

Abstract
The concept that obesity is an inflammatory state has changed our understanding of this condition and suggested that pharmacological interventions targeting inflammation may be useful strategies to improve metabolic complications of obesity. Phosphodiesterase 4 (PDE4) inhibitors exhibit profound antiinflammatory effects, but whether PDE4 inhibition suppresses obesity-induced inflammation is unknown. Among PDE4 isoforms, PDE4B is the major species mediating inflammatory responses. We therefore examined obesity-related phenotypes in mice deficient for PDE4B. Compared with wild-type littermates, PDE4B-null mice were leaner, with lower fat pad weights, smaller adipocytes, and decreased serum leptin levels on both chow and high-fat diets (HFDs). PDE4B deficiency suppressed TNF-α mRNA levels and macrophage infiltration in white adipose tissue in mice on HFD, but insulin sensitivity was unaltered. PDE4B-null mice on HFDs had increased locomotor activity. These results suggest a previously unappreciated role for PDE4B in the regulation of energy balance and that PDE4B inhibitors could have utility in treatment of obesity and for suppression of obesity-induced inflammation in white adipose tissue.
Mice deficient in phosphodiesterase 4B had reduced adiposity and high fat diet-induced adipose inflammation.
The global obesity epidemic poses a major public health issue (1,2) because obesity increases the risk of numerous health consequences, such as insulin resistance, type 2 diabetes, coronary heart diseases, and mortality (3,4). Great advances have been made in recent years in understanding the etiology of obesity and mechanisms underlying its various complications (5,6,7,8,9). Adipose tissue mass increases in obesity; importantly, however, adipose tissue is no longer considered solely a passive depot for energy storage; instead, it is an active metabolic and endocrine organ with critical roles in regulating systemic physiology, such as immunity and inflammation (10,11,12,13,14).
White adipose tissue (WAT) constitutively expresses the proinflammatory cytokine TNF-α, and its expression is markedly increased in obese rodents. In these animals, TNF-α can induce insulin resistance; conversely, its neutralization improves insulin sensitivity (15). This discovery provided a link, at the molecular level, between a proinflammatory cytokine that is overexpressed by expanded WAT and metabolic complications of obesity (15,16,17). Subsequently many other proinflammatory cytokines, such as IL-6, were also found to be overexpressed in obese animal models (18,19,20,21,22,23,24). Another important feature of obese WAT is that it is infiltrated with macrophages, which potentially contribute to the production of inflammatory mediators and the development of insulin resistance (23,24). The concept that obesity is an inflammatory state has changed our understanding of this condition and suggested that pharmacological interventions targeting inflammation may be useful strategies to improve metabolic complications of obesity (25,26,27,28,29,30,31,32).
Phosphodiesterase 4 (PDE4) is a family of enzymes that specifically catalyze the hydrolysis of cAMP, which plays key roles in the regulation of most cellular processes (33,34). The PDE4 family consists of four isoforms, encoded by distinct genes, PDE4A, PDE4B, PDE4C, and PDE4D (35,36). PDE4 inhibitors exhibit profound antiinflammatory effects both in vitro and in vivo (37,38,39), suggesting that some PDE4s are important modulators of inflammatory responses. Currently available PDE4 inhibitors are nonselective, limiting pharmacological approaches to examining functions of distinct PDE4s. Consequently, Conti and coworkers (40,41,42) generated mice deficient in PDE4A, PDE4B, or PDE4D and found that PDE4B is the major isoform mediating inflammatory responses. Indeed, in PDE4B-deficient (PDE4B−/−) mice, lipopolysaccharide-induced TNF-α production is largely suppressed in inflammatory cells (40). Because increased production of TNF-α has been implicated in many metabolic complications of obesity, we tested the hypothesis that PDE4B plays a role in obesity-induced inflammation. Consequently, in the current study, we examined various metabolic parameters in PDE4B−/− mice placed on either chow or high-fat diets (HFDs), and our results suggest a previously unappreciated role for PDE4B in the regulation of adipose tissue inflammation and energy balance.
Materials and Methods
Mice, HFD studies, and body composition analysis
PDE4B−/− mice were kindly provided by Dr. Marco Conti (Stanford University, Stanford, CA), and generation of these mice was as described (40). All animal protocols were approved by the Animal Care and Use Committee of the Beth Israel Deaconess Medical Center. Mice were housed at 22–24 C with a 14-h light, 10-h dark cycle and provided with ad libitum water and a chow diet (6% calories from fat, 8664; Harlan Teklad, Indianapolis, IN). Homozygous PDE4B−/− mice and wild-type (WT) littermates were obtained by mating mice heterozygous for the PDE4B gene disruption. Four-week-old male PDE4B−/− mice and WT littermates were placed on either a chow diet or a high-fat, high-sucrose diet (58% kcal from fat, 26% kcal from sucrose, D-12331; Research Diets, New Brunswick, NJ). All mice on diets were housed individually, and food intake and body weight were measured weekly. The fat and lean tissue masses were determined in living, nonanesthetized mice by using a magnetic resonance imaging (MRI) machine (Echo Medical Systems, Houston, TX).
RNA extraction and quantitative real-time PCR
To examine the expression pattern of PDE4B in various tissues, three WT 8-wk-old C57BL6 mice were used. Dissected tissues were immediately placed into RNAlater solution (Ambion, Austin, TX) for subsequent RNA extraction. Total RNA was isolated from tissues with RNeasy tissue minikit with deoxyribonuclease treatment (QIAGEN, Valencia, CA). One microgram of RNA was reverse transcribed to cDNA using random hexamers (Superscript; Ambion). Real-time PCR was performed on the ABI 7900 HT Fast real-time PCR system (Applied Biosystems, Foster City, CA), using Taqman based probes and Taqman universal PCR master mix (Applied Biosystems). Primer and probe sets for PDE4B, TNF-α, IL6, F4/80, and Cyclophillin were from Applied Biosystems. Relative expression levels were calculated by standard-curve method, and Cyclophillin was used as an internal control.
