

1. Introduction and Scope
Leaving groups have been defined as that part of a substrate that becomes cleaved by the action of a nucleophile.1 The IUPAC definition specifies a leaving group as a molecular fragment (charged or uncharged) that becomes “detached from an atom in what is considered to be the residual or main part of the substrate” in a given reaction.2 A leaving group that carries away an electron pair is called a nucleofuge.3 However, these terms are not synonymous since a nucleofuge may be a functional group that simply receives an electron pair during a nucleophilic attack without itself being cleaved away from the substrate.
The term “leaving group” seems to have entered the chemical literature4 in the early 1950’s although its mechanistic role was certainly understood much earlier. For example, in a 1951 paper detailing the study of nucleophilic displacement reactions of neopentyl tosylates, the term “leaving group” does not appear anywhere in the abstract or discussion.5 In this 1951 paper, the leaving group concept was expressed by naming the actual departing group in each example under consideration (such as halide, tosylate, etc.). In the following year and in the same journal, a paper describing a study of solvolysis reactions of norbornyl halide and arylsulfonate systems uses the term “leaving group” in the paper’s abstract. However, in the body of the six-page paper the term is used only once along with “departing ionizing group”.6 In a short 1953 communication on the solvolysis of various alkyl halides (< 600 words), the term “leaving group” appears three times.7 By the following year, the term seems to have gained general acceptance, appearing for the first time in the title of a paper.8 By the mid 1960’s the term “leaving group” appeared in the titles of over 25 papers and was used in over a 125 abstracts. Some undergraduate textbooks of organic chemistry also adopted the term by this time period.9
Leaving groups are ubiquitous in organic chemistry, playing a key role in a wide range of reactions, including nucleophilic substitution (aliphatic and aromatic), electrophilic substitution, and elimination reactions. A survey of 135 named organic reactions widely utilized in modern preparative organic chemistry10 reveals that 38 reactions involve heterolytic nucleofugal leaving groups at some stage of their mechanism. If this selection of named reactions is representative, then it could be inferred that leaving groups play an important role in as many as 25% of all organic reactions.
Despite the wide variety of available leaving groups, there is still a need to improve their performance in terms of selectivity, reaction rates, scalability, environmental compatibility, atom economy, and other parameters. Thus, research on leaving groups continues to be an area of fruitful endeavor. As described in the subsequent pages, variations on traditional leaving group motifs have been the principal means of developing groups with improved performance. This review therefore begins with a discussion of advances in sulfonate and carboxylate-based leaving groups. The use of chiral leaving groups in asymmetric synthesis is not a new area. However, an improved mechanistic understanding of asymmetric induction in a variety of reactions in recent years has inspired the design of new chiral leaving groups that are, in many cases, modifications of traditional groups such as sulfonates and carboxylates. These and other chiral leaving groups will be discussed in some detail. The field of organometallic chemistry has offered powerful new reagents and catalysts for small-molecule synthesis over the years. It is then no surprise that new organometallic leaving groups such as carboranes have recently emerged. Heterocyclic leaving groups have long been part of the synthesis chemist’s repertoire and new groups arise from this class of compounds with regularity. Many researchers look to biological systems for inspiration in the design of improved reagents. This trend is also seen in the development of nucleophile assisting leaving groups (NALGs) which stabilize a reaction transition state through chelating interaction with the incoming nucleophile. Several recent examples of this relatively new class of leaving group will be described in this review.
To our knowledge, the present article is the first review of heterolytic nucleofugal leaving groups broadly defined. Given the ubiquity of leaving groups in organic chemistry, it would seem nearly impossible to review this topic exhaustively except as an extensive monograph. Indeed, reviews in this area have concentrated on specific leaving groups types such as sulfonates,11 pyridines,12 2-thiopyridylcarbonates,13 and carbanionic14 to name a few. Thus, the present review will extend to research findings primarily from the last 15 years, although a limited number of leaving group technologies from earlier periods that have not been covered in previous reviews will also be discussed. Also, some leaving groups that have been briefly reviewed as part of a larger work have been revisited here in greater detail. New reactions involving existing leaving groups will not be discussed here. Instead, the focus of this review will be on the mechanisms and performance characteristics of relatively new leaving groups.
To further reduce the scope of this report to manageable proportions, heterolytic nucleofugal leaving groups involved in the vast field of acyl coupling chemistry has been excluded, especially since this area is reviewed on a routine basis.15 Also, reports describing the adaptation of existing leaving groups to the solid-phase will not be discussed here since this area has been reviewed in extensive surveys of solid phase organic synthesis techniques.16 Finally, new leaving groups in purely inorganic systems17 and in biological applications18 have been omitted here as these are somewhat tangential to the emphasis of this review on new leaving groups that facilitate the synthesis of organic small molecules.
2. Sulfonate and Carboxylate Leaving Groups
Sulfonyl chlorides such as tosyl and mesyl have found widespread use as reagents to convert alcohols into substrates for nucleophilic reactions. Significantly more reactive trifluoromethanesulfonate (triflate) leaving groups were subsequently introduced which exhibited several orders of magnitude increased reactivity.19 To gain some perspective, the relative reactivity of the most common leaving groups has been estimated to be substituted benzoates (1) < halides (~104–106) < sulfonates (~1010–1012) < perfluoroalkane sulfonates (~1015–1016).20 In the past 25 years, sulfonate and carboxylate leaving groups with intermediate reactivity have been prepared, often involving the introduction of electron-withdrawing substituents. In addition, new sulfonate leaving groups have been designed to suppress unwanted sulfur substitution and sulfene formation. Some of these sulfonate leaving group developments have been recently reviewed.11
2.1 Benzenedisulfonylimides
The conversion of alcohols to a sulfonate ester is a well established strategy to create substrates capable of undergoing nucleophilic substitution whereas the related conversion of amines to sulfonamides does not lead to a similar result. However, the addition of a second sulfonyl group, as in the case of benzenedisulfonylimides, provides a suitable leaving group. Using the bis-sulfonyl technique, amines have been converted to alcohols and azides with inversion of configuration.21 This conversion has been demonstrated with 1-phenyl and 1-cyclohexyl ethylamine (scheme 1). The reaction of either primary amine with benzene-1,2-disulfonyl chloride led to the corresponding benzenedisulfonylimide intermediates 1a and 1b in somewhat low yields. The reaction of 1a with KNO2 led to alcohol 2 in 83% yield and with 70% inversion of configuration. Using NaN3, azide product 3 was obtained with essentially complete inversion although the yield was fairy modest (53%) even after a reaction time of 4 days (scheme 1). Earlier studies involving dimesylimides and dinosylimides led to similarly high percent inversions but suffered from lower yields in both the imide formation and subsequent nucleophilic substitution steps.22 Disulfonylimide leaving group technology has also been used in the synthesis of aryl ethers starting from naphthyl disulfonylimide 4.23 The reaction of 4 with sodium phenoxide gave inverted aryl ether product 5 in 53% yield (scheme 1).
Scheme 1.

Conversion of primary amine to alcohols, azides, and aryl ethers via disulfonylimide intermediates.
2.2 Vinylogous sulfonates
Vinylogous sulfonates have been developed to serve as improved leaving groups for a variety of palladium-catalyzed allylic substitution reactions.24 Using a cis-phosphino-1,3-oxazine ligand, a variety of alkyl-substituted β-phenylcinnamyl substrates 6 are converted to malonate products 7 via Pd(II) catalysis. The stereochemical outcome was rationalized using a palladium allyl transition state involving the chiral oxazine ligand (scheme 2). This allylation reaction exhibits high enantioselectivity (95%) and increased catalyst turnover rates relative to traditional acetate leaving groups (scheme 2).25 However, this asymmetric method has only been demonstrated with β-phenylcinnamyl alcohol derivatives 6 with straight chain substituents at the carbinol position.
Scheme 2.

Vinylogous sulphonate leaving groups for asymmetric allylic alkylation.
2.3 Alkyl sulfonyls
Using 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene (10) as a palladium ligand, sulfonyl purine nucleoside 8 was coupled to a p-methoxyphenyl group to give product 9 in 81% yield via a Suzuki reaction (scheme 3).26 The sulfonyl purine derivative was obtained by an SNAr displacement of a halopurine precursor followed by a high-yielding oxidation. This methodology opens a new avenue for modification of purines at the C6 position.
Scheme 3.

Alkyl sulfonyl leaving group in Suzuki coupling reaction to give substituted nucleosides
2.4 Imidazole-1-sulfonates (imidazylates)
Imidazole-1-sulfonates (imidazylates) have been used with success in SN2 reactions involving mono and oligosaccharide substrates at primary and secondary positions.27 The imidazole-1-sulfonate can be prepared from an alcohol by reaction with sulfuryl chloride to form the chlorosulfate ester followed by reaction with imidazole or by reaction with N′,N-sulfuryldiimidazole.28 This leaving group has been applied primarily to the synthesis of mono and oligosaccharides. For example, diisopropylidene galactopyranose (11) was converted to derivative 12 with an imidazyl group at the 6-position. Imidazyl 12 was then converted to the corresponding 6-fluoro, iodo, and azido product 13 in good yields (scheme 4). An imidazylate intermediate has also been methylated (remote activation29) at the imidazole nitrogen leading to increased nucleofugacity (entry 3, scheme 4). The imidazylate leaving group has also been utilized in oligosaccharide derivatization.30
Scheme 4.

