

Abstract
Whereas hormone therapy is used for the treatment of menopausal symptoms, its efficacy in helping reduce the risk of other diseases such as Alzheimer’s disease has been questioned in view of the results of recent clinical trials that appeared inconsistent with numerous basic research studies that supported the beneficial effects of hormones. One possible explanation of this discrepancy may lie in the choice of hormone used. For example, we and others found that progesterone is neuroprotective whereas medroxyprogesterone acetate (MPA), the synthetic progestin used in hormone therapy, is not. Because our data suggest that progesterone-induced protection is associated with the induction of brain-derived neurotrophic factor (BDNF) levels and, importantly, can be blocked by inhibiting the neurotrophin signaling, we determined whether progesterone and medroxyprogesterone acetate differed in their ability to regulate BDNF levels in the explants of the cerebral cortex. We found that progesterone elicited an increase in both BDNF mRNA and protein levels, whereas medroxyprogesterone acetate did not. Furthermore, using both a pharmacological inhibitor of the progesterone receptor (PR) and PR knockout mice, we determined that the effects of progesterone were mediated by the classical PR. Our results underscore the fact that not all progestins have equivalent effects on the brain and suggest that the selection of the appropriate progestin may influence the success of hormone therapy formulations used in treating the menopause and/or reducing the risk for diseases associated with the postmenopausal period.
While progesterone is protective and induces an increase in brain-derived neurotrophic factor (BDNF) levels, the synthetic progestin, medroxyprogesterone acetate, is not protective and does not increase BDNF expression.
By 2010, the number of women between the ages of 45 and 64 yr will reach approximately 42 million, a marked increase from the value reported for 2000 (∼32 million) (U.S. Census Bureau. Projected population of the United States, by Age and Sex: 2000 to 2050, www.census.gov/ipc/www/usinterimproj/; Internet release date: March 18, 2004). This increasing number of peri- and postmenopausal women will consequently need to make decisions about the use of hormone therapy to treat not only their menopausal symptoms, but potentially, to maintain a healthy brain. Although numerous basic science studies, epidemiological studies, and some clinical trials have supported the potential benefit of hormone therapy in reducing the incidence of age-associated brain dysfunction (including reducing the risk for Alzheimer’s disease) (reviewed in Ref. 1), recent results, such as those from the Women’s Health Initiative Memory Study (2,3), have left the field unsettled as to the risk vs. reward aspect of hormone therapy, particularly for diseases of the brain including Alzheimer’s disease. Possible caveats, however, of the clinical trials included the possibility that the type of hormone (for example, progesterone vs. the synthetic hormone, medroxyprogesterone acetate) may have also influenced the outcomes (4,5). Our study focused on the differences in the neurobiology of two major progestins. Progesterone (P4) is the natural hormone produced in our body and medroxyprogesterone acetate (MPA), the most commonly used progestin in hormone therapy in the United States, is often used in conjunction with estrogen to reduce the risk of certain cancers (6,7).
We and others have shown progesterone is neuroprotective in various experimental models that mimic certain pathogenic aspects of brain dysfunction seen with the aging process. For example, progesterone pretreatment protected hippocampal neurons from toxicity associated with FeSO4− and amyloid-β (8). In addition, cerebral cortical explants and primary hippocampal neurons were protected from glutamate-induced cell death (9,10). Interestingly, the clinically used progestin, MPA, failed to protect hippocampal neurons against glutamate toxicity (10). Moreover, MPA attenuated estrogen-induced neuroprotection, whereas P4 did not (10).
The mechanisms underlying the differences in P4-induced neuroprotection and MPA’s negative effects on the nervous system are not completely understood. Because our previously published data suggested that brain-derived neurotrophic factor (BDNF) may be a relevant mediator of P4’s protective actions (9), we determined whether P4 and MPA differed in their ability to regulate BDNF.
The effects of P4 may be mediated by at least two classes of progesterone receptors (PRs), the classical PR and membrane PRs (11,12,13). The classical PR, like the estrogen receptor, is a nuclear transcription factor, acting through specific P4 response elements within the promoter region of target genes to regulate transcription (for review, see (Ref. 14) and is expressed in the cerebral cortex (15,16).
Candidates for putative membrane PRs include a single-transmembrane spanning receptor termed 25-Dx, whose neuroprotective function was associated with its influence on the maintenance of fluid homeostasis after traumatic brain injury (17). Another class of membrane PRs (α, β, and γ) (12,13) have a predicted seven-transmembrane spanning domain and, based on our data, is expressed in the cerebral cortex (data not shown). Given this multiplicity of receptor mediators of P4, we sought to determine which type of PR mediated the effect of P4 on BDNF. Collectively, the data underscore the fact that not all progestins are equivalent with respect to their effects on the brain and identify the classical PR as the relevant receptor involved in P4-induced BDNF expression.
Materials and Methods
Tissue culture
Organotypic explants were derived from about 360-μm-thick hemicoronal slices of postnatal d 3 (P3) frontal and cingulate cerebral cortex (day of birth = P1), obtained from pups born of C57BL/6, or PR knockout (PRKO) (18) and their wild-type counterpart mice and maintained as roller tube cultures (19) on rat tail collagen-coated/poly-l-lysine precoated glass coverslips, as previously described (20). The cultures were maintained in steroid-deficient and phenol red-free maintenance medium [25% heat inactivated horse serum (Sigma, St. Louis, MO), 22.5% Hanks’ balanced salt solution (Mediatech, Inc., Herndon, VA), 50% MEM (Sigma), 5.25 mg/ml of D(+)-glucose (Sigma), 2 mm l-glutamine (MediaTech), 50 μg/ml l-ascorbic acid, (Sigma)] supplemented with 2 nm 17-β estradiol (Steraloids, Newport, RI). Three hemicoronal slices were placed on each coverslip per Leighton tube. Antibiotics were not used.