Metabolic parameters
Leptin and insulin concentrations were measured by ELISA kits (CrystalChem, Chicago, IL). Blood glucose levels were measured by using a glucose monitor (glucometer; Bayer, Indianapolis, IN). For the glucose tolerance test, mice were fasted overnight and were then injected ip with glucose (1.5 mg/g body weight), and glucose levels were measured using blood collected through tail nicks at 0, 15, 30, 60, and 120 min. For the insulin tolerance test, mice were fasted for 3 h and then insulin (humulin R; Eli Lilly, Indianapolis, IN) was injected ip (1 mU/g body weight), and blood glucose levels were then measured as described above.
Histology and immunohistochemistry
Epididymal adipose tissue was fixed for 12–16 h at room temperature in zinc-formalin solution, embedded in paraffin, and sectioned at 5 μm. Hematoxylin and eosin (H&E) staining was performed using standard techniques. To measure adipocyte diameters, 16 mice (four for each genotype/diet combination) were used, and diameters of 100 cells from four sections for each group were measured by using the ImageJ software (National Institutes of Health, Bethesda, MD). To detect macrophages on adipose sections, immunohistochemistry was performed using an antibody against the macrophage marker F4/80 (eBioscience, San Diego, CA). Four mice for each genotype were used, and for each adipose depot, four fields per section were analyzed. Percentage of F4/80-positive cells was calculated as the number of nuclei of F4/80-positive cells divided by the total number of nuclei present in a field. The specificity of the F4/80 antibody was verified by performing immunohistochemistry by replacing the primary antibody with nonspecific IgG, and no positive staining was observed (data not shown). Cell counting was performed by an investigator blinded to genotype or diet treatments.
Adipocyte isolation and lipolysis
Adipocytes were isolated according to a slightly modified method of Rodbell (43,44). Briefly, epididymal fat pads were minced and suspended in Krebs Ringer HEPES-buffered solution [137 mm NaCl, 5 mm KCl, 4.2 mm NaHCO3, 1.3 mm CaCl2, 0.5 mm KH2PO4, 0.5 mm MgCl2, 0.4 mm MgSO4, 20 mm HEPES (pH 7.4)] containing 5 mm glucose, 1% BSA, and 1 mg/ml collagenase. The suspension was incubated at 37 C for 1 h with moderate shaking. To wash the digested tissues, a Pasteur pipet was inserted through the floating cell layer to remove the buffer, followed by resuspension using the same buffer without collagenase. After three washes, the cell layer, including undigested tissues, was filtered through nylon mesh (250 μm). Isolated adipocytes were washed into DMEM containing 10 mm HEPES, 1% BSA, 10% fatal bovine serum, and antibiotics (penicillin and streptomycin) and maintained at 37 C under sterile conditions. To perform lipolysis assay, adipocytes were suspended in Krebs Ringer HEPES buffer supplemented with 5 mm glucose and 1% BSA, with or without isoproterenol, and incubated at 37 C with moderate shaking. Glycerol released into the medium was measured by using a free glycerol reagent from Sigma (St. Louis, MO). Intracellular cAMP was measured by using a competitive enzyme immunoassay kit (R&D Systems, Minneapolis, MN). Glycerol and cAMP amounts were normalized by cellular protein content determined by Bio-Rad protein assay (Bio-Rad, Hercules, CA). Male 18- to 20-wk-old mice were used, and for each experiment, the data represent the mean value based on at least three mice for each genotype.
Energy expenditure
We used the comprehensive laboratory animal monitoring system (Columbus Instruments, Columbus, OH) to examine energy expenditure. Oxygen (O2) and carbon dioxide (CO2) contents were determined by O2 and CO2 sensors, and heat production on per-animal basis was calculated from the following equation: (3.82 + 1.23 × RER) × VO2, where RER is the respiratory exchange ratio (volume of CO2 produced/volume of O2 consumed per hour). Cumulative ambulatory activity counts were recorded every 30 min throughout the light and dark cycles. Weighed before each trial, mice were acclimatized to monitoring cages for 24 h before data collection and were housed individually in specially built Plexiglass cages maintained at 22 C under an alternating 12-h light, 12-h dark cycle (light period 0800–2000 h).
Statistical analysis
Data are expressed as the mean ± sem. Statistical significance was tested with unpaired two-tailed Student’s t tests unless otherwise indicated. Wilcoxon signed-rank tests were used to compare adipocyte diameter distributions between PDE4B−/− mice and WT littermates. The differences were considered statistically significant if P < 0.05.
Results
Expression pattern of PDE4B
PDE4B is highly expressed in the brain (45,46,47,48). To compare expression levels of PDE4B in other tissues, we performed real-time PCR in WT C57BL6 mice using Taqman probes for PDE4B. PDE4B was expressed in all the tissues that we examined, including WAT, brown fat, the stomach, the small intestine, the large intestine, the liver, the spleen, the hypothalamus, and the frontal cortex (supplemental Fig. 1, published as supplemental data on The Endocrine Society’s Journals Online web site at http://endo.endojournals.org). Consistent with previous reports, PDE4B was highly expressed in the brain, such as the frontal cortex and the hypothalamus, and PDE4B was expressed at a similar level in the spleen. In addition, PDE4B was also abundantly expressed in the liver, WAT, and brown fat.