Galactopyranose derivatization via imidazylates.
More recently, the reaction of an imidazylate of a 4-deoxyarabinoside with tetrabutylammonium cyanide (prepared in situ from Bu4NF and TMSCN) gave the cyanide product in an exceptionally high yield (81%).31 Highly regioselective elimination reactions involving imidazylate intermediates have been reported. As a representative example, pyranose 14 was converted to the corresponding imidazylate intermediate which underwent an elimination reaction in situ to form enopyranoside 15 in 95% yield (scheme 5).32
Scheme 5.
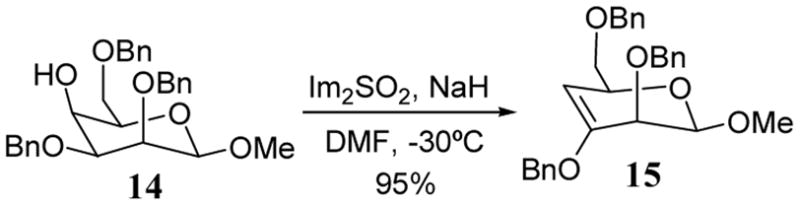
Highly regioselective imidazylate elimination to give an enopyranoside.
2.5 Pyridine and quinoline sulfonates
The secondary esters of 2-pyridine and 8-quinoline sulfonic acid decompose cleanly at moderate temperatures to give olefins in high yields (scheme 6).33 A variety of substrates were successfully converted to the olefin using this pyrolysis method including cyclohexanol sulfonate ester 16 (scheme 6), exo-norbornol, menthol, neomenthol, and 2-octanol. Negligible differences in product yield were observed between the pyridine and quinoline sulfonate esters. Product studies indicated that the pyrolysis did not proceed via a concerted E2-type mechanism. Instead, an E1 mechanism was invoked in which the reaction was facilitated by the basic nitrogen of the quinoline sulfonate leaving group.
Scheme 6.
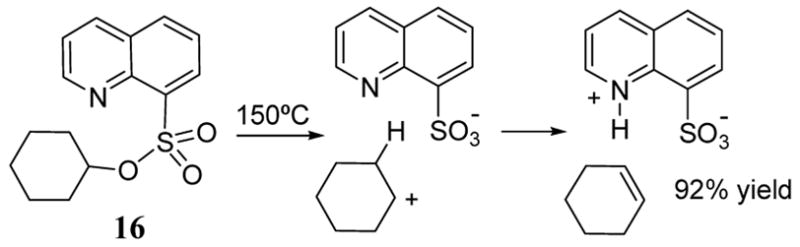
Pyrolysis of 8-quinoline sulfonate 16 to give cyclohexene.
2.6 Fluorocarboxylates
Perfluorobutyrate, conveniently prepared from a precursor perfluoro alcohol, has been shown to possess a reactivity similar to that of halides in solvolysis reactions.20 A product and isotope distribution study for the reaction of 1-adamantly perfluorobutyrate (17) in 80:20 ethanol/H218O demonstrated exclusive alkyl-O cleavage to give the ethyl ether and 18O-alcohol products in a 94% combined yield (scheme 7). The 1- and 3-homoadamantyl perfluorobutyrate analogs of 17 exhibited faster solvolysis rates due to the increased flexibility of the hydrocarbon skeleton.
Scheme 7.
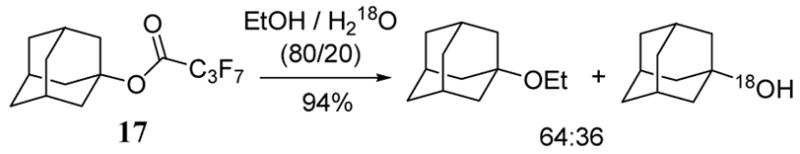
Perfluorobutyrate leaving group in solvolysis.
Quinoline-2,4(1H,3H)-diones give the Perkow reaction product or lead to enol ether depending on the nature of the carboxyl leaving group at the 3-position.34 The use of the trifluoroacetate leaving group in substrate 18 led to the Perkow enol phosphite product 19 upon treatment with triethyl phosphite (scheme 8). With the novel fluoroiodoacetyl leaving group, enol ether 20 was obtained in 68% yield.
Scheme 8.
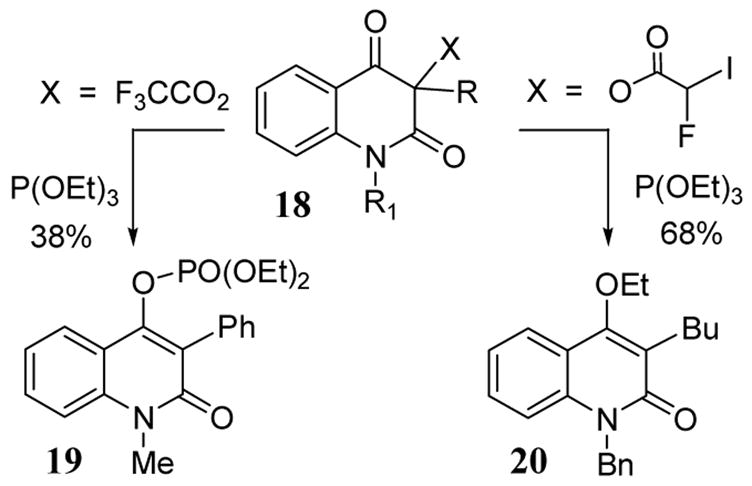
Perkow reaction involving fluorocarboxylates.
2.7 Alkynoate
A novel Hg(OTf)2 catalyzed glycosylation procedure has been developed using alkynoic acid residues as the leaving group under mild reaction conditions and efficient catalytic turnover.35 This leaving group is remotely activated in the presence of Hg(II) which complexes the alkyne moiety such as in 21 (scheme 9). Intermediate 21 underwent endocyclization with the mercury/alkyne complex leading to oxonium cation 22 and to methylene lactone 23 (regenerating the mercury catalyst). Addition of alcohols to intermediate 21 then gave glycosyl products 24. This procedure was particularly efficient for the glycosylation of hindered alcohols such as menthol, adamantanol and fenchol (scheme 9). Due to its remote activation mechanism, this alkynoate leaving group can also be categorized as an activation-deactivation strategy which is discussed in section 6 of this report.
Scheme 9.

Alkynoate leaving group for glycosylation.
2.8 Vinylogous carboxylates and carbonates
Glycosylation using vinylogous carboxylates and carbonates (enol ethers) as leaving groups have been described.36 The reaction of tetra-O-benzyl glucopyranose (25) with 2 equiv. of methyl-2-butynoate in the presence of a catalytic amount of tri-n-butylphosphine gave corresponding glycosyl donor 26 (scheme 10). In this synthesis, glycosyl donor 26 was obtained exclusively in the E-configuration due to the steric bulk imposed by the pyranose moiety during the formation of the enol ether. Treatment of donor 26 with alcohol 27 at room temperature gave disaccharide 28 in 82% yield (α/β = 9:2). A large excess of TMSOTf (9–12 eq) was required to obtain an optimal yield and anomeric ratio. With a smaller amount of TMSOTf (1 eq) at room temperature, the coupling yield to give 28 was reduced to 19%.
Scheme 10.

Synthesis of disaccharide 28 using a vinylogous carbonate leaving group.
3. Chiral Leaving Groups
Methods for asymmetric synthesis have been classified into four types, each based either on the chirality of the substrate, auxiliary, added reagent, or catalyst.37 Although research to develop new and more efficient chiral catalysts currently appears to dominate the field of asymmetric methodology,38 the development of new chiral auxiliary-based methods continues. The conversion of a prochiral substrate to a chiral center using a chiral leaving group provides an attractive alternative to traditional chiral auxiliary-based methods. Chiral leaving groups such as menthoxy have been used in the highly efficient syntheses of planar chiral binapthyl compounds, an area extensively reviewed.39 Importantly, chiral leaving groups do not require a removal step which is a common drawback in many chiral auxiliary methods.
3.1 BINOL derivatives
A method for face-selective alkylation of prochiral olefins using BINOL derivatives as chiral leaving groups under acidic conditions has been developed.40 In this strategy, a Lewis acid-activated C–O bond (as in 29) is cleaved at the moment of electrophilic addition releasing the BINOL leaving group (scheme 11). Lewis acid/LG complex 29 can be thought of as a cationic synthon possessing a chiral environment analogous to Lewis acid-assisted chiral Brønsted acid (LBA) used in enantioselective protonation reactions.41
Scheme 11.

Lewis acid-activated alkene addition reaction involving chiral leaving group LG*.
Analogous to the LBA method, asymmetric methods for alkoxymethylation of silyl enol ethers using a non-racemic BINOL leaving group has been developed. For example, the reaction of trimethylsilyl enol ether 30 with BINOL derivative 31 in the presence of SnCl4 led to product 32 in 91% yield and 75% ee (scheme 12). This result was an improvement over an earlier reported asymmetric hydroxymethylation reaction.42
Scheme 12.

Enantioselective alkoxymethylation of enol ether 30 with BINOL derivative 31 using SnCl4.
The observed stereochemical outcome of the alkoxymethylation reaction can be explained in terms of an acyclic extended transition state similar to one proposed by Noyori.43 NMR studies indicate that the BINOL leaving group in 31 coordinates with SnCl4 at the aryl ether oxygens (scheme 13).40 The introduction of silyl enol ether 30 is thought to give transition state 33 which can be represented using a Newman-type diagram (scheme 13). As indicated in the scheme, transition state 33 is stabilized by a π-π attractive interaction between phenyl group of 30 and one of the napthyl groups of BINOL 31. The other possible approach of enol ether 30 to the 31a complex would give transition state 34 involving a destabilizing steric repulsion between the R and the phenyl group of 30 (scheme 13).
Scheme 13.

Proposed extended transition states for enantioselective alkoxymethylation reactions.
The chiral naphthol leaving group of 31 has also been utilized in enantioselective Prins-type reactions with trisubstituted alkenes in the presence of SnCl4. Rapid additions of SEM (CH2OCH2CH2SiMe3) were observed with several alkenes with selectivity for the less substituted carbon of the double bond. Chlorinated side products were also observed. These side products could be converted to the SEM-alkene on treatment with collidine. In the case of 2-methyl-2-pentene, electrophilic SEM addition occurred at the 3-postion giving desired allyl SEM product 35 and chloro product 36 in quantitative yield. Subsequent treatment of the mixture with base gave 85% yield of 35 in 81% ee (scheme 14).40
Scheme 14.

Enantioselective Prins-type reaction of 2-methyl-2-pentene with 31.
The first biomimetic asymmetric synthesis of (+)-limonene was accomplished in 1983 (up to 77% ee) using (R)-BINOL as a leaving group in an intramolecular cyclization of a mononeryl ether with bulky organoaluminium reagents.44 More recently, improved enantioselectivities have been realized using BINOL derivatives activated by SnCl4 (scheme 15). Thus neryl-BINOL derivative 37 was converted to (+)-limonene (38) and α-terpinyl chloride (39) in 91% yield as a 4:5 mixture.40 The transformation was highly enantioselective (93% ee) in chlorinated solvents such as CH2Cl2 and propyl chloride. Heating the mixture of 38 and 39 in 2,4,6-collidine under reflux conditions converted chloride 38 to limonene in 71% yield (scheme 15).
Scheme 15.

Enantioselective cyclization of BINOL derivative 36 to give limonene (37).
3.2 Chiral amines
Chiral amine leaving groups derived from proline have been used to direct cuprate additions to cycloalkanones 42. The treatment of 42 with n-Bu2CuLi·LiBr and added salts (LiBr or ZnBr2) led to non-racemic 3-substituted 2-exo-methylenecycloalkanones 43. In most cases, reaction yields were between 80–90% with good enantioselectivities observed (scheme 16).45 Enones 42 were conveniently prepared from the reaction of α-(nitromethyl) enones 40 with (S)-(+)-(methoxymethyl) pyrrolidine (41) in acetonitrile (scheme 16).46 The use of ZnBr2 as an additive led to lower enantioselectivities especially in the case of cyclopentanone (scheme 16, entry 2).
Scheme 16.

Synthesis of non-racemic 3-butyl-2-exo-methylenealkanones.
A transition-state model to explain the enantioselectivity for the selective Bu2CuLi additions has been proposed (scheme 17).45 Chiral enone 42 initially forms weak tridentate chelation complex 44 with Bu2CuLi by coordination of three heteroatoms to the lithium, followed by additional d-π * complex formation between the copper atom and the conjugated enone moiety. This chelation d-π * complexation occurs at the Si face, since the Re face is shielded by the extruding pyrrolidine ring. Added ZnBr2 gave complex 45 which led to lower enantioselectivities (scheme 16, entries 2, 4, and 6) by impeding Li+-directed chelation π-complexation between Bu2CuLi and 42 leading to fast 1,4-addition via a kinetic or nonchelated d-π * complex due to the strong Lewis acidity of ZnBr2.
Scheme 17.

Transition-State models of Bu2CuLi addition to 42 with LiBr and ZnBr2 additives.
Proline-derived leaving groups have been used to induce asymmetry in addition-elimination reactions of chiral nitroenamines.47 A class of compounds with unique reactivity,48 nitroenamines have been prepared as chiral reagents through an amine exchange reaction between morpholino enamines 46 and amines 47 to give chiral enamines 48 in 90% yields with exclusive E configuration (scheme 18).49 Nitroenamines 48 were reacted with zinc enolates of α-methyl substituted lactone 49 in THF at −78°C to give adduct 50 containing a chiral quaternary carbon in high enantioselectivity and yield (scheme 18).50 The zinc countercation was chosen since initial reports indicated that this metal led to superior enantioselectivities and chemical yields relative to analogous Li+ and Cu+ enolates.51 Extensive optimization indicated that four equivalents of zinc enolate were essential for high enantioselectivities. The enantioselectivity depended on the bulkiness of R in the chiral nitroenamines, namely, the order was Ph > Et > Me, which indicated that increase in bulk of the chiral auxiliary increased the enantioselectivity of the reaction. The effects of substituents at the 1-position (R2) were also recently explored using an achiral nitroenamine.52
Scheme 18.