All procedures that involved the use of animals were approved by the University of North Texas Health Science Center’s Institutional Animal Use and Care Committee.
Treatment of cultures
The cultures were maintained in vitro for 6 d as previously described (20). On the sixth day in vitro a 24-h hormone washout was performed, consisting of replacing the maintenance medium with media that did not contain exogenously added 17β-estradiol. On the seventh day in vitro, cultures were spiked with the appropriate treatment (see below) for the specified amount of time. Controls were also hormone deprived and sham (vehicle)-treated to account for any consequences of procedural manipulation of the explants.
P4 (4-pregnen-3, 20-dione; Sigma), MPA (6α-methyl-17α-hydroxy-progesterone acetate, Depo-provera; Sigma), and RU486 (mifepristone; Sigma) were dissolved in dimethylsulfoxide. Vehicle controls were performed in parallel such that control cultures were exposed to 0.1% dimethylsulfoxide. A stock (240 μm) of P4-BSA from Steraloids was dissolved in sterile water. Free (unconjugated) progesterone was removed from the P4-BSA solution using a 10-kDa cutoff Microcon cartridge (Millipore, Billerica, MA). The concentrations of l-glutamate used in the studies described here were based on concentration-response curves performed in cortical explants (data not shown) identifying 5–15 mm as optimal concentrations for inducing significant increases in lactate dehydrogenase (LDH) release, an index of cell viability. The concentrations of progesterone and MPA chosen for the studies presented herein were based on prior studies conducted in our laboratory, in the same experimental model, in which a concentration of 100 nm progesterone resulted in optimal protection (9). Furthermore, given that MPA has been reported to have an approximately 3-fold higher affinity for the PR (21), we did not assess concentrations higher than the highest concentration of P4 tested (1 μm).
Assessment of cell viability
P4’s protective effects were evaluated based on its ability to reduce l-glutamate-induced LDH release into the culture media. P4 was applied 24 h before treatment with l-glutamic acid (Sigma) for 6 h. This duration of glutamate ensures minimal degradation of the released LDH in the conditioned media [t1/2 of LDH in solution has been reported to be ∼9 h (CytoTox-ONE homogeneous membrane integrity assay technical bulletin no. TB306; Promega, Madison, WI)]. A fluorometric assay (CytoTox-One homogenous membrane integrity assay kit; Promega) was used for the measurement of LDH released from damaged or dying cells. The assay itself is based on the ability of LDH to catalyze the conversion of the assay substrate, resazurin, to the fluorescent product, resorufin. Briefly, after aliquoting 100 μl of conditioned media into a 96-well plate, the substrate was added and incubated for 10 min at room temperature. After termination of the enzymatic reaction, resulting fluorescence was measured [560 nm (excitation)/590 nm (emission)] using a Viktor3 ELISA plate reader (PerkinElmer, Boston, MA). Relative fluorescent units were normalized to the amount of protein associated with the explant culture (assessed using Bio-Rad DC protein assay kit; Bio-Rad Laboratories, Inc., Hercules, CA) from which the conditioned media was derived. These values were expressed as a percentage of vehicle-treated control.
Western blot analysis
Harvested tissue samples were dissected and immediately immersed into lysis buffer containing protease and phosphatase inhibitors as described previously (22). After homogenization, samples were centrifuged at 99,000 × g for 15 min at 4 C, and the resulting supernatants were evaluated for total protein concentrations using the Bio-Rad DC (Bio-Rad Laboratories) protein assay kit [based on the method of Lowry et al. (23)]. Equal amount of protein (90 μg) from each sample was electrophoresed on SDS-PAGE (10% polyacrylamide gel) followed by transfer to polyvinylidene difluoride membrane (0.22 μm pore size; Bio-Rad Laboratories). The membrane was blocked overnight with 3% BSA [dissolved in 0.2% Tween 20 containing Tri buffered saline solution (TBS-T)] before application of the primary antibody. The membrane was immunoblotted using commercially available polyclonal antibodies to the phosphorylated form of ERK1/2 (rabbit antiphospho-ERK1/2, 1:1000; Millipore) or total ERK (goat anti-ERK1, goat anti-ERK2, 1:500 each; Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Antibody binding to the membrane was detected using a secondary antibody (donkey antirabbit or rabbit antigoat IgG) conjugated to horseradish peroxidase (1:10,000; Pierce Chemical Co., Rockford, IL) and visualized after incubation with enzyme-linked chemiluminescence (SuperSignal West Pico chemiluminescent substrate; Pierce) using the UVP image analysis system (UVP, Upland, CA).
BDNF ELISA
An ELISA was used to detect and quantify total cellular BDNF levels (Promega). A 96-well Nunc MaxiSorp surface polystyrene flat-bottom immunoplate was precoated with an anti-BDNF monoclonal antibody [diluted 1:1000 in coating buffer (25 mm sodium bicarbonate and 25 mm sodium carbonate [pH 9.7])]. After rinsing off unbound antibody with TBS-T buffer [20 mm Tris-HCl (pH 7.6), 150 mm NaCl and 0.05% (vol/vol) Tween 20] and blocking the plate to minimize nonspecific binding, 100 μg of sample lysate (in a final volume of 100 μl) or volume of appropriate BDNF standard (ranging in concentration from 7.8 to 500 pg/ml) were added. After five washes with TBS-T, the captured BDNF was then incubated with the polyclonal antihuman BDNF antibody. The amount of specifically bound polyclonal antibody was then detected through the use of the anti-IgY-horseradish peroxidase tertiary antibody, which when exposed to the chromogenic substrate [TMB (3,3′,5,5′-tetramethylbenzidine) one solution; Promega], changes color in proportion to the amount of BDNF present in the sample. The color intensity was quantified by measuring the absorbance at 450 nm using a Viktor3 ELISA plate reader (PerkinElmer). Only values that were within the linear range of the standard curve were considered valid. BDNF levels were normalized to protein content within each sample and reported as percent of vehicle control. Control experiments demonstrated that our percent recovery with this ELISA was calculated to be 72.3 ± 3.2%.