Reduced fat content and fat pad weights in PDE4B−/− mice
PDE4B−/− mice and WT littermates showed similar body weights on both chow and HFD. However, there was a tendency for PDE4B−/− mice to have lower body weights, and the difference in body weight became larger over time (Fig. 1A), consistent with a previous report (49). Nevertheless, at 7 months of age, the difference was still not statistically significant. No significant difference in food intake was observed (data not shown). Importantly, at 24 wk old, PDE4B−/− mice on both chow and HFD showed significant reduction in fat content that was determined by MRI (Fig. 1B). On chow diet, there was an approximately 11% decrease (P < 0.05), whereas for mice on HFD, there was 15% decrease (P < 0.01) in fat content. Consistently, weights of total fat pads showed more than 20% decrease in 28-wk-old mice either on chow (P < 0.01) or HFD (P < 0.01) (Fig. 1C). Weights of epididymal fat pads, perirenal fat pads, and sc fat pads also showed significant decrease (Fig. 1, D–F). The weight decrease appeared to be specific to WAT because in brown fat, the heart, and the liver, there was no significant weight difference between PDE4B−/− mice and WT littermates on either chow or HFD (Fig. 1, G–I).
Figure 1.

Reduced fat content and fat pad weights in PDE4B−/− mice. Body weight curves (A), fat content determined by MRI for mice at 24 wk of age (B), and weights of total fat pads (C), epididymal fat pads (D), perirenal fat pads (E), sc fat pads (F), brown fat (G), heart (H), and liver (I) for PDE4B−/− (KO) and WT mice on either chow or HFD at 28 wk of age (n = 6–8/group). Data are expressed as mean ± sem. *, P < 0.05; #, P < 0.01.
Reduced size of adipocytes in PDE4B−/− mice
Because PDE4B−/− mice had reduced fat content, we examined the size differences of adipocytes between PDE4B−/− mice and WT littermates at 28 wk of age. H&E staining was performed using epididymal adipose tissue sections, and there was a clear difference in adipocyte sizes by visual inspections (Fig. 2A). To quantify the difference, diameters of 100 adipocytes from four mice per genotype on each diet were determined. Adipocyte diameters followed a normal distribution; however, compared with WT mice, the adipocyte diameter distribution of PDE4B−/− mice shifted toward smaller sizes (Fig. 2, B and C). Wilcoxon signed-rank tests showed that adipocyte diameters in PDE4B−/− mice were significantly smaller than those in WT mice (P < 0.01) on both chow and HFD.
Figure 2.

Reduced size of adipocytes in PDE4B−/− mice. A, Representative H&E staining of epididymal adipose tissue sections from PDE4B−/− (KO) and WT mice on either chow or HFD at 28 wk of age. B, Distribution of adipocyte diameters for mice on chow diet. C, Distribution of adipocyte diameters for mice on HFD. Dotted lines denote the mean of adipocyte diameters. Diameters of 100 adipocytes from four mice were quantified per genotype on each diet. Wilcoxon signed-rank tests were used to compare adipocyte diameter distributions. #, P < 0.01.
Reduced leptin levels in PDE4B−/− mice
Leptin is an adipokine that plays pivotal roles in energy homeostasis, and its levels correlate with WAT mass (50,51). We therefore examined serum leptin levels in these mice. At 10 wk old, on either chow or HFD, leptin levels were not significantly different (data not shown). However, at 25 wk of age, consistent with reduced fat content, on chow diet, serum leptin levels were about 20% less in PDE4B−/− mice (P < 0.05), whereas on HFD, leptin levels were 25% less in PDE4B−/− mice (P < 0.05) (Fig. 3A). We also examined serum levels of adiponectin, an adipokine whose levels are decreased in obesity (52,53,54). As expected, in WT mice HFD treatment lowered adiponectin levels (P < 0.05); in contrast, in PDE4B−/− mice, its levels were not significantly decreased. Consistently, PDE4B−/− mice on HFD had higher adiponectin levels than WT littermates (P < 0.05) (Fig. 3B). In these mice, levels of fasting serum glucose and insulin did not show significant differences (Fig. 3, C and D). Glucose tolerance test and insulin tolerance test were performed and also showed no significant differences (data not shown). These results suggest that insulin sensitivity was not improved in PDE4B−/− mice.
Figure 3.

Reduced serum leptin levels in PDE4B−/− mice. Serum levels of leptin (A), adiponectin (B), fasting glucose (C), and fasting insulin (D) in PDE4B−/− (KO) and WT mice. Serum samples were taken from mice at 25 wk of age (n = 6–8/group). Data are expressed as mean ± sem. *, P < 0.05.
Reduced WAT TNF-α mRNA and macrophage infiltration in PDE4B−/− mice on HFD
Because obesity is associated with increased production of WAT proinflammatory cytokines, such as TNF-α, we did real-time PCR to examine TNF-α mRNA levels in epididymal adipose tissue from mice at 28 wk of age. In PDE4B−/− mice on HFD, TNF-α mRNA levels were significantly suppressed (Fig. 4A). The TNF-α levels were more than 3-fold higher in the WT than in PDE4B−/− mice (P < 0.05). We also examined WAT IL6 levels. IL6 levels were about 2-fold higher in WT mice treated with HFD than in PDE4B−/− mice, although the difference was not statistically significant (data not shown).
Figure 4.