Synthesis of chiral nitroenamines 48 and asymmetric addition of α-methyl substituted lactone enolates 49.
A possible transition state model to explain the enantioselective product outcome and the role of multiple equivalents the zinc enolate has been proposed (scheme 19). Two equivalents of zinc enolate were consumed to form complex 51 by coordination with nitroenamine. Zinc enolate can then attack 51 from topside or underside. Attack of 51 from the topside requires two equivalents of zinc enolate. However, the steric bulk of the R groups (phenyl as shown in scheme 19) inhibit zinc enolate approach from the topside. Although Re face attack of complex 51 requires a third zinc enolate, the approach of the nucleophilic entails less steric impedance.
Scheme 19.
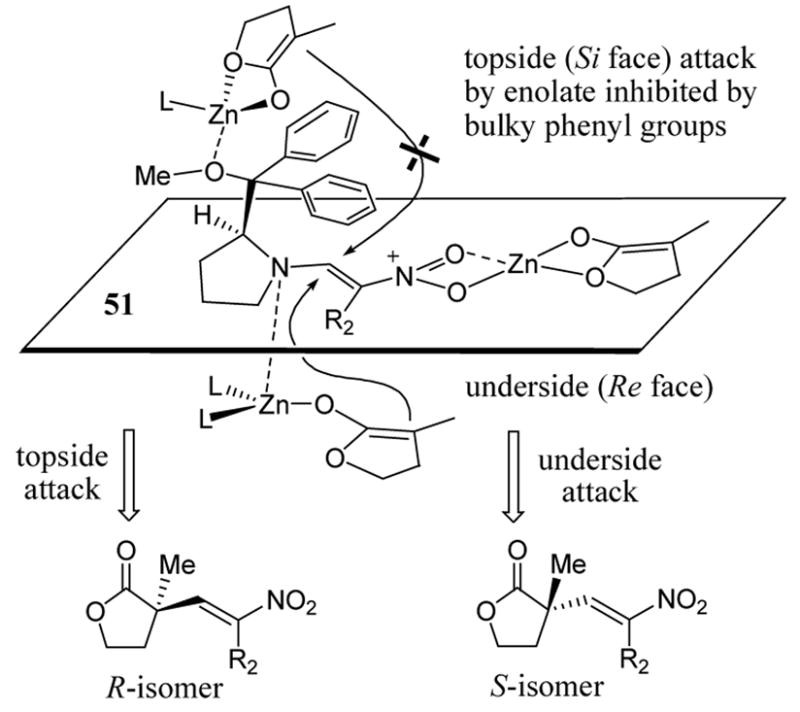
Transition state model rationalizing enantioselective formation of nitro-olefination product.
Asymmetric nitro-olefinations have been utilized in the efficient synthesis of a variety of indole alkaloid natural products containing quaternary carbon stereocenters. These syntheses have been previously reviewed.47,53 A recent total synthesis of spirotryprostatin B is illustrative of asymmetric nitro-olefination synthetic approaches (scheme 20).54 Nitro-olefination of an indole enolate gave intermediate 52 in 97% ee. Synthetic manipulation of the highly versatile vinyl nitro group gave advanced intermediate 53 after several steps. The remaining key transformations to obtain spirotryprostatin B involved the introduction of proline as a peptidic linkage to 53 followed by heteroatom substitution at the allylic position (scheme 20).
Scheme 20.

Application of nitro-olefination in the total synthesis of spirotryprostatin B.
More recently, a Baylis-Hillman type reaction has been developed to give non-racemic propargyl ethers 56 using stoichiometric quinidine as a chiral leaving group (scheme 21). Propargyl alcohol was added to bromomethyl enoates 54 via quinidinium intermediates 55 to give adducts 56 in modest enantioselectivities (25–40% ee) and yields (32–47%).55
Scheme 21.

Enantioselective synthesis of propargyl ethers 56 using quinidine as a chiral amine leaving group.
3.3 Chiral Sulfonates
The methyl ester of N-diphenylmethylene glycine (58) has been lithiated with LDA and treated with non-racemic furanose-derived methylating agent 57 to give alanine in 52% yield and 40% ee after hydrolysis (scheme 22).56
Scheme 22.

Enantioselective alkylation of protected glycine using a chiral sulfate leaving group.
Chiral camphor-10-sulfonate (casylate) leaving groups have been studied in solvolysis reactions.57 The solvolysis rate of the two diastereomers formed from D-2-octyl L-casylate and from L-2-octyl L-casylate was measured. These studies revealed a difference in the rate arguing for an SN2-type transition state. The approach of the ethanol nucleophile was thought to be influenced in part due to van der Waals interactions between the chiral leaving group and the octanyl substrate as in proposed transition state 59 (scheme 23). Reactions proceeding through a solvent-separated ion pair mechanism as in the case of a cholesterol D- and L-casylates gave nearly identical rates of solvolysis in aqueous ethanol.58 In neither the octyl nor the cholesterol casylate solvolysis reactions was the enantioselectivity measured although it was anticipated to be low.
Scheme 23.
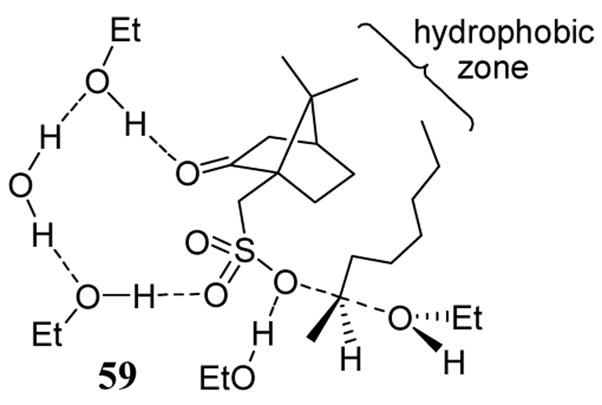
Proposed low energy transition state for the ethanolysis of the camphor-10-sulfonate of 2-octanol.
3.4 Chiral carboxylates
The use of chiral carbamates as leaving groups in allylic substitution is an area of active investigation and has been recently reviewed.59 However, analogous reactions with chiral carboxylates have been little explored. Enantioselective copper-mediated SN2′ substitution reactions involving ortho-diphenylphosphanylferrocene carboxylate (o-DPPF) as chiral leaving groups have been recently reported (scheme 24).60 DPPF, which is conveniently prepared,61 was initially developed as a catalyst-directing group in rhodium based hydroformulation reactions.62 The high enantioselectivity of product 61 (95%) observed in the addition of copper catalyst to 60 was rationalized on the basis of a strong phosphine-copper liganding interaction in transition state 62 (scheme 24). Interestingly, the use of a chiral ferrocene thiol as a copper ligand gave product 61 in only 64% ee when reacted with the precursor allylic acetate.63 In this case a nearly stoichiometric amount of the ferrocene thiol ligand was required to achieve a reasonable enantioselectivity.
Scheme 24.

Enantio- and regioselective addition of n-BuMgBr to o-DPPF ester 60.
4. Organometallic Leaving Groups
Most leaving groups contain a heteroatom or halogen atom at the point of cleavage from the substrate. A general strategy for improving the nucleofugacity of such leaving groups is to incorporate an electron withdrawing functional group as part of the departing moiety. Organometallic leaving groups offer the possibility of improved nucleofugacity through redox processes not available to second-row heteroatoms typically found in leaving groups. For example, the phenyliodonio leaving group exhibits a nucleofugacity 106 times greater than the triflate group allowing for SN2 substitutions on vinyl substrates.64 Surveys of nucleophilic reactions involving this leaving group have recently been published.65
4.1 Triarylbismuth
In the presence of a bulky amine base, methyltriphenylbismuthonium tetrafluoroborate (63) reacts with benzyl alcohol to give the corresponding methyl ether in 95% yield (scheme 25).66 Kinetic studies of this methylation reaction revealed a two-fold rate enhancement relative to the analogous transformation involving methyl triflate, a powerful leaving group. These studies indicate that triphenylbismuth is an exceptionally good leaving group. Other weak nucleophiles have also been successfully methylated with 63 including benzene sulfinate, benzoate, DMF, and thioacetamide (scheme 25).
Scheme 25.

Reaction of bismuthonium 63 with benzyl alcohol and synthesis of other methylation reaction products.
The excellent nucleofugal properties of triarylbismuth have also been utilized in the direct alkenylation of β-dicarbonyl and phenol compounds in the presence of 1,1,3,3-tetramethylguanidine (TMG) (scheme 26).67 For example, 2-oxocyclohexanecarboxylate (64) reacted with tritolylbismuthonium 65 to give vinylated product 66 in 88% yield. The reaction is thought to proceed via a pentacoordinate bismuth intermediate such as 67. Under the same conditions, ethyl oxobutanoate and 2,6-dimethyl phenol substrates led to vinylation products 68 and 69, respectively (scheme 26). The use of hypervalent bismuth reagents in the α-arylation and vinylation of silyl enol ethers68 and enones69 are also known. Related vinylation reactions with alkenyllead triacetates have also been reported.70
Scheme 26.

Reaction of alkenyltriarylbismuthonium salt 65 with 2-oxocyclohexanecarboxylate (64) to give vinylated product 66.
4.2 Carboranes
A new class of potent alkylating agents based on carborane leaving group (CB11Me5Br6)− has been recently developed. Carborane anions are among the least nucleophilic anions presently known. They also offer improved leaving group characteristics over the better known fluoroantimonates in that they are non-oxidizing and are not a source of fluorides which may participate in unwanted nucleophilic side reactions. In solution, the carborane reagents exist as equilibrating isomers with the alkyl group at the 7–11 or 12 bromide position of the CB11 icosahedral anion (scheme 27). A crystal structure analysis revealed that the alkyl-carborane bond is covalent.71
Scheme 27.
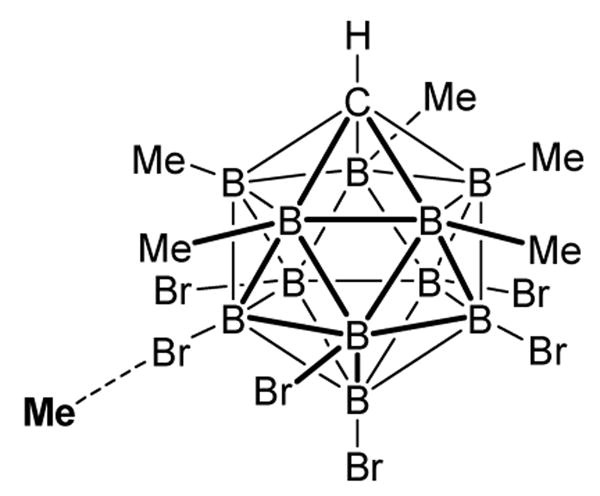
7-isomer of Me(CB11Me5Br6)
In the case of Me(CHB5Me5Br6) (70), the high electrophilic reactivity of this reagent has been illustrated in three methylation reactions71 that do not occur with methyl triflate, a traditional and powerful methylating agent (scheme 28). Benzene was methylated using stoichiometric amounts of 70 to give the toluenium ion. Remarkably, carborane 70 also reacted with several linear and branched alkanes containing four to six carbons to form tertiary carbocations at or below room temperature via hydride abstraction. Highly electron deficient phosphorus compounds such as 71 were methylated in the presence of carborane 70 in liquid SO2 while remaining unreactive toward neat boiling methyl triflate.
Scheme 28.