Genotyping
DNA from tail snips was extracted using Sigma’s REDExtract-N-Amp tissue PCR kit. PCR was done using REDExtract-N-Amp PCR mix (Sigma-Aldrich). PCR was performed by denaturing the DNA at 94 C for 3 min, followed by 30 cycles of amplification: 94 C for 1 min, 55.7 C for 2 min, 72 C for 3 min, and a final extension step at 72 C for 10 min. The following PR-specific primers were used: P1 (5′-TAG ACA GTG TCT TAG ACT CGT TGT TG-3′), P3 (5′-GAT GGG CAC ATG GAT GAA ATC-3′), and L1 (5′-CTT CAC CCA CCG GTA CCT TAC GCT TC-3′). The PCR product was separated by 1.3% agarose gel electrophoresis and visualized with ethidium bromide staining using a 2-Log DNA ladder (New England Biolabs, Ipswitch, MA). A 590-bp DNA band indicated WT (P1/P3 primers), a 110-bp band indicated PRKO (P1/LI primers), and visualization of both bands identified heterozygous animals.
RNA isolation and cDNA synthesis
Total RNA was extracted from explant cultures and deoxyribonuclease treated using the RNeasy lipid kit (QIAGEN, Valencia, CA) according to manufacturer’s instructions. RNA concentrations of extracted RNA were calculated from the absorbance at 260 nm. The quality of RNA was assessed by absorption at 260 and 280 nm (A260 to A280 ratios of 1.9–2.10 were considered acceptable) and by assessing the integrity of the 18S and 28S rRNA bands after ethidium bromide staining of samples separated on agarose gels. Total RNA (1.5 μg) was reverse transcribed into cDNA in a total volume of 50 μl using the high-capacity DNA archive kit (Applied Biosystems, Foster City, CA) according to manufacturer’s instructions.
Primers and probes for quantitative real-time RT-PCR
PCR primers and probes for the target gene, BDNF, and the endogenous control, 18S rRNA, were purchased as Assays-On-Demand (Applied Biosystems). The assays were supplied as 20× mix of PCR primers (900 nm) and TaqMan probes (200 nm). The BDNF assay (Mm00432069_m1) contained 6-carboxy-fluorescein phosphoramidite dye label at the 5′ end of the probes and minor groove binder and nonfluorescent quencher at the 3′ end of the probes. The 18S rRNA assays (4319413E) contained VIC-labeled probes (Applied Biosystems). The assays are optimized for use on ABI prism sequence detection system (Applied Biosystems) using the default machine settings.
Quantitative real-time RT-PCR
The reaction mixture containing water, 2× quantitative PCR master mix (Eurogentec, Herstal, Belgium), and 20× Assay-On-Demand for BDNF was prepared. A separate reaction mixture was prepared for the endogenous control, 18S rRNA. The reaction mixture was aliquoted in a 96-well plate and cDNA (11 ng of RNA converted to cDNA) was added to give a final volume of 30 μl. Each sample was analyzed in triplicate. Two nontemplate controls (ribonuclease free water) were included on each plate for reaction mixture containing BDNF and 18S rRNA assays. Amplification and detection were performed using the ABI 7300 sequence detection system (Applied Biosystems) with the following profile: 2 min hold at 50 C [UNG (uracil-N-glycosylase) activation], 10 min hold at 95 C, followed by 40 cycles of 15 sec at 95 C (denaturation), and 1 min at 60 C (annealing and extension). Sequence detection software 1.3 (Applied Biosystems) was used for data analysis.
The comparative cycle threshold (CT) method (2−ΔΔCT) was used to calculate the relative changes in target gene expression (24). In the comparative CT method, the amount of target, normalized to an endogenous control (18S) and relative to a calibrator (untreated control), is given by the 2−ΔΔCT equation. Quantity is expressed relative to a calibrator sample that is used as the basis for comparative results. Therefore, the calibrator was the baseline (control) sample and all other treatment groups were expressed as an n fold (or percent) difference relative to the calibrator. The average and sd of 2−ΔΔCT was calculated for the values from at least three independent experiments, and the relative amount of target gene expression for each sample was plotted in bar graphs using GraphPad Prism 4 software (GraphPad, San Diego, CA).
Statistical analysis
Data from at least three independent experiments were subjected to ANOVA, followed by a single degree of freedom F test within the main effect of ANOVA (analysis performed using Systat; Systat, Inc., San Jose, CA). For statistical consideration of BDNF mRNA, single sample t test was used to compare the effects of progesterone against vehicle-treated control. The data are presented as bar graphs depicting the means ± sem, using GraphPad Prism 4 software.
All procedures involving animals were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee here at UNT Health Science Center.
Results
P4, but not MPA, protects against glutamate-induced cytotoxicity
We have previously shown that P4 protects organotypic explants from glutamate-induced toxicity (9). Because MPA, and not progesterone, is more commonly used in hormone therapy regimens, we determined whether MPA is also able to protect against glutamate-induced toxicity. Cerebral cortical explants were pretreated with P4 (100 nm) or MPA (100 nm) for 24 h before treatment with l-glutamate (15 mm, 6 h). Pretreatment of cerebral cortical cultures with P4 significantly reduced glutamate-induced LDH release (Fig. 1A), whereas an equimolar concentration of MPA did not (Fig. 1B).