Reduced TNF-α expression and macrophage infiltration in WAT of PDE4B−/− mice. A, Expression of TNF-α mRNA in epididymal adipose tissue. Representative immunohistochemical staining of epididymal adipose tissue sections using an antibody against the macrophage marker F4/80 for WT (B) and PDE4B−/− (KO) (C) mice on HFD at 28 wk of age. D, Quantification of percentage of macrophages present in epididymal adipose tissue sections. E, Expression of F/80 mRNA in epididymal adipose tissue for mice on HFD. For macrophage quantification, four adipose tissue sections for each of the four PDE4B−/− or WT littermates were counted. Percentages of F4/80-positive cells were calculated as the number of nuclei of F4/80-positive cells divided by the total number of nuclei. For mRNA expression, Taqman-based real-time PCR was performed and Cyclophilin was used as an internal control (n = 6–8/group). Data are expressed as mean ± sem. *, P < 0.05. AU, Arbitrary units.
Obesity is characterized by infiltration of macrophages into WAT (23,24). We therefore examined macrophage infiltration by doing immunohistochemistry using an antibody against the macrophage marker F4/80 on epididymal adipose tissue sections. There was a clear difference of macrophage staining results between WT and PDE4B−/− mice by visual inspection. Many adipocytes were surrounded by infiltrating macrophages; in WT mice these cells frequently aggregated, whereas in PDE4B−/− mice, they were isolated (Fig. 4, B and C). To quantify macrophage infiltration, we calculated the percentage of F4/80-positive cells as the number of nuclei of F4/80-positive cells divided by the total number of nuclei present in a field. Indeed, for mice on HFD, there was a 30% decrease in the percentage of infiltrating macrophages (P < 0.05) (Fig. 4D). Consistently, quantitative PCR showed F4/80 mRNA was also significantly decreased in WAT of PDE4B−/− mice (P < 0.05) (Fig. 4E).
Increased isoproterenol-induced cAMP levels in PDE4B−/− adipocytes
Although PDE4s are cAMP-specific phosphodiesterases, different PDE4 isoforms have distinct functions in different tissues. To determine whether PDE4B is involved in cAMP regulation in adipocytes, we isolated adipocytes from epididymal fat pads and treated these cells with the β-adrenergic agonist isoproterenol (ISO). Levels of cAMP peaked at 2 min after ISO treatment in both WT and PDE4B−/− adipocytes; however, at 5 and 10 min after ISO treatment, cAMP levels were significantly higher in PDE4B−/− adipocytes (Fig. 5A). This result suggests that PDE4B plays a role in mediating cAMP degradation in adipocytes. Previous results showed that treating adipocytes with PDE4 inhibitors increased lipolysis rate (55,56,57,58). Because these inhibitors are nonselective, it is not clear which isoform is responsible for the increased lipolysis caused by PDE4 inhibition. We then examined lipolysis in isolated adipocytes, which were treated with ISO, and glycerol released into the medium was measured to quantitate lipolysis. No significant difference was observed between PDE4B−/− and WT adipocytes at any time point (Fig. 5B) and with different ISO concentrations (data not shown). This result suggests that PDE4B is not a major PDE4 isoform in regulation of adipocyte lipolysis.
Figure 5.
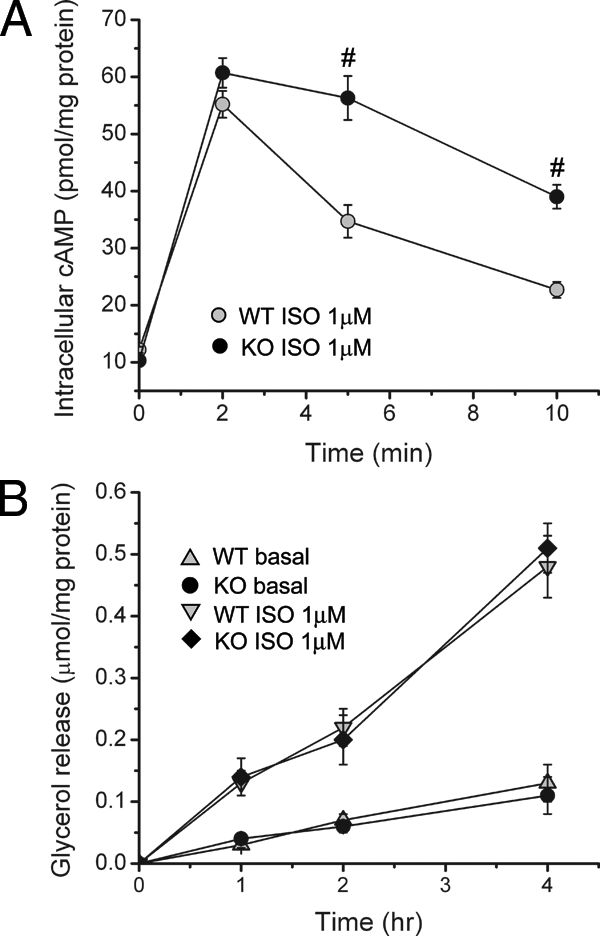
Increased ISO-induced intracellular cAMP levels in PDE4B−/− mice. A, Intracellular cAMP levels in adipocytes treated with 1 μm ISO at the indicated time points. Adipocytes were isolated from either PDE4B−/− (KO) or WT mice. B, Adipocyte lipolysis as assessed by glycerol released into the medium for adipocytes treated with 1 μm ISO at the indicated time points. #, P < 0.01.