Methylation reactions involving carborane 70.
5. Heterocyclic Leaving Groups
If leaving groups employed in activated ester reactions for amide synthesis are excluded, pyridines are among the most widely-used heterocyclic leaving groups in organic synthesis, an area extensively explored and reviewed elsewhere.12 However, a limited number of other heterocyclic leaving groups have appeared in the intervening years involving more specialized applications.
5.1 Benzoisothiazole-3-ones
Mercaptobenzoates containing a 1,2-benzoisothiazole-3-one at the sulfur position are useful substrates for nucleophilic attack of anilines and primary amines to give N-substituted sulfenamides. As a representative example, mercaptobenzoate 72 reacted with aniline to give sulfenamide 73 (scheme 29).72 Subsequent treatment of 73 with sodium methoxide in methanol led to N-substituted 1,2-benzoisothiazole-3-one 74.
Scheme 29.

Representative synthesis of N-substituted 1,2-benzoisothiazole-3-ones.
The benzoisothiazole-3-one leaving group also functions well with activated methylene nucleophiles leading to products 75 and, to a lesser extent, with Grignard reagents to give aryl thioether 76 (scheme 30).73 A related benzothiazole leaving group has been employed in the stereospecific syn conjugate attack of a 2,3-enopyranoside leading to an unsaturated sugar.74
Scheme 30.

Reaction of benzoisothiazolin-3-one 63 with activated methylene nucleophiles and Grignard reagent.
5.2 Indole
Dinucleoside methylphosphinite 78 has been prepared by the attack of a 5′-hydroxyl of a protected thymidine on substrate 77 containing a tricoordinate phosphorous reagent with an indole leaving group (scheme 31).75 Similar but lower yielding coupling reactions involving tetracoordinate phosphorous with p-nitrophenol leaving groups have also been described.76
Scheme 31.

Use of indole as a leaving group in dinucleoside synthesis.
5.3 3,5-dichloropyridine
In the presence of organolithium and magnesium reagents, 3,5-dichloro-4-pyridinecarbonitrile behaved primarily as an electrophilic substrate. Thus alkyl or aryl nucleophiles have been reported to produce substitution products with displacement of the cyano group at the 4-position or the chloro group at the 5-position. However, 3,5-dichloro-4-pyridinecarboxaldehyde (79) underwent addition with phenyl lithium at the carbonyl carbon to yield anion intermediate 80 (scheme 32). Aqueous basic workup of this intermediate led to the formation of benzaldehyde in low yields most likely through the expulsion of the 3,5-dichloro-4-pyridyl residue as a carbanionic leaving group leading to 3,5-dichloropyridine (81) after protonation.77
Scheme 32.

Use of 3,5-dichloro-4-pryidyl anion as leaving group in aldehyde synthesis.
5.4 Quinolines
Resin-bound 4-phenyl-1,2-dihydroquinoline (DHQ) 82 has been synthesized in high yields using an aza Diels-Alder cycloaddition reaction for applications as a safety catch linker in the synthesis of amides and carboxylic acids.78 While this solid-phase technique has been previously reviewed as a parallel synthesis strategy,79 it is included in this report since it represents a new type of heterocyclic leaving group.
The use of resin 82 in the synthesis of 4-fluorobenzoyl amide 85 began with the treatment of this resin with 4-fluorobenzoyl chloride to give resin-bound intermediate 83 (scheme 33). Activation of 83 was achieved with several oxidizing agents to give quinolinium 84. In the case of DDQ and CPh3BF4, resin 83 was treated with 3 equiv of oxidizing agents at room temperature for 10h and 3h, respectively, to promote the formation of the quinolinium species. Subsequent addition of benzylamine then cleaved the benzoyl substrate from solid-support to give amide product 85. With the CAN oxidant (entry 4), cleavage was performed in an aqueous acetonitrile solution at room temperature to give 4-fluorobenzoic acid in 62% yield in high purity.
Scheme 33.

Use of 4-phenyl-1,2-dihydroquinoline as safety catch linker.
5.5 Pentafluorophenyl
Reactions that result in the cleavage of carbon-carbon bonds are relatively rare. Most of these reactions involve alkoxide fragmenting to give ketones and generally require stabilized carbanion leaving groups.80 Some common examples include the haloform reaction and the retro-aldol reaction. However, examples involving less stabilized81 or even non-stabilized carbanions are known, though extreme steric crowding or high temperatures are often required.82
Pentafluorophenyl anion leaving groups have been observed in carbon-carbon bond cleavage reactions of cis-1-pentafluorophenyl-4-tert-butylcyclohexanol (86) to give ketone 87 (scheme 34).83 This fragmentation reaction was performed using alkali metal methoxides in DMSO since the alkoxide of 86 was unreactive in ethereal solvents (as in the Grignard reaction used to prepare 86 in the first instance). A nucleophilic aromatic substitution reaction of the pentafluoro leaving group occurred subsequent to the fragmentation of 86 to give observed coproduct 88. The fragmentation has also been observed with other tertiary alcohols (89–91) and secondary alcohol 92 (scheme 34). Although no isolated yields are reported, NMR studies indicate that the conversions are nearly quantitative.
Scheme 34.

Pentafluorophenyl anions in the fragmentation of alcohols to give carbonyl products.
6. Activation-deactivation Leaving Groups
The synthesis of oligosaccharides through iterative glycosylation reactions of thioglycosides has been applied with success over a number of years due to the stability of the anomeric thio functional group towards a wide range of reaction conditions.84 The use of leaving groups that are activated at an atom that is not directly attached to the anomeric center of a glycosyl donor has been defined as remote activation (recently reviewed).85 In recent years, a new glycosylation strategy that allows chemoselective activation of an S-thiazolyl (STaz) leaving group of a glycosyl substrate has been developed. This principle can be illustrated with donor glycosyl substrate 93 (scheme 35). The reaction of 93 with a promoter such as Cu(OTf)2 gave activated substrate 94. Significantly, this activation can occur in the presence of a glycosyl substrate containing an STaz group (such as 95) that has been deactivated (capped) by complexation with PdBr2 or other agent.86 The removal of the Pd(II) cap from intermediate 96 following the coupling step to give product 97 was then accomplished with NaCN. An attractive feature of this strategy is that it does not require protecting group manipulation (armed/disarmed techniques87) to control the leaving group ability. Overall yields disaccharide syntheses using this activation-coupling-deactivation strategy were high regardless of the protecting group choice (scheme 35).
Scheme 35.

Schematic of activation/deactivation concept and representative synthesis examples.
The selective activation of an S-benzoxazolyl (SBox) leaving group of glycosyl donor 99 in the presence of acceptor 98 containing a thioethyl group at the anomeric center has been reported (scheme 36).88 Disaccharide product 100 was obtained as a single diastereomer (α only) in nearly quantitative yield. Importantly, the SBox leaving group is stable towards protecting group manipulations and it is activated for glycosyl coupling under mild conditions using silver triflate. The SBox leaving group in combination with certain saccharide protecting group schemes has been shown to offer a degree of chemoselectivity in coupling reactions not envisioned by the armed-disarmed principle.89 Hexofuranosyl 1-phosphates have also been efficiently prepared in non-protected form using the SBox leaving group with phosphoric acid activation.90
Scheme 36.

Selective activation of SBox glycoside in the presence of an anomeric thioethyl group.
A more flexible oligosaccharide synthesis approach involved the selective activation of glycosyl substrate 101 which contained either an STaz leaving group (R = Taz) or an SEt (R = Et) at the anomeric position (scheme 37).91 When 101 contained a thioethane leaving group, it was coupled to monosaccharide 102 to give product 104 in good yield (93%). This coupling was accomplished by the selective activation of the STaz leaving group of 102 using silver triflate in the presence of the thioethane group of substrate 101 (scheme 37). Alternatively, when 101 contained an STaz leaving group, it was coupled to 103 to give disaccharide 105 in 85%. The selective coupling was accomplished by activation of the thioethane group using N-iodosuccinimide (NIS) with catalytic triflic acid. This selective activation strategy has been extensively optimized and its versatility in multistep oligosaccharide synthesis demonstrated.92
Scheme 37.

Selective activation of STaz over SEt and vice versa in disaccharide synthesis.
Taking advantage of the unique reactivities of the Sbox and STaz leaving groups, a one-pot, 3-step, synthesis of tetrasaccharide 111 was achieved in 73% overall yield (scheme 38).93 Thus donor 106 was activated in the presence of thioethyl-containing 98 in the presence of AgOTf to give dimer 107. In the same pot, STaz-containing receptor 108 was added.
Scheme 38.

One-pot synthesis of tetrasaccharide 111 using selective activation of SBox, SEt, and STaz.
In the presence of NIS/TfOH, the thioethyl group of disaccharide 107 was selectively activated leading to coupling product 109. In a final step, the STaz group of trisaccharide 109 was activated with AgOTf to promote coupling with receptor 110 leading to tetrasaccharide 111 in an overall isolated yield of 73% (scheme 38). The SBox and STaz leaving group strategies have also been extended to the synthesis of oligosaccharides containing α-sialosides.94
7. Nucleophile Assisting Leaving Groups (NALGs)
Nucleophile assisting leaving groups (NALGs) may be defined as a leaving group that contains a chelating unit capable of stabilizing the transition state of a nucleophilic reaction. The rate enhancement afforded by a NALG is not necessarily due to an increase in the nucleofugacity of the leaving group upon cation chelation as observed in the activation-deactivation method described in the previous section. Instead, a NALG should also interact with nucleophiles in the course of a reaction to lower the transition state energy of the rate limiting step. For example, in the case of substrate 112, it is expected that the negative charge imparted to the leaving group moiety (LG) by an incoming nucleophile (Nuc) in transition state 114 should provide a more favorable chelation complex relative to its neutral precursor ligand 113 (scheme 39). Depending on the specific nucleophilic reaction mechanism, cation chelation in the transition state should reduce the energy of activation (Ea1) of NALG substrates relative to substrates containing traditional leaving groups. Without the added stabilizing effect of nucleophilic salt chelation with a nearby multidentate ligand, the energy of activation (Ea2) for reactions involving traditional leaving groups (leading to a transition state such as 115) is expected to be higher (Ea2 > Ea1). As described in the following paragraphs, differential metal ion stabilization of transition states relative to reactants in acetyl transfer reactions has been observed in numerous systems.95
Scheme 39.

Rationale for rate enhancement observed with nucleophile assisting leaving groups (NALGs).
The reaction of 1-chloroanthraquinone (116) with a variety of alkanols in NaH failed to give the desired nucleophilic aromatic substitution product in refluxing THF. However, the addition of catalytic amounts of oligo ethylene glycols led to successful alkanol substitution products. In the case of n-hexadecanol, substitution product 117 was obtained in 80% yield using a catalytic amount (24%) of triethylene glycol (scheme 40).96 Initially acting as nucleophile, triethylene glycol added to 116 to produce intermediate 118 which coordinated the sodium cation as suggested by X-ray crystal structure data. This cation-complexed intermediate most likely played a key role in both coordinating the stoichiometric alkoxide nucleophile (RO−) as well as stabilizing the negative charge forming on the oxygen of the leaving group. In this chloroanthraquinone study, metal chelated oligo-glycol oxides were superior nucleophilic agents relative to straight chain alkanoxide anions. However, a separate group demonstrated that phenolate anions containing oligo ether units in the ortho position exhibited no improved nucleophilic properties relative to their non-chelating analogs.97
Scheme 40.