Figure 1.
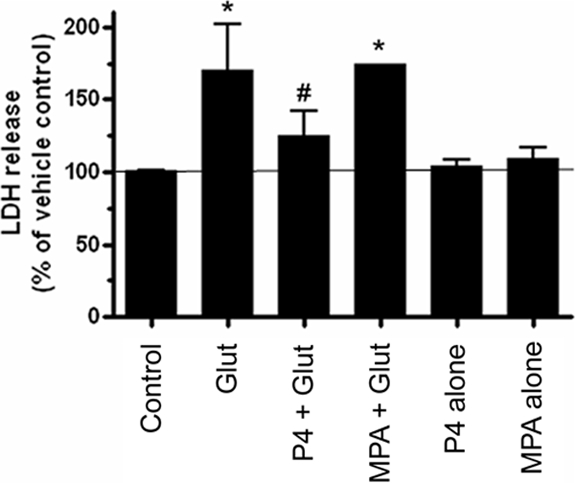
P4 but not MPA protects against glutamate-induced cytotoxicity. Cerebral cortical explants were pretreated with P4 (100 nm) for 24 h before the administration of l-glutamate (Glut; 15 mm for 6 h). P4 prevented glutamate-induced LDH release, insofar as these values were not statistically different from vehicle-treated control. MPA, however, failed to prevent glutamate-induced LDH release. LDH release is expressed as a percentage of that seen in the vehicle-treated control (set at 100%), with the latter represented by the solid horizontal line. The graph shown represents data from three independent experiments. Data are presented as mean ± sem. *, P < 0.01 relative to vehicle-treated control; #, P < 0.05 vs. MPA + Glut group.
Differential regulation of both BDNF mRNA and protein by MPA and P4
Recently we have shown that P4’s protective effects against glutamate-induced excitotoxic/oxidative insult are associated with an induction of BDNF (9). Furthermore, the cytoprotection was blocked by inhibiting neurotrophin signaling (9). In this study, we hypothesized that the differences in the neuroprotective capabilities of MPA and P4 were based on the differences in their ability to regulate BDNF mRNA and protein levels. As we had seen previously, P4, at a concentration that protected against glutamate-induced excitotoxic/oxidative insult (100 nm), elicited an increase in the expression of BDNF mRNA, whereas MPA, failed to do so. Using real-time RT-PCR, our results showed that P4 (100 nm) induces and approximately 79% increase in BDNF mRNA expression, whereas treatment with MPA (100 nm) resulted in a 42% decrease in BDNF mRNA levels (Fig. 2).
Figure 2.
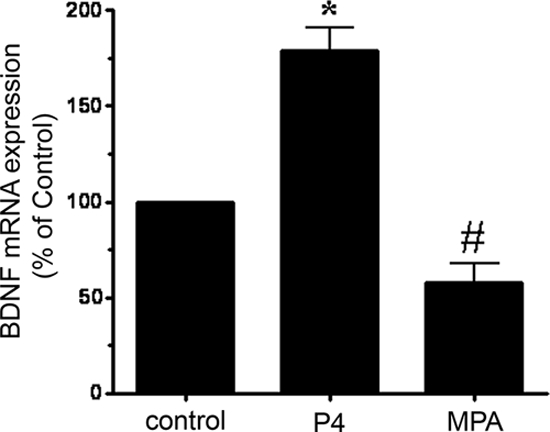
Differential regulation of BDNF mRNA by P4 and MPA. BDNF mRNA was assessed in cerebral cortical explants treated with P4 (100 nm) or MPA (100 nm) for 18 h. Using real-time RT-PCR, we found that P4 but not MPA elicited an increase in BDNF mRNA. Data are presented as mean ± sem of five independent experiments for P4 treatments and four independent experiments for MPA treatments. *, P ≤ 0.01 vs. control; #, P < 0.05 vs. control.
We also assessed the levels of BDNF protein and found that progesterone induced an increase in cellular BDNF levels in a concentration-dependent manner (Fig. 3A). In contrast, MPA (10 nm, 100 nm, 1 μm) failed to increase BDNF protein levels. In fact, there was a net reduction in the total BDNF content in the cultures treated with 10 nm (74% decrease) and 100 nm (59% decrease) MPA (Fig. 3B).
Figure 3.
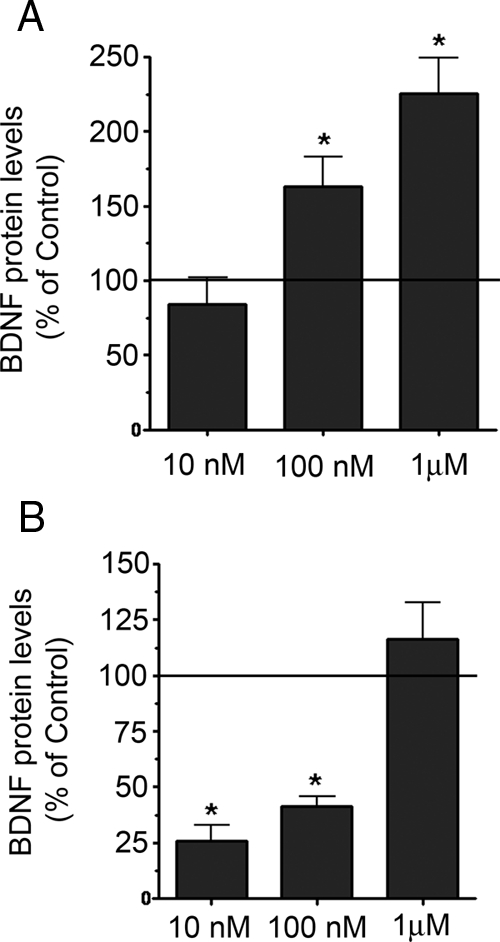
Differential regulation of total cellular BDNF content by MPA and P4. BDNF protein levels were assessed in cerebral cortical explants treated with P4 or MPA for 18 h using an ELISA. A, P4 induced an increase in BDNF expression at concentrations of 100 nm and 1 μm. B, MPA does not induce an increase in BDNF expression. There was a decrease in total cellular BDNF in cultures treated with 10 and 100 nm MPA. BDNF levels are presented as a percentage of the vehicle-treated control, represented by the solid horizontal line. Data are represented as mean ± sem. *, P < 0.01 relative to vehicle-treated control.