Increased locomotor activity in PDE4B−/− mice on HFD
It is possible that decreased fat content of PDE4B−/− mice resulted from increased energy expenditure. We therefore examined energy expenditure in these mice using the comprehensive laboratory animal monitoring system. In WT mice, HFD treatment decreased locomotor activity, but PDE4B deficiency almost completely abolished this decrease. Although on a chow diet, no difference was observed; on HFD, PDE4B−/− mice had increased locomotor activity than WT mice during both light and dark cycles (P < 0.05) (Fig. 6A). We also examined oxygen consumption, which showed a tendency toward increased oxygen consumption in PDE4B−/− mice (Fig. 6B), although this difference was not statistically significant. Respiratory exchange ratio and heat product did not show significant differences between genotypes (data not shown).
Figure 6.
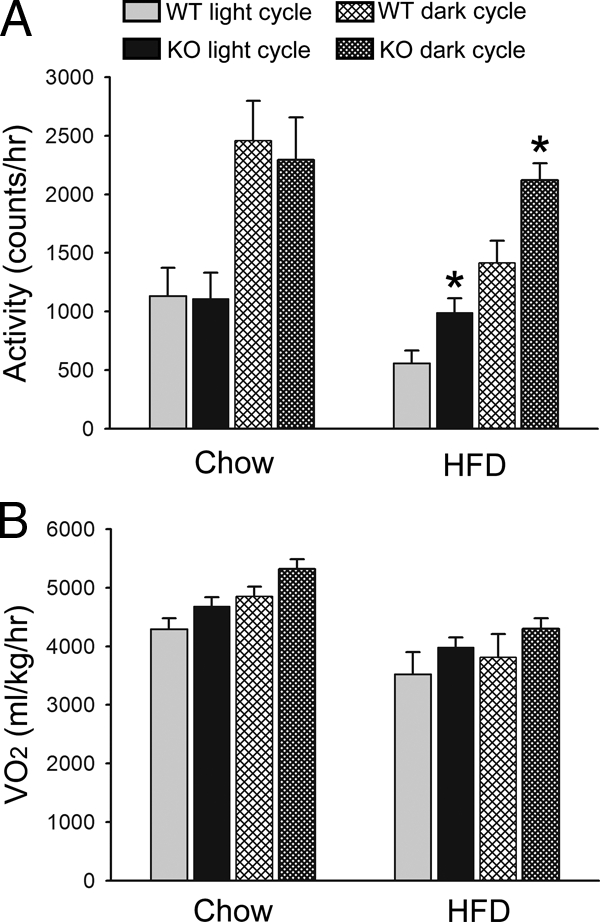
Increased locomotor activity of PDE4B−/− mice on HFD. Locomotor activity (A) and oxygen consumption (B) of PDE4B−/− (KO) and WT mice on either chow or HFD during light and dark cycles (n = 6–8/group). Data are expressed as mean ± sem. *, P < 0.05.
Discussion
An important advance in obesity research has been the understanding that obesity is associated with a state of chronic, low-grade inflammation, with WAT being an important element in this process, as evidenced by both increased expression of inflammatory markers and infiltration of inflammatory cells (15,23,24,28). In the current study, we examined the impact of PDE4B deficiency on WAT inflammation and other metabolic parameters. Compared with WT littermates, PDE4B−/− mice, unexpectedly, were leaner. On both chow and HFD, these mice had lower fat pad weights, and consistently, had smaller adipocytes and lower serum leptin levels. In addition, on HFD, WAT TNF-α mRNA levels and macrophage infiltration were reduced in PDE4B−/− mice. These results suggest that PDE4B deficiency can suppress many important WAT inflammatory responses induced by HFD treatment. Nevertheless, insulin sensitivity was not improved in these mice because no difference was observed in glucose and insulin levels or in glucose tolerance and insulin tolerance tests. Some previous studies showed that solely blocking TNF-α action did not improve insulin resistance in humans (59,60,61,62). Therefore, one possible reason for unchanged insulin sensitivity is that PDE4B deficiency only suppressed WAT TNF-α production but did not significantly affect other inflammatory markers.
Although PDE4s are cAMP-specific hydrolyzing enzymes, different PDE4s have distinct and overlapping functions in different cell types (33,34). For instance, in mouse embryonic fibroblasts, a recently study showed that PDE4D plays a major role in regulating cAMP accumulation (63). We isolated adipocytes from PDE4B−/− and WT mice and examined the role of PDE4B in adipocyte cAMP regulation. Although basal cAMP levels were comparable, PDE4B−/− adipocytes had increased cAMP levels 5 and 10 min after ISO treatment. This result suggests that PDE4B is involved in cAMP regulation in adipocytes. By using PDE4 inhibitors, previous reports showed that PDE4s are involved in adipocyte lipolysis (55,56,57,58). In these experiments, use of PDE4 inhibitors generally increased lipolysis rate. Because currently available PDE4 inhibitors are nonselective, e.g. rolipram inhibits all isoforms with comparable potencies, it is not clear which specific PDE4 isoform is mainly responsible for adipocyte lipolysis. Our results show that PDE4B−/− adipocytes had no significant change in lipolysis rate, suggesting that PDE4B is not a critical isoform mediating adipocyte lipolysis. To further examine distinct functions of different PDE4s, it would be necessary to examine cAMP regulation and lipolysis in adipocytes deficient for PDE4A, PDE4B, or PDE4D.