Podand-catalyzed nucleophilic aromatic substitution reaction of 1-chloroanthroquinone.
This podand-catalyzed method gave optimal results with long chain n-alkanols (C9 and C16) most likely due to their improved solubility in THF relative to smaller alkanols. Similar experiments involving 15-crown-5 methyl alcohol gave significantly poorer results. Although a more effective sodium cation chelating agent than triethylene glycol, the crown ether analog was thought to draw the sodium cation into its cavity away from the anthraquinone residue.98 This presumably limited the ability of the cation to stabilize the negative charge building up on the oxygen of the leaving group in the transition state.
Crown ether-based phenolate and carboxylate nucleophile assisting leaving groups (NALGs) exhibited enhanced nucleofugacity in methylation and acylation reactions. Although this area has been previously reviewed,99 aspects of this research will be readdressed in the present paper to provide a theoretical basis and backdrop for more recent NALG research.
The rates of nucleophilic displacement of the electrophilic methyl group of substrates 119, 120, and 121 (scheme 41) by benzyl thiolate varied markedly depending on the metal counter ion.100 With potassium benzyl thiolate, the metal was sequestered using a well known potassium cation chelating agent 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane (K222) leading to a loss of rate enhancement. To help assess the relative contribution of the metal counter cation, all methylation reactions in the study were compared to experiments involving K222 to give relative rates (krel).
Scheme 41.

Methylating agents containing crown ether leaving groups and structure of K222.
In the case of benzo-18-crown-5 substrate 119, the peak reactivity occurred with potassium thiolate giving a relative rate of 826 when compared to the potassium sequestered experiment involving K222 (scheme 42). This effect was far less marked with analogous 27-crown-8 substrate 120 where the maximum relative rate was 53. Presumably, the much larger crown ether system of 120 provided a poor fit for the cations used in the study. However, in both 119 and 120, the methylation rate was enhanced relative to an electronically similar substrate, 2,6-dimethyl anisole (122). Rate studies of 122 showed a slight rate depression relative to the potassium cation sequestration experiment involving K222 (scheme 42) suggesting a weak association of the alkali metal ions with the thiolate nucleophile.97
Scheme 42.

Relative methylation reaction rates of 119–121 containing crown ether leaving groups (LG) and control substrates 122 and 123.
Substrate 121 containing an 18-crown-5 benzoate moiety also showed a methylation rate enhancement in the presence of various metal cations with the optimal relative rate of 47 occurring in the presence of sodium benzyl thiolate (scheme 42). The preference for sodium cation in the case of 121 has been rationalized in terms of the decreased pore size of the crown ether cavity relative to 119. Due to the bulkier methoxycarbonyl group, the cavity of 121 was expected to be somewhat smaller than the corresponding cavity of 119 and therefore more suitable to host the smaller sodium cation.
Metal cation effects on the rate of benzyl thiolate methylation were nearly an order of magnitude larger with crown-anisole substrate 119 relative to crown-benzoate 121. This effect has been explained on the basis of a strong electrostatic interaction between the chelated metal and the aryl ether oxygen in transition state 124 (scheme 43). With crown-benzoate substrate 121, the negative charge from the incoming thiolate nucleophile is delocalized over the two oxygens of the carboxylate functional group, as in 125, and therefore not as effectively stabilized by the chelated metal.
Scheme 43.

Transition state of 124 and 125 in the methylation of a benzyl thiolate metal salt.
In the presence of metal methoxide in methanol, an aryl acetate substrate bearing a tetra(oxyethylene) chain in the ortho position, compound 126, exhibited substantially accelerated acetyl transfer reactions (scheme 44).101 In general, the mechanism of acyl-transfer reactions is known to proceed by a two-step process, the first of which involves the rate-determining formation of a tetrahedral intermediate. In the case of 126, the presence of a nearby multidentate ligand was expected to stabilize a metal cation interaction with the methoxide addition product as depicted in transition state 127 (scheme 44). Acetyl transfer reactions of 126 were carried out in methanol solutions of Me4NOMe of varying concentrations and the reaction rates were calculated. These rates were then compared to reactions carried out at the same concentration of Me4NOMe but containing added metal bromide salts. Rate enhancements were observed with several metal bromides (scheme 44).
Scheme 44.
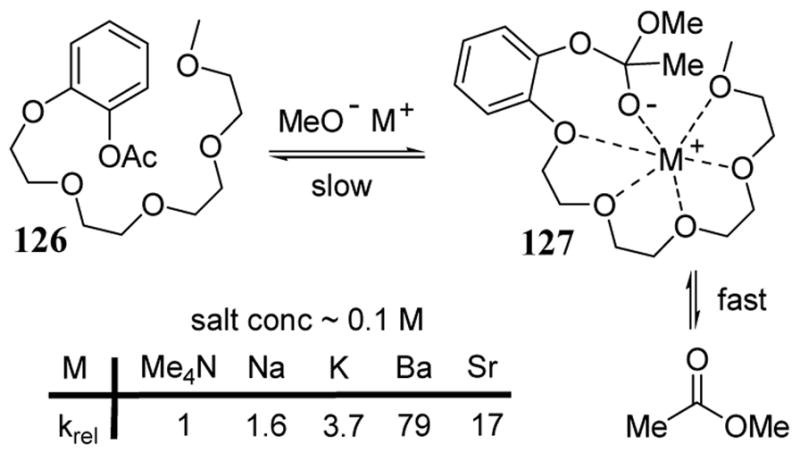
Rate acceleration of acetyl transfer with substrate 126 containing an oligoether NALG.
In the case of added BaBr2 (at 0.1 M), the relative rate was determined to be 79. Under the same conditions, the relative rate of acetyl transfer with phenyl acetate (with no chelating arm) was determined to be 3. Although considered a fairly weak chelating unit, the oligo(oxyethylene) chain of 126 was responsible for a net 25-fold rate enhancement. The improved rate enhancement of the alkaline earth metals Ba2+ and Sr2+ was attributed to both a larger electrostatic stabilization of the transition state leading to the tetrahedral intermediate as well as to a better utilization of the coordinative interactions with the oligoether unit.95
Improved metal catalyzed rate accelerations of acetyl transfer reactions to methoxide were observed with all of the homologs of crown ether 128 in the presence of barium salts relative to the same reactions using only Me4NOMe (scheme 45).95,102 Interestingly, Ba2+ gave the optimal acetyl transfer rates despite the fact that its cation binding affinities for 128 are an order of magnitude lower than the alkali cations. This increased acceleration of Ba2+ relative to the alkali cations may be partially explained by improved electrostatic stabilization of divalent over monovalent cations.
Scheme 45.

Barium cation catalyzed acetylation reactions with crown ether NALG 128.
When considering the differences of Ba2+ catalysis among crown ether substrates 128, there is no correlation between binding affinity (as measured by KS) and relative rate. Indeed, the highest observed rate (krel = 760) occurred with the 18-crown-5 analog of substrate 128 (n = 3) where no appreciable binding affinity (< 10 M−1) for the barium cation was measured (scheme 45). While larger ring sizes better accommodate the barium cation (for example KS = 260 M−1 when n = 5), the improved fit of the cation in the crown ether pore may shift the cation away from the reactive center and diminish its ability to stabilize the forming negative charge in the transition state through electrostatic interaction. Similar rate studies involving acetyl transfer to ethoxide instead of methoxide revealed a more pronounced metal catalysis effect with substrates 128.103 Both methoxide and ethoxide studies underscore the concept that effective rate enhancement of acetyl transfer reactions by metal cations does not require strong metal-binding affinity in the reactant state.
The rate enhancement effect in acyclic NALG systems is competitive with more rigid macrocyclic entities (scheme 46). In rate studies of acetyl transfer reactions to ethoxide in the presence of barium salts, the relative rate of 129 was over 10-fold greater than macrocycle 130 for smaller ring sizes (n = 2 and 3). The conformational rigidity of these smaller macrocycles destabilizes interactions with the metal catalyst. Only when macrocycle 130 was of sufficient size (n = 4) did the rate enhancement supersede that of acyclic NALG 129.104 Further improvement in acetyl transfer were realized with calix[4]arene 131 which contained a phenoxide anion capable of providing an additional binding interaction with the Ba2+ catalyst.99b
Scheme 46.
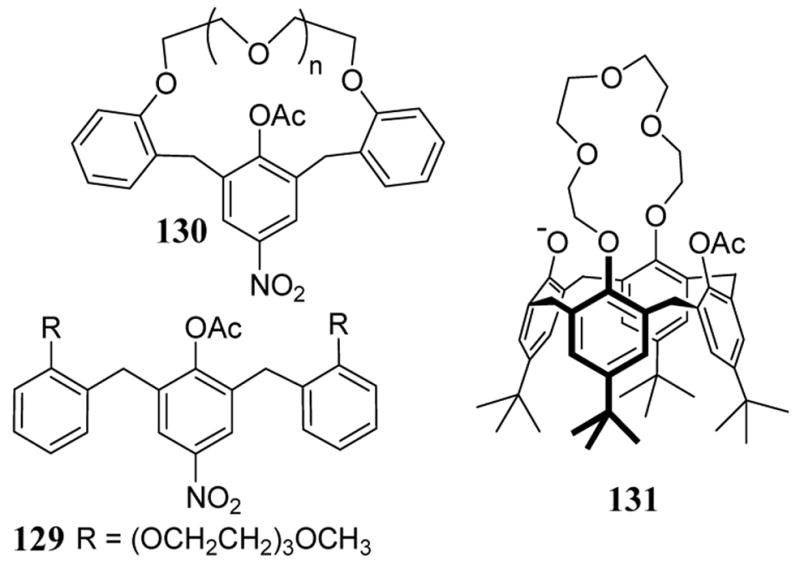
Other acyclic and macrocyclic acetylating agents accelerated by BaBr2.
There are numerous examples of leaving group stabilization by a metal cation in substrates containing a chelating unit. While these examples involve traditional leaving groups and are therefore outside the scope this review, they further illustrate the concept of cation-mediated electrophilic assistance in nucleophilic reactions. For example, the addition of sodium alkoxide to a chlorocyclophosphazene led to regioselective addition of alkoxide to give product 133 (scheme 47).105 These nucleophilic substitution reactions also proceed at enhanced rates. Both the acceleration and regioselectivity were rationalized in terms of transition state 132 involving sodium cation stabilization of the partial negative charge forming on the chloride leaving group.
Scheme 47.

Cation assisted addition of sodium alkoxide to a macrocyclic substrate.
Alkyl bromide products were obtained in good yields and under mild conditions with substrates 134 containing a 2-pyridyl sulfonate leaving group. This leaving group exhibited enhanced reactivity towards magnesium bromide, a divalent metal salt (scheme 48). Pre-coordination of 134 with the nucleophilic salt leading to complex 135 provided an internal activation effect.106 The bromination reactions were remarkably fast with unactivated secondary substrates. In the case of two norbornyl substrates, 2-pyridyl sulfonate was displaced by bromide to give complete conversion to the corresponding inverted bromide products in 30 seconds (scheme 48). The reaction rates with these norbornyl substrates were significantly higher than the tosylates (70 min) and 8-quinolylsulfonates (120 min).106 Inversion of configuration was observed for all substrates 134.
Scheme 48.