P4 regulates BDNF expression through the classical PR
Given that mRNA for both the classical PR and the membrane PR are expressed in cerebral cortical explants (data not shown), we wanted to evaluate whether the induction of BDNF mRNA and protein by P4 was mediated by the classical (intracellular PR) or the membrane PR.
To determine whether the effect of P4 on BDNF induction was dependent on the classical PR, cerebral cortical explants were pretreated with the pharmacological antagonist, RU486 (1 μm), before the treatment with 100 nm P4. Real-time RT-PCR revealed that RU486 prevented the increase in BDNF mRNA expression (Fig. 4A) induced by progesterone. RU486 also blocks the effect of progesterone on BDNF protein to an equivalent extent (Fig. 4B).
Figure 4.
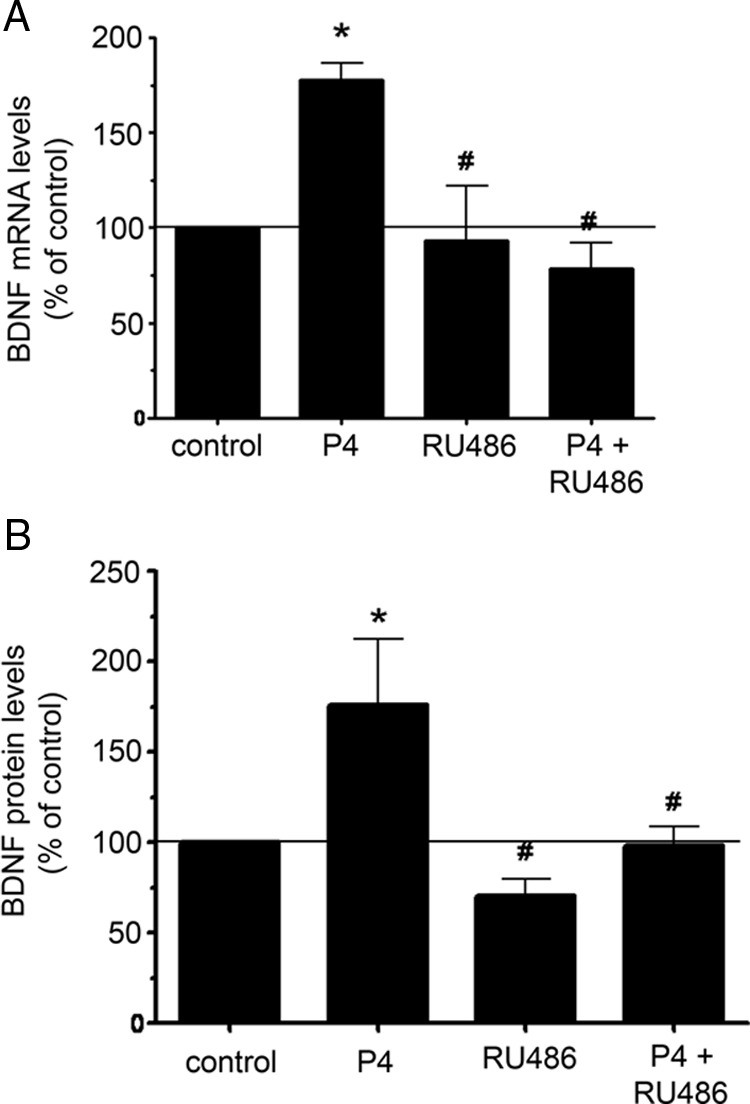
P4-induced BDNF expression is inhibited by RU486. Cerebral cortical explants were treated with P4 (100 nm) and the PR antagonist, RU486 (1 μm). BDNF mRNA was assessed in cerebral cortical explants after 18 h. A, Using real-time RT-PCR, we found that P4 induced an increase in BDNF mRNA, whereas RU486 inhibited the P4-induced response. The graph shown represents data from seven independent experiments for samples treated with P4 or P4 in presence of RU486 and five independent experiments for RU486 treatments. *, P < 0.0001 relative to vehicle-treated control; #, P < 0.0001 relative to P4-treated samples. B, BDNF protein levels were assessed in cerebral cortical explants using an ELISA. BDNF levels are presented as a percentage of the vehicle-treated control, represented by the solid horizontal line. The graph shown represents data from four independent experiments for samples treated with P4. There were five independent experiments for samples treated with P4 in presence of RU486 and four independent experiments for RU486 treatments. *, P < 0.05 relative to vehicle-treated control; #, P < 0.05 relative to P4-treated samples.