Our results provide additional evidence to suggest that PDE4B is involved in controlling locomotor activity. A previous report showed that PDE4B−/− mice had an exaggerated locomotor response to amphetamine (49). In our study, PDE4B−/− mice on HFD had higher locomotor activity than WT littermates. In WT mice, HFD treatment decreased locomotor activity, but PDE4 deficiency almost completely blunted this reduction. So far, at least four studies have been done examining phenotypes of PDE4B−/− mice, and the phenotypes appear to focus on the brain and the immune system. PDE4B−/− mice were shown to have anxiogenic-like behaviors (64) and decreased striatal dopamine and serotonin activities (49). In inflammatory cells, PDE4B was demonstrated to be essential for lipopolysaccharide-induced TNF-α production (40,41). Our results suggest a novel role of PDE4B in WAT. For instance, PDE4B deficiency can reduce adiposity and suppress some important obesity-induced inflammatory changes in WAT. Although the mechanism is not clear, increased locomotor activity of PDE4B−/− mice on HFD, presumably consequent to actions in the central nervous system, is likely to in part explain the reduced adiposity in these mice.
In summary, PDE4B−/− mice had reduced adiposity, with lower fat pad weights, smaller adipocytes, and lower serum leptin levels, on both chow and HFD. Importantly, PDE4B deficiency suppressed WAT TNF-α mRNA levels and macrophage infiltration induced by HFD. Furthermore, PDE4B−/− mice on HFD had increased locomotor activity. These results suggest unexpected roles for PDE4B in the regulation of energy balance via changes in locomotor responses to HFD and changes in adipose tissue inflammation in response to HFD. PDE4s have received wide attention as drug targets for treating chronic inflammatory diseases (65,66,67,68), and PDE4B-selective inhibitors may retain antiinflammatory properties with fewer side effects (41). Our results also suggest that therapeutic interventions with PDE4 inhibitors, especially those specific for PDE4B, may suppress obesity-induced inflammatory changes in WAT and could reduce adiposity through changes in physical activity that alter energy balance.
Supplementary Material
Acknowledgments
We thank Dr. Marco Conti for providing PDE4B-deficient mice. We also thank Katherine Kurgansky for technical assistance in MRI measurements and cell counting and Fenfen Liu for technical assistance in comprehensive laboratory animal monitoring system studies. We acknowledge helpful discussions with Drs. Hang Shi, Cheol Son, Nabila Aboulaich, and Dong Kong.
Footnotes
This work was supported by National Institutes of Health Grants DK R37 28082 (to J.S.F.) and RO1 DK56113 and PO1 DK56116 (to E.M.-F.). The comprehensive laboratory animal monitoring system support was provided by the physiology core of Grant PO1 DK56116 (to E.M.-F.).
Disclosure Summary: The authors have nothing to disclose.
First Published Online April 9, 2009
Abbreviations: H&E, Hematoxylin and eosin; HFD, high-fat diet; ISO, isoproterenol; MRI, magnetic resonance imaging; PDE4, phosphodiesterase 4; WAT, white adipose tissue; WT, wild type.
References
- Taubes G 1998 As obesity rates rise, experts struggle to explain why. Science 280:1367–1368 [DOI] [PubMed] [Google Scholar]
- Popkin BM, Doak CM 1998 The obesity epidemic is a worldwide phenomenon. Nutr Rev 56:106–114 [DOI] [PubMed] [Google Scholar]
- Allison DB, Fontaine KR, Manson JE, Stevens J, VanItallie TB 1999 Annual deaths attributable to obesity in the United States. JAMA 282:1530–1538 [DOI] [PubMed] [Google Scholar]
- Billington CJ, Epstein LH, Goodwin NJ, Hill JO, Pi-Sunyer JX, Rolls BJ, Stern J, Wadden TA, Weinsier RL, Wilson GT, Wing RR, Yanovski SZ, Hubbard VS, Hoofnagle JH, Everhart J, Harrison B 2000 Overweight, obesity, and health risk. National Task Force on the Prevention and Treatment of Obesity. Arch Intern Med 160:898–904 [DOI] [PubMed] [Google Scholar]
- Spiegelman BM, Flier JS 2001 Obesity and the regulation of energy balance. Cell 104:531–543 [DOI] [PubMed] [Google Scholar]
- Flier JS 2004 Obesity wars: molecular progress confronts an expanding epidemic. Cell 116:337–350 [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte Jr D, Seeley RJ, Baskin DG 2000 Central nervous system control of food intake. Nature 404:661–671 [DOI] [PubMed] [Google Scholar]
- Barsh GS, Farooqi IS, O'Rahilly S 2000 Genetics of body-weight regulation. Nature 404:644–651 [DOI] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM 2000 Molecular regulation of adipogenesis. Annu Rev Cell Dev Biol 16:145–171 [DOI] [PubMed] [Google Scholar]
- Ahima RS, Flier JS 2000 Adipose tissue as an endocrine organ. Trends Endocrinol Metab 11:327–332 [DOI] [PubMed] [Google Scholar]
- Mohamed-Ali V, Pinkney JH, Coppack SW 1998 Adipose tissue as an endocrine and paracrine organ. Int J Obes Relat Metab Disord 22:1145–1158 [DOI] [PubMed] [Google Scholar]
- Trayhurn P, Beattie JH 2001 Physiological role of adipose tissue: white adipose tissue as an endocrine and secretory organ. Proc Nutr Soc 60:329–339 [DOI] [PubMed] [Google Scholar]
- Fruhbeck G, Gomez-Ambrosi J, Muruzabal FJ, Burrell MA 2001 The adipocyte: a model for integration of endocrine and metabolic signaling in energy metabolism regulation. Am J Physiol Endocrinol Metab 280:E827–E847 [DOI] [PubMed] [Google Scholar]