Transformation of 2-pyridyl sulfonates 134 to alkyl bromides with inversion of configuration.
The 2-pyridylsulfonate leaving group has also been employed in the alkoxyaminocarbonylation of β-dicarbonyl compounds (scheme 49). Activated amine reagent 136 containing the 2-pyridylsulfonate leaving group reacted with enolates of β-dicarbonyl compounds following a Lossen-type rearrangement (most likely via intermediate 137) to give tricarbonyl hydroxamates 138 (scheme 49).107
Scheme 49.

Alkoxyaminocarbonylation using a 2-pyridylsulfonate leaving group.
Studies involving nucleophilic reactions with traditional sulfonate leaving groups revealed a strong rate dependence on cation identity and coordinative availability. In these studies, nucleophilic reactions of mesylates and tosylates were accelerated by the addition of polyether and macrocyclic ether ligands in low polarity solvents.108 In the case of n-octyl mesylate (139), treatment with metal iodide salts in the presence of several chelating reagents led to a rate acceleration relative to the benchmark reaction using tetraoctyl ammonium iodide (scheme 50). With polyethylene glycol 140 and 15-crown-5 141, the acceleration effect was most marked with lithium iodide which was explained on the basis of the stronger Lewis acid character of Li+ relative to sodium and potassium cations. A similar but less pronounced rate acceleration effect was also observed in this same system with some alkali earth metals.109 Phosphinic ester substrates also exhibit accelerated rates of alkylation with metal iodides.110 In the presence of added ligand 142 (a cryptand), the acceleration affect of the addition of metal iodide to octyl mesylate is minimal relative to the benchmark reaction and shows no cation preference (scheme 50). The strong chelation properties of cryptand 142 were thought to have limited the ability of the cation to provide electrophilic assistance in the transition state of the nucleophilic reaction.
Scheme 50.
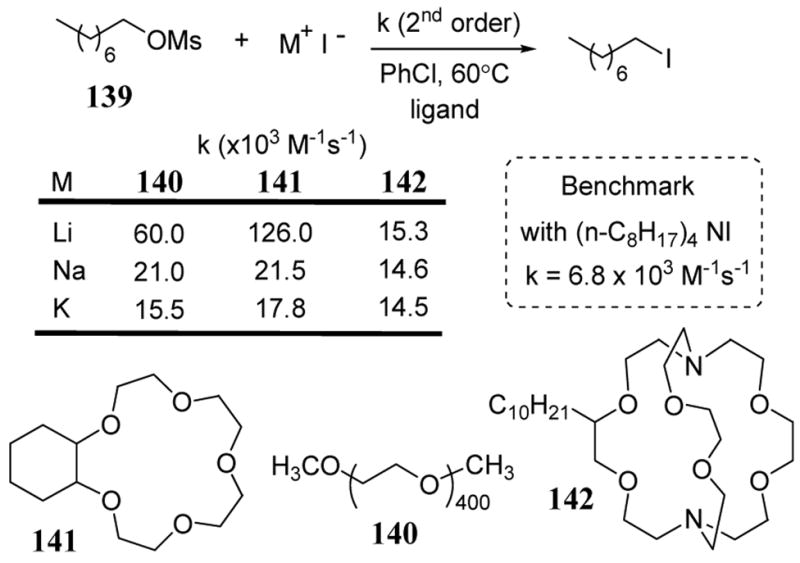
Rate acceleration of iodide addition to mesylate 139 with added ligands 140–142.
Sulfonate leaving groups containing an oligoether metal-chelating moiety have been recently reported.111 The chelating units were designed to stabilize developing negative charge on the oxygens of the sulfonate leaving group in the transition state analogous to previously described work involving aryloxy and arylcarboxyl leaving groups that contained metal chelating units (scheme 41).99 Sulfonylating agents 143 required for the various NALGs used in the study were prepared from sulfobenzoic acid anhydride by treatment with phosphorous pentachloride followed by the addition of methyl-oligo-ethylene oxides (scheme 51). Arylsulfonyl chlorides 143 were then reacted with 3-phenylpropanol to give the corresponding sulfonate esters 144 in yields ranging from 72–96%. Sulfonate esters 144 (except for n = 0) were exceptionally stable to silica gel chromatography. Sulfonate esters 144 were reacted with lithium, sodium, and potassium bromide giving 3-phenyl-1-bromopropane as the substitution product (scheme 51). Methyl ester sulphonate 144a (n = 0) served as a baseline to help determine the rate enhancing role of the various oligoether side chains in compounds 144b–144e.
Scheme 51.
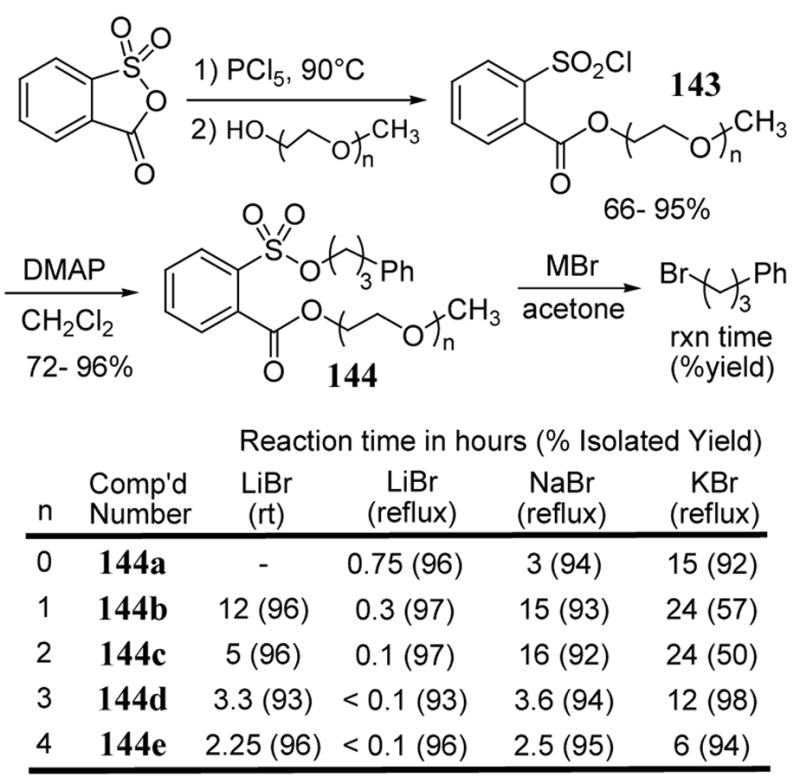
Rates of metal bromide additions to arylsulfonates 144 to give 3-phenyl-1-bromopropane.
The reaction of LiBr with 144a gives nearly complete conversion to product at 4 times the rate of NaBr. The increasing number of ethylene oxide units correlates well with a decrease in reaction time required for LiBr to convert substrates 144 to product. In the case of 144e, which contains a tetraethylene oxide unit, the rate of reaction with LiBr is approximately 15 times faster than with 144a (n = 0). The increased reaction rate of LiBr relative to NaBr and KBr for all NALGs 144 appear to parallel earlier intermolecular studies involving alkylmesylate reactions with various metal iodide salts.108 The presence of ethylene oxide units increase the relative rate of LiBr versus NaBr additions to 50 fold for 144b (n = 1) and 160 fold 144c (n = 2). The trend seems to level off with additional ethylene oxide units. Significant rate enhancements toward metal halide addition have also been observed with secondary sulphonate substrates derived from reagent 143.111
Macrocyclic ether-containing sulfonates also exhibited a rate enhancement and salt selectivity effect in metal bromide addition reactions (scheme 52). The reaction rates of LiBr with substrates 145a and 145b were nearly 50 times faster than the baseline methylester sulfonate 144a (scheme 51). In terms of selectivity, the 12-crown-4 containing 145a reacts with LiBr in acetone to form product in 1.3 h at room temperature. However, with sodium or potassium bromide, the displacement reaction required 4 h and 8 h under refluxing conditions, respectively. This corresponds to a selectivity of greater than 200-fold for lithium bromide over sodium and potassium bromide.111
Scheme 52.
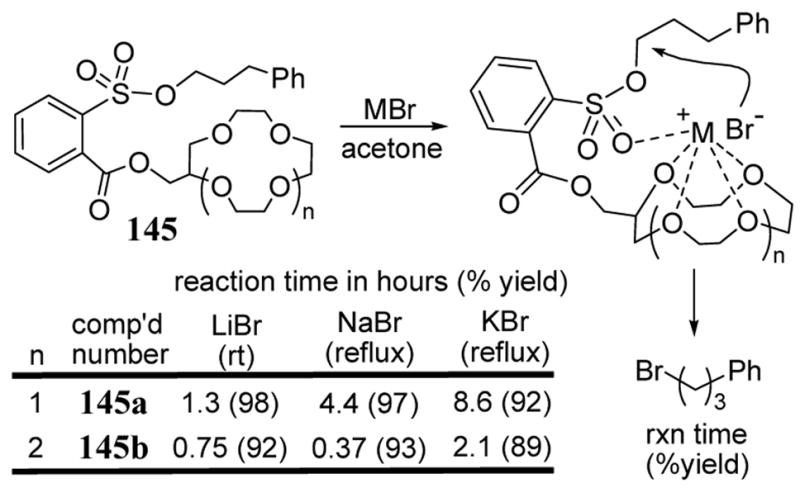
Rates of metal bromide additions to crown ether-containing sulfonates.
Crystallographic studies of triethylene oxide-containing NALGs derived from 144d revealed that three of the side arm ether oxygens of the NALG and one of the sulfonyl oxygens are coordinated to metal cation. Interestingly, the ester carbonyl of the NALG was nearly orthogonal and therefore poorly conjugated to the phenyl ring.111 To avoid this non-ideal chelation geometry, a series of NALGs related to 144 but without the carbonyl group have been prepared (scheme 53).112 Starting from aryl ethers 146, chlorosulfonylation followed by the addition of phenylpropyl alcohol led to sulfonates 147. The reaction of 147 with LiBr in acetone at room temperature to give 3-phenyl-1-bromopropane exhibited enhanced rates (5 to 10-fold) relative to NALGs 144 which possess an ester group. As in the ester NALG series 144, increased length of chelating side chain corresponded to heightened reactivity towards a nucleophilic metal salt.
Scheme 53.

Rates of metal bromide additions to NALGs 147 containing oligo-ethylene oxide chelating units without a carbonyl group.
Limited studies of NALGs derived from 143 suggest that the nucleophilic displacement reactions may occur (at least in some cases) via an SNi-type mechanism. For example, 2-adamantyl NALG substrate 148 was expected to exhibit little or no reactivity towards metal halide salts since backside nucleophilic displacement is essentially precluded due to steric crowding and ionization mechanisms are disfavored due to the constrained ring geometry.113 However, treatment of this substrate with LiBr in refluxing acetone gave the 2-bromoadamantane product in 98% yield after 16 h (scheme 54). By contrast, the corresponding 2-adamantly tosylate gave no reaction even after 70 h in refluxing acetone. The triflate of 2-adamantol also failed to give bromide product when treated with LiBr.
Scheme 54.