To further define the role of the classical PR in progesterone’s effects on BDNF, we assessed the effect of progesterone in cerebral cortical cultures derived from the PRKO mouse (18). The results showed that P4 failed to elicit an increase in BDNF mRNA in the cerebral cortical cultures derived from PRKO mice but induced a robust increase in BDNF levels in cultures derived from their wild-type counterparts (Fig. 5). The enhanced response of the wild-type cultures to P4 may be due to differences in background strain of the mice from which the cortical cultures were derived (C57BL/6 vs. the dual background C57BL/6 × 129 SvEv strains associated with the PRKO and their wild-type counterparts). Such pharmacogenetic influences on the effectiveness of P4 warrant further study. We also found that the membrane-impermeable progesterone, P4-BSA (which is expected to have limited, if any, interaction with the intracellular PR), failed to induce an increase in BDNF expression (Fig. 6A) relative to the BSA-alone-treated control, whereas it did elicit an increase in ERK phosphorylation (Fig. 6B).
Figure 5.
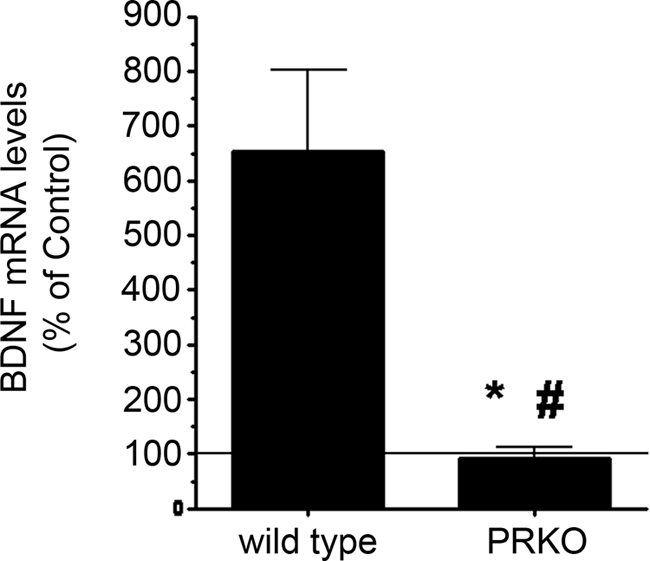
P4 fails to elicit an increase in BDNF expression in PRKO Mice. Cerebral cortical explants from P3 mice were treated for 18 h with P4. Using real-time RT-PCR, we found that P4 induced an increase in BDNF mRNA in cerebral cortical explants from wild-type but not PRKO mice. BDNF levels within the cultures derived from each genotype are presented as a percentage of their respective vehicle-treated controls (depicted by the solid horizontal line). Data are presented as mean ± sem from four independent experiments. *, P < 0.05; #, P < 0.01.
Figure 6.
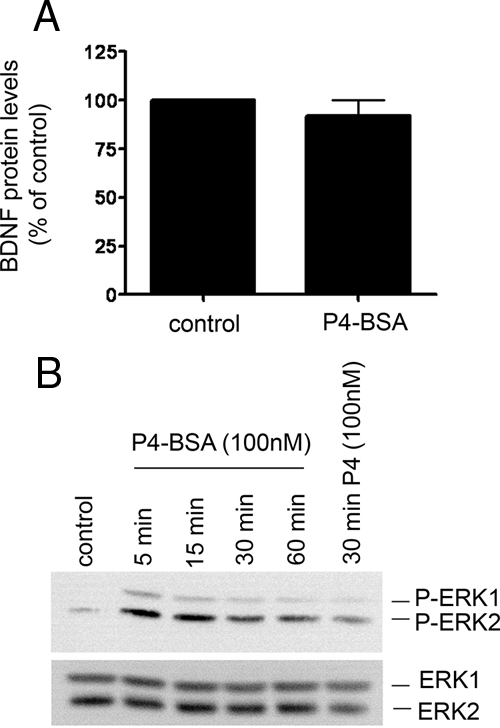
P4, but not its membrane impermeant form (P4-BSA), induces an increase in BDNF protein expression. A, BDNF mRNA was assessed in cerebral cortical explants treated with P4-BSA (100 nm) or an equimolar concentration of BSA alone (control) for 18 h. We found that P4-BSA did not induce statistically significant increase in BDNF cellular content. Data are presented as mean ± sem of three independent experiments. B, Western blot analysis revealed that P4-BSA elicited a time-dependent increase in ERK phosphorylation (P-ERK; upper panel), achieving maximal levels between 5 and 15 min and declining thereafter. P4 treatment (30 min) was used as the positive control and yielded ERK phosphorylation levels similar to that observed with P4-BSA at the same time point. In separate control experiments, we found that equimolar concentrations of BSA alone did not elicit ERK phosphorylation at similar time points (data not shown). The lower panel represents reprobing the phospho-blot for total ERK protein to ensure equal loading across lanes.
Discussion
Our data describe an important difference between the effects of P4 and the clinically used progestin, MPA, on neuroprotection and suggest that differences in the regulation of BDNF may underlie, at least in part, the differences in neuroprotective effects of these two progestins. The differences in the neuroprotective effects of P4 and MPA reported here are consistent with that described by Nilsen and Brinton (10), who showed that P4, and not MPA, protected primary hippocampal neurons from glutamate-induced toxicity.
One mechanism underlying the protective effects of progesterone is its ability to elicit ERK phosphorylation. For example, we have previously shown that in organotypic explants of the cerebral cortex, the protective effects of P4 against glutamate-induced cytotoxicity were dependent on the ERK/MAPK signaling pathway (9). This requirement of the ERK/MAPK pathway in P4’s protective effects was also noted in hippocampal neurons (25). Interestingly, we and others have found that MPA, despite its inability to exert neuroprotective effects, does elicit ERK phosphorylation. Further exploration by Nilsen and Brinton (25), however, revealed that although the temporal pattern of ERK phosphorylation was similar between P4 and MPA, only P4 promoted the nuclear translocation of ERK. Thus, one possible reason for the difference in the ability of P4 and MPA to protect against cytotoxicity may be related to the subcellular localization of the phosphorylated ERK. Therefore, the phosphorylation of ERK is necessary but not sufficient for cytoprotection.
