi: 10.1021/ja077162g. [DOI] [PubMed] [Google Scholar]
- xxxiii.a) Chiacchio U, Liguori A, Rescifina A, Romeo G, Rossano F, Sindona G, Uccella N. Tetrahedron. 1992;48:123. [Google Scholar]; b) Chiacchio U, Casuscelli F, Liguori A, Romeo G, Sindona G, Uccella N. Heterocycles. 1993;36:585–600. [Google Scholar]; c) Casuscelli F, Chiacchio U, Rescifina A, Romeo R, Romeo G, Tommasini S, Uccella N. Tetrahedron. 1995;51:2979. [Google Scholar]
- xxxiv.Canterbury DP, Frontier AJ, Um JM, Cheong PHY, Goldfeld DA, Huhn RA, Houk KN. Org Lett. 2008 doi: 10.1021/ol8019154. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxxv.Kingsbury CA, Draney D, Sopchik A, Rissler W, Durham D. J Org Chem. 1976;41:3863. [Google Scholar]
- xxxvi.Kazuhiro Y, Takayuki S. Tetrahedron. 2007;63:6259. [Google Scholar]