Evidence in support of an SNi-type mechanism with 2-adamantyl NALG substrate 148.
Arylsulfonates of hindered secondary alcohols derived from reagent 149 were also converted to the corresponding alkyl chlorides very rapidly and in the presence of titanium tetrachloride at low temperatures.114 These chlorination reactions are thought to proceed via an SNi-type mechanism leading to exclusive retention of configuration. Thus the treatment of sulfonate esters 149 with TiCl4 in methylene chloride at −78 °C led to high yields of the corresponding alkyl chlorides (scheme 55). In each case, the reaction was complete in less than 2 minutes.
Scheme 55.
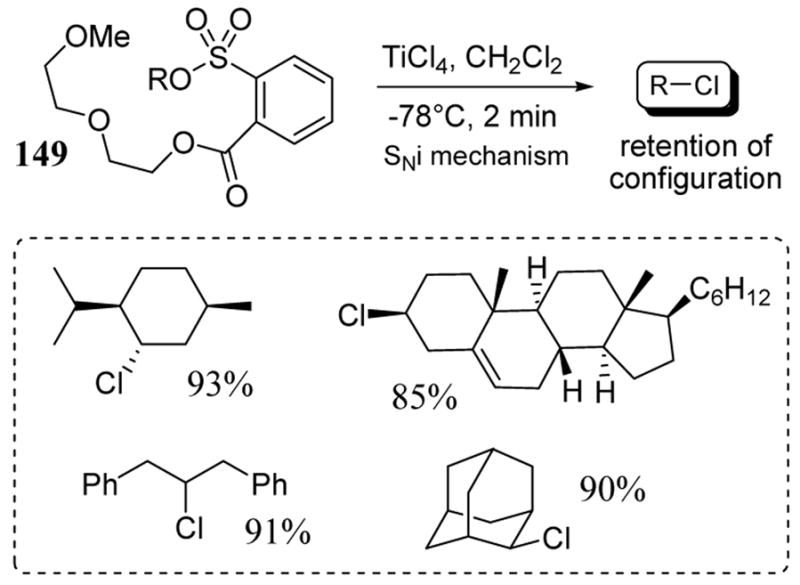
Reaction of sulphonate esters 149 with TiCl4 leading to alkyl chlorides with retention of configuration.
With NALG 149 derived from 1,3-diphenyl-2-propanol, the respective chloride product was obtained in 91% yield upon treatment with TiCl4. Tosyl esters of this substrate gave nearly exclusive elimination product in the presence of alkali metal chloride agents. NALG sulfonates 149 of cholesterol and menthol were converted to the corresponding chlorides almost instantaneously at −78 °C (scheme 55). In the 2-adamantyl system, a 90% conversion to the chloride product was observed with no side product arising from rearrangement of the adamantine nucleus.115 Recently this method has been extended to stereospecific bromination and azidations using TiBr4 and Ti(N3)4 respectively.116
8. Conclusions
In this report, we have provided an overview of recent developments in heterolytic leaving group chemistry. Although leaving groups have been an area of active research since at least the mid 1950’s, progress in this field continues at an unabated pace. Indeed, it might be argued that the increasing molecular complexity of modern synthesis targets has spurred the development of leaving groups in recent years. This has certainly been true in the area of oligosaccharide synthesis which has moved forward in large part due to improved anomeric leaving groups such as activation-deactivation groups. In more established leaving group classes such as sulfonates and carboxylates, we have shown in this report that many useful variations are still being made to improve their performance. Non-traditional leaving groups such as carboranes and chiral ferrocenyl-containing carboxylates can be seen as early offerings from the organometallic field. It is expected that, just as this field has made powerful contributions to the area of organic reaction catalysis, so will new and more useful organometallic leaving groups emerge. Finally, nucleophile assisting leaving groups (NALGs), including those inspired by biological systems, offer impressive degrees of selectivity and rate enhancement. We anticipate that future advances in this area will lead to new classes of “designer” leaving groups which will facilitate the synthesis of molecular targets of ever-increasing complexity. Hopefully, this review will stimulate further development in this exciting field.
Acknowledgments
The authors wish to thank the National Institutes of Mental Health (66963-01) and General Medical Science (073621-01), and NSF (0311369) for their support of our research in this area.
Biographies
 Salvatore D. Lepore was born in Philadelphia, PA and received his B.S. in Chemical Engineering with honors in 1992 from the University of South Florida. He worked for a year as an engineer for an environmental consulting firm before deciding to pursue graduate studies at Purdue University. Salvatore received his doctoral degree in 1997 under the guidance of Andrus B. Merritt in the area of natural product total synthesis. He then spent three years as a postdoctoral fellow at Eli Lilly in Indianapolis under the direction of Michael R. Wiley. Salvatore’s studies at Lilly focused on the development of new solid phase synthesis methods. In 2000, he joined the chemistry faculty at Florida Atlantic University as an Assistant Professor and was promoted to the Associate level in 2006. His current research efforts are in the development of new leaving groups with applications in radiotracer synthesis. He is also developing new asymmetric methods for the preparation of conjugated allenyl carbonyl compounds with applications in natural product total synthesis. Salvatore’s research has been funded by a number of agencies including the ACS PFR, NIMH, NIGMS, NSF, and Florida Centers of Excellence program. In 2006, he received his university’s Researcher of the Year award.
Salvatore D. Lepore was born in Philadelphia, PA and received his B.S. in Chemical Engineering with honors in 1992 from the University of South Florida. He worked for a year as an engineer for an environmental consulting firm before deciding to pursue graduate studies at Purdue University. Salvatore received his doctoral degree in 1997 under the guidance of Andrus B. Merritt in the area of natural product total synthesis. He then spent three years as a postdoctoral fellow at Eli Lilly in Indianapolis under the direction of Michael R. Wiley. Salvatore’s studies at Lilly focused on the development of new solid phase synthesis methods. In 2000, he joined the chemistry faculty at Florida Atlantic University as an Assistant Professor and was promoted to the Associate level in 2006. His current research efforts are in the development of new leaving groups with applications in radiotracer synthesis. He is also developing new asymmetric methods for the preparation of conjugated allenyl carbonyl compounds with applications in natural product total synthesis. Salvatore’s research has been funded by a number of agencies including the ACS PFR, NIMH, NIGMS, NSF, and Florida Centers of Excellence program. In 2006, he received his university’s Researcher of the Year award.
 Deboprosad (Debo) Mondal was born in Diamond Harbour (India) in 1981. He received his B.S. in Chemistry from Calcutta University in 2002 and an M.S. from the Indian Institute of Technology, Kanpur in 2004. He is currently a doctoral student under the guidance of Professor Salvatore D. Lepore in the Department of Chemistry at Florida Atlantic University. Debo’s main research involves the design of new leaving groups to solve some critical problems in organic synthesis. He is also developing new titanium IV reagents for stereoselective halogenations and azidations.
Deboprosad (Debo) Mondal was born in Diamond Harbour (India) in 1981. He received his B.S. in Chemistry from Calcutta University in 2002 and an M.S. from the Indian Institute of Technology, Kanpur in 2004. He is currently a doctoral student under the guidance of Professor Salvatore D. Lepore in the Department of Chemistry at Florida Atlantic University. Debo’s main research involves the design of new leaving groups to solve some critical problems in organic synthesis. He is also developing new titanium IV reagents for stereoselective halogenations and azidations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Smith MB, March J. Advanced Organic Chemistry. 5. John Wiley; New York: 2001. p. 275. [Google Scholar]
- 2.IUPAC Compendium of Chemical Terminology. 2. 1997. [Google Scholar]
- 3.Mathieu J, Allais A, Valls J. Angew Chem. 1960;72:71–74. [Google Scholar]
- 4.Based on a CAS database search using SciFinder Scholar 2006 searching the term “leaving group” with the two words closely associated.
- 5.Bordwell FG, Pitt BM, Knell M. J Am Chem Soc. 1951;73:5004. [Google Scholar]
- 6.Winstein S, Morse BK, Grunwald E, Jones HW, Corse J, Trifan D, Marshall H. J Am Chem Soc. 1952;74:1127–1132. [Google Scholar]
- 7.Swain CG, Dittmer DC. J Am Chem Soc. 1953;75:4627. [Google Scholar]
- 8.Lewis ES, Boozer CE. J Am Chem Soc. 1954;76:791–793. [Google Scholar]
- 9.Including the popular undergraduate text by Morrison, R. T.; Boyd, R. N. Organic Chemistry (2nd ed.), Allyn and Bacon, Inc, Boston 1966.
- 10.Laue T, Plagens A. Named Organic Reactions. 2. John Wiley; New York: 2005. p. IX. [Google Scholar]
- 11.Netscher T. Rec Res Develop Org Chem. 2003;7:71–83. [Google Scholar]
- 12.(a) Katritzky AR. Tetrahedron. 1980;36:679–699. [Google Scholar]; (b) Katritzky AR, Musumarra G. Chem Soc Rev. 1984:4768. [Google Scholar]
- 13.Lou B, Eckhardt E, Hanessian S. Prep Carbohydrate Chem. 1997:449–466. [Google Scholar]
- 14.Capozzi MAM, Cardellicchio C, Naso F. Eur J Org Chem. 2004:1855–1863. [Google Scholar]
- 15.For a recent comprehensive review see: Montalbetti CAGN, Falque V. Tetrahedron. 2005;61:10827–10852.
- 16.For a recent comprehensive review see: Dolle RE. J Comb Chem. 2005;7:739–798. doi: 10.1021/cc050082t.
- 17.For representative examples: Bulluss GH, Knott KM, Ma ESF, Aris SM, Alvarado E, Farrell N. Inorg Chem. 2006;45:5733–5735. doi: 10.1021/ic060741m.Parsons S, Smith AA, Winpenny REP. Chem Comm. 2000;7:579–580.Pasini A, D’Alfonso G, Manzotti C, Moret M, Spinelli S, Valsecchi M. Inorg Chem. 1994;33:4140–4148.