Given our previous finding that the protective effect of progesterone on the cerebral cortex was associated with an induction of both the mRNA and protein levels of BDNF and that inhibition of neurotrophin signaling attenuated P4’s protective effects (9), we determined whether the difference in progesterone’s and MPA’s protective effects may also have been attributed to differences in the regulation of BDNF. In this study, we found that P4 elicited an increase in both BDNF mRNA and protein levels whereas MPA at equimolar concentrations failed to do so. Interestingly, MPA suppressed BDNF expression, both at the level of mRNA and protein. These data suggest that MPA may be depleting the cellular stores of BDNF and as such could prevent the protective effects of other protective factors whose mechanism of action may involve the synthesis of neurotrophins. Consistent with this hypothesis is the observation that MPA antagonizes the protective effects of estradiol (10), a hormone with well-documented effects on BDNF levels (26,27). Thus, limiting the ability of other factors such as estradiol to induce the expression of BDNF may limit and/or antagonize the protective effects of estradiol.
P4 can exert its effects on cell function through several mechanisms. The dogmatic view is that P4 binds to, and activates a member of the nuclear hormone receptor family of transcription factors, resulting in the regulation of gene transcription. More recently, however, alternative receptors such as the recently described membrane progesterone receptors have been implicated in mediating the effects of P4 (12,13). To determine which receptor was involved in the effects of P4 on the regulation of BDNF levels, we used complementary pharmacological and genetic approaches. Our results support the role of the classical PR in mediating the effects of P4 on BDNF. We also determined that the membrane-impermeant P4-BSA, which would minimally interact with the intracellular PR, failed to elicit an increase in BDNF mRNA or protein levels. However, P4-BSA did elicit ERK phosphorylation, suggesting that P4’s effects on ERK phosphorylation may be mediated by a membrane PR. Moreover, given the importance of both ERK activation and the induction of BDNF in P4’s ability to promote cell viability, these data suggest that the membrane-mediated mechanism (presumably the membrane PR) and the classical PR act in a complementary manner to promote neuroprotection. Ongoing studies in our laboratory will define the role of the putative membrane receptor in P4e’s protective effects in greater detail.
In Alzheimer’s disease, decreased expression of BDNF mRNA levels in the cortex and hippocampus have been reported (28). In addition, Fahnestock et al. (29) described a 50% decrease in the BDNF mRNA levels in the nucleus basalis of Meynert when compared with age-matched controls. Furthermore, an up to 40% reduction of pro-BDNF protein levels (the protein precursor of the neurotrophic mature BDNF) was noted within the parietal cortex (29,30). Thus, the ability of P4 to induce BDNF may have therapeutic value for treating and preventing such neurodegenerative diseases as Alzheimer’s disease. However, as our data suggest, the choice of progestin may be critical in terms of its therapeutic efficacy. In fact, our study highlights the fact that biology of P4 and MPA are different. Even though P4 and the synthetic progestin, MPA (which is most commonly used progestin in hormone therapy), may be equally effective at reducing the uterotrophic effects of unopposed estrogen treatment, their effects on the brain are clearly not identical.
Collectively, our findings reported herein identify a mechanism through which P4 protects cortical neurons and, further, may explain some of the negative effects associated with MPA treatment. Future studies that further delineate differences in the neurobiology of such related progestins may offer insight into the selection of the appropriate progestin used in hormone therapy formulations.
Footnotes
This work was supported by National Institutes of Health Grants AG022550, AG023330, and AG027956; a grant from the Alzheimer’s Association; and a gift from the Garvey Texas Foundation (to M.S.).
Disclosure Summary: The authors have nothing to disclose.
First Published Online March 26, 2009
Abbreviations: BDNF, Brain-derived neurotrophic factor; CT, cycle threshold; LDH, lactate dehydrogenase; MPA, medroxyprogesterone acetate; P, postnatal day; P4, progesterone; P4-BSA, membrane impermeant form of P4; PR, P4 receptor; PRKO, PR knockout; TBS-T, Tween-20 containing Tris-buffered saline solution.