- Flier JS 1995 The adipocyte: storage depot or node on the energy information superhighway? Cell 80:15–18 [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM 1993 Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science 259:87–91 [DOI] [PubMed] [Google Scholar]
- Feinstein R, Kanety H, Papa MZ, Lunenfeld B, Karasik A 1993 Tumor necrosis factor-α suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. J Biol Chem 268:26055–26058 [PubMed] [Google Scholar]
- Hotamisligil GS, Spiegelman BM 1994 Tumor necrosis factor α: a key component of the obesity-diabetes link. Diabetes 43:1271–1278 [DOI] [PubMed] [Google Scholar]
- Fried SK, Bunkin DA, Greenberg AS 1998 Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab 83:847–850 [DOI] [PubMed] [Google Scholar]
- Shimomura I, Funahashi T, Takahashi M, Maeda K, Kotani K, Nakamura T, Yamashita S, Miura M, Fukuda Y, Takemura K, Tokunaga K, Matsuzawa Y 1996 Enhanced expression of PAI-1 in visceral fat: possible contributor to vascular disease in obesity. Nat Med 2:800–803 [DOI] [PubMed] [Google Scholar]
- Yudkin JS, Stehouwer CD, Emeis JJ, Coppack SW 1999 C-reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arterioscler Thromb Vasc Biol 19:972–978 [DOI] [PubMed] [Google Scholar]
- Mohamed-Ali V, Goodrick S, Rawesh A, Katz DR, Miles JM, Yudkin JS, Klein S, Coppack SW 1997 Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-α, in vivo. J Clin Endocrinol Metab 82:4196–4200 [DOI] [PubMed] [Google Scholar]
- Lundgren CH, Brown SL, Nordt TK, Sobel BE, Fujii S 1996 Elaboration of type-1 plasminogen activator inhibitor from adipocytes. A potential pathogenetic link between obesity and cardiovascular disease. Circulation 93:106–110 [DOI] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H 2003 Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante Jr AW 2003 Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112:1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Goldfine AB 2006 Inflammation and insulin resistance. J Clin Invest 116:1793–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornier MA, Dabelea D, Hernandez TL, Lindstrom RC, Steig AJ, Stob NR, Van Pelt RE, Wang H, Eckel RH 2008 The metabolic syndrome. Endocr Rev 29:777–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandona P, Aljada A, Bandyopadhyay A 2004 Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol 25:4–7 [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS 2006 Inflammation and metabolic disorders. Nature 444:860–867 [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hotamisligil GS 2005 Inflammation, stress, and diabetes. J Clin Invest 115:1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel RH, Grundy SM, Zimmet PZ 2005 The metabolic syndrome. Lancet 365:1415–1428 [DOI] [PubMed] [Google Scholar]
- Pickup JC 2004 Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care 27:813–823 [DOI] [PubMed] [Google Scholar]
- Moller DE 2000 Potential role of TNF-α in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol Metab 11:212–217 [DOI] [PubMed] [Google Scholar]
- Houslay MD, Adams DR 2003 PDE4 cAMP phosphodiesterases: modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem J 370:1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti M, Richter W, Mehats C, Livera G, Park JY, Jin C 2003 Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J Biol Chem 278:5493–5496 [DOI] [PubMed] [Google Scholar]
- Houslay MD, Sullivan M, Bolger GB 1998 The multienzyme PDE4 cyclic adenosine monophosphate-specific phosphodiesterase family: intracellular targeting, regulation, and selective inhibition by compounds exerting anti-inflammatory and antidepressant actions. Adv Pharmacol 44:225–342 [DOI] [PubMed] [Google Scholar]
- Conti M, Jin SL 1999 The molecular biology of cyclic nucleotide phosphodiesterases. Prog Nucleic Acids Res Mol Biol 63:1–38 [DOI] [PubMed] [Google Scholar]
- Hatzelmann A, Schudt C 2001 Anti-inflammatory and immunomodulatory potential of the novel PDE4 inhibitor roflumilast in vitro. J Pharmacol Exp Ther 297:267–279 [PubMed] [Google Scholar]
- Houslay MD, Schafer P, Zhang KY 2005 Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today 10:1503–1519 [DOI] [PubMed] [Google Scholar]
- Teixeira MM, Gristwood RW, Cooper N, Hellewell PG 1997 Phosphodiesterase (PDE)4 inhibitors: anti-inflammatory drugs of the future? Trends Pharmacol Sci 18:164–171 [DOI] [PubMed] [Google Scholar]
- Jin SL, Conti M 2002 Induction of the cyclic nucleotide phosphodiesterase PDE4B is essential for LPS-activated TNF-α responses. Proc Natl Acad Sci USA 99:7628–7633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SL, Lan L, Zoudilova M, Conti M 2005 Specific role of phosphodiesterase 4B in lipopolysaccharide-induced signaling in mouse macrophages. J Immunol 175:1523–1531 [DOI] [PubMed] [Google Scholar]
- Jin SL, Richard FJ, Kuo WP, D'Ercole AJ, Conti M 1999 Impaired growth and fertility of cAMP-specific phosphodiesterase PDE4D-deficient mice. Proc Natl Acad Sci USA 96:11998–12003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honnor RC, Dhillon GS, Londos C 1985 cAMP-dependent protein kinase and lipolysis in rat adipocytes. I. Cell preparation, manipulation, and predictability in behavior. J Biol Chem 260:15122–15129 [PubMed] [Google Scholar]