References
- Simpkins JW, Singh M 2008 More than a decade of estrogen neuroprotection. Alzheimers Dement 4:S131–S136 [DOI] [PubMed] [Google Scholar]
- Shumaker SA, Legault C, Rapp SR, Thal L, Wallace RB, Ockene JK, Hendrix SL, Jones 3rd BN, Assaf AR, Jackson RD, Kotchen JM, Wassertheil-Smoller S, Wactawski-Wende J 2003 Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 289:2651–2662 [DOI] [PubMed] [Google Scholar]
- Shumaker SA, Legault C, Kuller L, Rapp SR, Thal L, Lane DS, Fillit H, Stefanick ML, Hendrix SL, Lewis CE, Masaki K, Coker LH 2004 Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA 291:2947–2958 [DOI] [PubMed] [Google Scholar]
- Singh M, Sumien N, Kyser C, Simpkins JW 2008 Estrogens and progesterone as neuroprotectants: what animal models teach us. Front Biosci 13:1083–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins JW, Singh M 2004 Consortium for the Assessment of Research on Progestins and Estrogens (CARPE), Fort Worth, Texas, August 1–3, 2003. J Womens Health (Larchmt) 13:1165–1168 [DOI] [PubMed] [Google Scholar]
- Hirvonen E 1996 Progestins. Maturitas 23(Suppl):S13–S18 [DOI] [PubMed] [Google Scholar]
- Gambrell Jr RD 1986 The role of hormones in the etiology and prevention of endometrial cancer. Clin Obstet Gynaecol 13:695–723 [PubMed] [Google Scholar]
- Goodman Y, Bruce AJ, Cheng B, Mattson MP 1996 Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid β-peptide toxicity in hippocampal neurons. J Neurochem 66:1836–1844 [DOI] [PubMed] [Google Scholar]
- Kaur P, Jodhka PK, Underwood WA, Bowles CA, de Fiebre NC, de Fiebre CM, Singh M 2007 Progesterone increases brain-derived neuroptrophic factor expression and protects against glutamate toxicity in a mitogen-activated protein kinase- and phosphoinositide-3 kinase-dependent manner in cerebral cortical explants. J Neurosci Res 85:2441–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen J, Brinton RD 2002 Impact of progestins on estrogen-induced neuroprotection: synergy by progesterone and 19-norprogesterone and antagonism by medroxyprogesterone acetate. Endocrinology 143:205–212 [DOI] [PubMed] [Google Scholar]
- Krebs CJ, Jarvis ED, Chan J, Lydon JP, Ogawa S, Pfaff DW 2000 A membrane-associated progesterone-binding protein, 25-Dx, is regulated by progesterone in brain regions involved in female reproductive behaviors. Proc Natl Acad Sci USA 97:12816–12821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Bond J, Thomas P 2003 Identification, classification, and partial characterization of genes in humans and other vertebrates homologous to a fish membrane progestin receptor. Proc Natl Acad Sci USA 100:2237–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Rice CD, Pang Y, Pace M, Thomas P 2003 Cloning, expression, and characterization of a membrane progestin receptor and evidence it is an intermediary in meiotic maturation of fish oocytes. Proc Natl Acad Sci USA 100:2231–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conneely OM, Lydon JP 2000 Progesterone receptors in reproduction: functional impact of the A and B isoforms. Steroids 65:571–577 [DOI] [PubMed] [Google Scholar]
- MacLusky NJ, McEwen BS 1980 Progestin receptors in rat brain: distribution and properties of cytoplasmic progestin-binding sites. Endocrinology 106:192–202 [DOI] [PubMed] [Google Scholar]
- Kato J, Hirata S, Nozawa A, Yamada-Mouri N 1994 Gene expression of progesterone receptor isoforms in the rat brain. Horm Behav 28:454–463 [DOI] [PubMed] [Google Scholar]
- Meffre D, Delespierre B, Gouezou M, Leclerc P, Vinson GP, Schumacher M, Stein DG, Guennoun R 2005 The membrane-associated progesterone-binding protein 25-Dx is expressed in brain regions involved in water homeostasis and is up-regulated after traumatic brain injury. J Neurochem 93:1314–1326 [DOI] [PubMed] [Google Scholar]
- Lydon JP, DeMayo FJ, Funk CR, Mani SK, Hughes AR, Montgomery Jr CA, Shyamala G, Conneely OM, O’Malley BW 1995 Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev 9:2266–2278 [DOI] [PubMed] [Google Scholar]
- Gahwiler B 1981 Organotypic monolayer cultures of nervous tissue. J Neurosci Methods 4:329–342 [DOI] [PubMed] [Google Scholar]
- Singh M, Setalo Jr G, Guan X, Frail DE, Toran-Allerand CD 2000 Estrogen-induced activation of the mitogen-activated protein kinase cascade in the cerebral cortex of estrogen receptor-alpha knock-out mice. J Neurosci 20:1694–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitruk-Ware R 2004 Pharmacological profile of progestins. Maturitas 47:277–283 [DOI] [PubMed] [Google Scholar]
- Singh M, Setalo Jr G, Guan X, Warren M, Toran-Allerand CD 1999 Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: convergence of estrogen and neurotrophin signaling pathways. J Neurosci 19:1179–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ 1951 Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275 [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD 2001 Analysis of relative gene expression data using real-time quantitative PCR and the 2[−ΔΔC(T)] method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- Nilsen J, Brinton RD 2003 Divergent impact of progesterone and medroxyprogesterone acetate (Provera) on nuclear mitogen-activated protein kinase signaling. Proc Natl Acad Sci USA 100:10506–10511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Meyer EM, Simpkins JW 1995 The effect of ovariectomy and estradiol replacement on brain-derived neurotrophic factor messenger ribonucleic acid expression in cortical and hippocampal brain regions of female Sprague-Dawley rats. Endocrinology 136:2320–2324 [DOI] [PubMed] [Google Scholar]
- Sohrabji F, Miranda RC, Toran-Allerand CD 1995 Identification of a putative estrogen response element in the gene encoding brain-derived neurotrophic factor. Proc Natl Acad Sci USA 92:11110–11114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger RM, Schnarr J, Henry P, Castelo VT, Fahnestock M 2000 Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: decreased levels in Alzheimer’s disease. Brain Res Mol Brain Res 76:347–354 [DOI] [PubMed] [Google Scholar]
- Fahnestock M, Garzon D, Holsinger RM, Michalski B 2002 Neurotrophic factors and Alzheimer’s disease: are we focusing on the wrong molecule? J Neural Transm Suppl 241–252 [DOI] [PubMed] [Google Scholar]
- Michalski B, Fahnestock M 2003 Pro-brain-derived neurotrophic factor is decreased in parietal cortex in Alzheimer’s disease. Brain Res Mol Brain Res 111:148–154 [DOI] [PubMed] [Google Scholar]