- Rodbell M 1964 Metabolism of isolated fat cells. I. Effects of hormones on glucose metabolism and lipolysis. J Biol Chem 239:375–380 [PubMed] [Google Scholar]
- Cherry JA, Davis RL 1999 Cyclic AMP phosphodiesterases are localized in regions of the mouse brain associated with reinforcement, movement, and affect. J Comp Neurol 407:287–301 [PubMed] [Google Scholar]
- Iona S, Cuomo M, Bushnik T, Naro F, Sette C, Hess M, Shelton ER, Conti M 1998 Characterization of the rolipram-sensitive, cyclic AMP-specific phosphodiesterases: identification and differential expression of immunologically distinct forms in the rat brain. Mol Pharmacol 53:23–32 [DOI] [PubMed] [Google Scholar]
- Lobban M, Shakur Y, Beattie J, Houslay MD 1994 Identification of two splice variant forms of type-IVB cyclic AMP phosphodiesterase, DPD (rPDE-IVB1) and PDE-4 (rPDE-IVB2) in brain: selective localization in membrane and cytosolic compartments and differential expression in various brain regions. Biochem J 304(Pt 2):399–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Torres S, Miro X, Palacios JM, Cortes R, Puigdomenech P, Mengod G 2000 Phosphodiesterase type 4 isozymes expression in human brain examined by in situ hybridization histochemistry and[3H]rolipram binding autoradiography. Comparison with monkey and rat brain. J Chem Neuroanat 20:349–374 [DOI] [PubMed] [Google Scholar]
- Siuciak JA, McCarthy SA, Chapin DS, Martin AN 2008 Behavioral and neurochemical characterization of mice deficient in the phosphodiesterase-4B (PDE4B) enzyme. Psychopharmacology (Berl) 197:115–126 [DOI] [PubMed] [Google Scholar]
- Friedman JM, Halaas JL 1998 Leptin and the regulation of body weight in mammals. Nature 395:763–770 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM 1994 Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432 [DOI] [PubMed] [Google Scholar]
- Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y 1999 Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257:79–83 [DOI] [PubMed] [Google Scholar]
- Hu E, Liang P, Spiegelman BM 1996 AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem 271:10697–10703 [DOI] [PubMed] [Google Scholar]
- Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF 1995 A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 270:26746–26749 [DOI] [PubMed] [Google Scholar]
- Wang H, Edens NK 2007 mRNA expression and antilipolytic role of phosphodiesterase 4 in rat adipocytes in vitro. J Lipid Res 48:1099–1107 [DOI] [PubMed] [Google Scholar]
- Gronning LM, Baillie GS, Cederberg A, Lynch MJ, Houslay MD, Enerback S, Tasken K 2006 Reduced PDE4 expression and activity contributes to enhanced catecholamine-induced cAMP accumulation in adipocytes from FOXC2 transgenic mice. FEBS Lett 580:4126–4130 [DOI] [PubMed] [Google Scholar]
- Snyder PB, Esselstyn JM, Loughney K, Wolda SL, Florio VA 2005 The role of cyclic nucleotide phosphodiesterases in the regulation of adipocyte lipolysis. J Lipid Res 46:494–503 [DOI] [PubMed] [Google Scholar]
- Nakamura J, Okamura N, Kawakami Y 2004 Augmentation of lipolysis in adipocytes from fed rats, but not from starved rats, by inhibition of rolipram-sensitive phosphodiesterase 4. Arch Biochem Biophys 425:106–114 [DOI] [PubMed] [Google Scholar]
- Bernstein LE, Berry J, Kim S, Canavan B, Grinspoon SK 2006 Effects of etanercept in patients with the metabolic syndrome. Arch Intern Med 166:902–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofei F, Hurel S, Newkirk J, Sopwith M, Taylor R 1996 Effects of an engineered human anti-TNF-α antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes 45:881–885 [DOI] [PubMed] [Google Scholar]
- Paquot N, Castillo MJ, Lefebvre PJ, Scheen AJ 2000 No increased insulin sensitivity after a single intravenous administration of a recombinant human tumor necrosis factor receptor: Fc fusion protein in obese insulin-resistant patients. J Clin Endocrinol Metab 85:1316–1319 [DOI] [PubMed] [Google Scholar]
- Dominguez H, Storgaard H, Rask-Madsen C, Steffen Hermann T, Ihlemann N, Baunbjerg Nielsen D, Spohr C, Kober L, Vaag A, Torp-Pedersen C 2005 Metabolic and vascular effects of tumor necrosis factor-α blockade with etanercept in obese patients with type 2 diabetes. J Vasc Res 42:517–525 [DOI] [PubMed] [Google Scholar]
- Bruss MD, Richter W, Horner K, Jin SL, Conti M 2008 Critical role of PDE4D in β2-adrenoceptor-dependent cAMP signaling in mouse embryonic fibroblasts. J Biol Chem 283:22430–22442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HT, Huang Y, Masood A, Stolinski LR, Li Y, Zhang L, Dlaboga D, Jin SL, Conti M, O'Donnell JM 2008 Anxiogenic-like behavioral phenotype of mice deficient in phosphodiesterase 4B (PDE4B). Neuropsychopharmacology 33:1611–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Ducharme Y, Macdonald D, Robichaud A 2001 The next generation of PDE4 inhibitors. Curr Opin Chem Biol 5:432–438 [DOI] [PubMed] [Google Scholar]
- Dastidar SG, Rajagopal D, Ray A 2007 Therapeutic benefit of PDE4 inhibitors in inflammatory diseases. Curr Opin Investig Drugs 8:364–372 [PubMed] [Google Scholar]
- Lipworth BJ 2005 Phosphodiesterase-4 inhibitors for asthma and chronic obstructive pulmonary disease. Lancet 365:167–175 [DOI] [PubMed] [Google Scholar]
- Giembycz MA 2000 Phosphodiesterase 4 inhibitors and the treatment of asthma: where are we now and where do we go from here? Drugs 59:193–212 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.