- 18.For representative examples: Wood TC, Johnson KL, Naylor S, Weinshilboum RM. Drug Metab Dispos. 2002;30:1123–1128. doi: 10.1124/dmd.30.10.1123.Hlasta DJ, Ackerman JH, Court JJ, Farrell RP, Johnson JA, Kofron JL, Robinson DT, Talomie TG, Dunlap RP, Franke CA. J Med Chem. 1995;38:4687–4692. doi: 10.1021/jm00023a008.Krantz A, Copp LJ, Coles PJ, Smith RA, Heard SB. Biochemistry. 1991;30:4678–4687. doi: 10.1021/bi00233a007.
- 19.Crossland RK, Wells WE, Shiner VJ. J Am Chem Soc. 1971;93:4217–4219. [Google Scholar]
- 20.Farcasiu D, Jaehme J, Ruechardt C. J Am Chem Soc. 1985;107:5717–5722. [Google Scholar]
- 21.Sorbye K, Tautermann C, Carlsen P, Fiksdahl A. Tetrahedron Asymm. 1998;9:681–689. [Google Scholar]
- 22.Heggvik L, Fiksdahl A. Tetrahedron Asymm. 1997;8:2189–2192. [Google Scholar]
- 23.Said SA, Fiksdahl A. Tetrahedron: Asymm. 1999;10:2627–2633. [Google Scholar]
- 24.Evans PA, Brand TA, Robinson JE. Tetrahedron Lett. 1999;40:3105–3108. [Google Scholar]
- 25.Evans PA, Brandt TA. Org Lett. 1999;1:1563–1565. [Google Scholar]
- 26.Liu J, Robins MJ. Org Lett. 2005;7:1149–1151. doi: 10.1021/ol050063s. [DOI] [PubMed] [Google Scholar]
- 27.Vatele J, Hanessian S. Prep Carbohydrate Chem. 1997:127–149. [Google Scholar]
- 28.(a) Hanessian S, Vatele JM. Tetrahedron Lett. 1981;22:3579–3582. [Google Scholar]; (b) Vatele JM, Hanessian S. Tetrahedron. 1996;52:10557–10568. [Google Scholar]
- 29.(a) Hanessian S, Bacquet C, LeHong N. Carbohydrate Res. 1980;80:C17. [Google Scholar]; (b) Hanessian S. Prep Carbohydrate Chem. 1997:381–388. [Google Scholar]
- 30.David S, Malleron A, Dini C. Carbohydrate Res. 1989;188:193–200. [Google Scholar]
- 31.Best WM, MacDonald JM, Skelton BW, Stick RV, Tilbrook DMG, White AH. Can J Chem. 2002;80:857–865. [Google Scholar]
- 32.Attolino E, Catelani G, D’Andrea F. Tetrahedron Lett. 2002;43:1685–1688. [Google Scholar]
- 33.Corey EJ, Posner GH, Atkinson RF, Wingard AK, Halloran DJ, Radzik DM, Nash JJ. J Org Chem. 1989;54:389–393. [Google Scholar]
- 34.Paleta O, Pomeisl K, Kafka S, Klasek A, Kubelka V. Beilstein J Org Chem. 2005;1:17. doi: 10.1186/1860-5397-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Imagawa H, Kinoshita A, Fukuyama T, Yamamoto H, Nishizawa M. Tetrahedron Lett. 2006;47:4729–4731. [Google Scholar]
- 36.Osa Y, Takeda K, Sato T, Kaji E, Mizuno Y, Takayanagi Tetrahedron Lett. 1999;40:1531–1534. [Google Scholar]
- 37.(a) Mackie RK, Smith DM, Aitken RA. Guidebook to Organic Synthesis. 3. Longman; UK: 1999. pp. 302–303. [Google Scholar]; (b) Seyden-Penen J. Chiral Auxiliaries and Ligands in asymmetric Synthesis. John Wiley; New York: 1995. [Google Scholar]
- 38.Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis. Vol. 3 Springer-Verleg; New York: 1999. [Google Scholar]
- 39.(a) Bringmann G, Mortimer AJP, Keller PA, Gresser MJ, Garner J, Breuning M. Angew Chem Int Ed. 2005;44:5384–5427. doi: 10.1002/anie.200462661. [DOI] [PubMed] [Google Scholar]; (b) Wallace TW. Org Biomolecular Chem. 2006;4:3197–3210. doi: 10.1039/b608470m. [DOI] [PubMed] [Google Scholar]
- 40.Nakamura H, Ishihara K, Yamamoto H. J Org Chem. 2002;67:5124–5137. doi: 10.1021/jo020165l. [DOI] [PubMed] [Google Scholar]
- 41.Ishihara K, Ishibashi H, Yamamoto H. J Am Chem Soc. 2001;123:1505–1506. and references cited therein. [Google Scholar]
- 42.Fujii M, Sato Y, Aida T, Yoshihara M. Chem Express. 1992;7:309. [Google Scholar]
- 43.Murata S, Suzuki M, Noyori R. J Am Chem Soc. 1980;102:3248–3249. [Google Scholar]
- 44.Sakane S, Fujiwara J, Maruoka K, Yamamoto H. J Am Chem Soc. 1983;105:6154–6155. [Google Scholar]
- 45.Tamura R, Watabe K, Ono N, Yamamoto Y. J Org Chem. 1992;57:4895–4903. [Google Scholar]
- 46.Tamura R, Katayama H, Watabe K, Suzuki H. Tetrahedron. 1990;46:7557–7568. [Google Scholar]
- 47.The use of chiral leaving groups in nitro-olefination reactions has been briefly reviewed: Berner OM, Tedeschi L, Enders D. Eur J Org Chem. 2002;12:1877–1894.
- 48.Rajappa S. Tetrahedron. 1999;55:7065–7114. [Google Scholar]
- 49.Faulques M, Rene L, Royer R. Synthesis. 1982;4:260–261. [Google Scholar]
- 50.Yang X, Wand R. Tetrahedron Asymm. 1997;8:3275–3281. [Google Scholar]
- 51.Fugi K, Node M, Nagasawa H, Naniwa Y, Terada S. J Am Chem Soc. 1986;8:3855–3856. [Google Scholar]
- 52.Forzato C, Nitti P, Pitacco G, Valentin E, Morganti S, Rizzato E, Spinelli D, Dell’Erba C, Petrillo G, Tavani C. Tetrahedron. 2004;60:11011–11027. [Google Scholar]
- 53.Fuji K. ACGC Chem Res Comm. 2000;11:32–37. [Google Scholar]
- 54.Bagul TD, Lakshmaiah G, Kawabata T, Fuji K. Org Lett. 2002;4:249–251. doi: 10.1021/ol016999s. [DOI] [PubMed] [Google Scholar]
- 55.Basavaiah D, Kumaragurubaran N, Sharada DS, Reddy RM. Tetrahedron. 2001;57:8167–8172. [Google Scholar]
- 56.(a) Duhamel P, Valnot JY, Eddine J. J Tetrahedron Lett. 1982;23:2863–2866. [Google Scholar]; (b) Duhamel P, Ple G, Valnot JY. Heterocycles. 1994;37:751–757. [Google Scholar]
- 57.McManus SP, Roberts FE, Lam DH, Hovanes B. J Org Chem. 1982;47:4386–4388. [Google Scholar]
- 58.McManus SP, Safavy K, Kamkar, Roberts FE. J Org Chem. 1982;47:4388–4389. [Google Scholar]
- 59.(a) Alexakis A, Malan C, Lea L, Tissot-Croset K, Polet D, Falciola C. Chimia. 2006;60:124–130. [Google Scholar]; (b) Karlström ASE, Bäckvall J-E. Copper-Mediated Enantioselective Substitution reactions. In: Krause N, editor. Modern Organocopper Chemistry. Wiley-VCH; 2002. pp. 262–269. [Google Scholar]
- 60.Breit B, Breuninger D. Synthesis. 2005:147–157. [Google Scholar]
- 61.Breit B, Breuninger D. Synthesis. 2005:2782–2786. [Google Scholar]
- 62.Breit B, Breuninger D. J Am Chem Soc. 2004;126:10244–10245. doi: 10.1021/ja0467364. [DOI] [PubMed] [Google Scholar]
- 63.Karlström ASE, Huerta FF, Meuzelaar GJ, Bäckvall JE. Synlett. 2001:923–926. [Google Scholar]
- 64.Okuyama T, Takino T, Sueda T, Ochiai MJ. Am Chem Soc. 1995;117:3360–3367. [Google Scholar]
- 65.(a) Ochiai M. J Organometallic Chem. 2000;611:494–508. [Google Scholar]; (b) Stang PJ. J Org Chem. 2003;68:2997–3008. doi: 10.1021/jo030022e. [DOI] [PubMed] [Google Scholar]
- 66.Matano Y. Organomet. 2000;19:2258–2263. [Google Scholar]
- 67.Matano Y, Imahori H. J Org Chem. 2004;69:5505–5508. doi: 10.1021/jo0492721. [DOI] [PubMed] [Google Scholar]
- 68.Ooi T, Goto R, Maruoka KJ. Am Chem Soc. 2003;125:10494–10495. doi: 10.1021/ja030150k. [DOI] [PubMed] [Google Scholar]
- 69.Koech PK, Krische MJJ. Am Chem Soc. 2004;126:5350–5351. doi: 10.1021/ja048987i. [DOI] [PubMed] [Google Scholar]
- 70.Moloney MG, Pinhey JTJ. Chem Soc Chem Comm. 1984;15:965–966. [Google Scholar]
- 71.Kato T, Stoyanov E, Geier J, Grutzmacher H, Reed CA. J Am Chem Soc. 2004;126:12451–12457. doi: 10.1021/ja047357d. [DOI] [PubMed] [Google Scholar]
- 72.Shimizu M, Sugano Y, Konakahara T, Gama Y, Shibuya I. Tetrahedron. 2002;58:3779–3783. [Google Scholar]
- 73.Bao M, Shimizu M, Shimada S, Inoue J, Konakahara T. Tetrahedron. 2004;60:11359–11366. [Google Scholar]
- 74.Al Neirabeyeh M, Rollin P. J Carbohydrate Chem. 1990;9:471–478. [Google Scholar]
- 75.Wang JC, Just G. Tetrahedron Lett. 1997;38:2241–2244. [Google Scholar]
- 76.Lesnikowski ZJ, Jaworska M, Stec WJ. Tetrahedron Lett. 1987;28:5535–5538. [Google Scholar]
- 77.Picci N, Pocci M, Gugliuzza A, Puoci F, De Munno A, Iemma F, Bertini V. Heterocycles. 2001;55:2075–2084. [Google Scholar]
- 78.Arseniyadis S, Wagner A, Mioskowski C. Tetrahedron Lett. 2004;45:2251–2253. [Google Scholar]
- 79.(a) Dolle RE. J Comb Chem. 2005;7:739–798. doi: 10.1021/cc050082t. [DOI] [PubMed] [Google Scholar]; (b) Fraser HL, Floyd MB, Sosa ACB. Prog Heterocyclic Chem. 2005;17:261–303. [Google Scholar]
- 80.Wietzerbin K, Bernadou J, Meunier B. Eur J Inorg Chem. 2000:1391–1406. [Google Scholar]
- 81.Benkeser RA, Siklosi MP, Mozdzen E. C J Am Chem Soc. 1978;100:2134–2139. [Google Scholar]
- 82.(a) Lomas JS, Dubois JE. J Org Chem. 1984;49:2067–2069. [Google Scholar]; (b) Arnett EM, Small LE, McLver RT, Miller JS. J Org Chem. 1978;43:815–817. [Google Scholar]; (c) Zook HD, March J, Smith DFJ. Am Chem Soc. 1959;81:1617–1620. [Google Scholar]
- 83.Garner CM, Fischer HC. Tetrahedron Lett. 2006;47:7405–7407. [Google Scholar]
- 84.Codee JDC, Litjens REJN, Van den Bos LJ, Overkleeft HS, Van der Marel GA. Chem Soc Rev. 2005;34:769–782. doi: 10.1039/b417138c. [DOI] [PubMed] [Google Scholar]
- 85.Hanessian S, Lou B. Chem Rev. 2000;100:4443–4463. doi: 10.1021/cr9903454. [DOI] [PubMed] [Google Scholar]
- 86.Pornsuriyasak P, Gangadharmath UB, Rath NP, Demchenko AV. Org Lett. 2004;6:4515–4518. doi: 10.1021/ol048043y. [DOI] [PubMed] [Google Scholar]
- 87.Mootoo DR, Konradsson P, Udodong UE, Fraser-Reid B. J Am Chem Soc. 1988;110:5583–5585. [Google Scholar]
- 88.Demchenko AV, Malysheva NN, De Meo CD. Org Lett. 2003;5:455–458. doi: 10.1021/ol0273452. [DOI] [PubMed] [Google Scholar]
- 89.Kamat MN, Demchenko AV. Org Lett. 2005;7:3215–3218. doi: 10.1021/ol050969y. [DOI] [PubMed] [Google Scholar]
- 90.Euzen R, Ferrieres V, Plusquellec D. J Org Chem. 2005;70:847–855. doi: 10.1021/jo0484934. [DOI] [PubMed] [Google Scholar]
- 91.Demchenko AV, Pornsuriyasak P, De Meo C, Malysheva NN. Angew Chem, Int Ed. 2004;43:3069–3072. doi: 10.1002/anie.200454047. [DOI] [PubMed] [Google Scholar]
- 92.Pornsuriyasak P, Demchenko AV. Chem Eur J. 2006;12:6630–6646. doi: 10.1002/chem.200600262. [DOI] [PubMed] [Google Scholar]
- 93.Pornsuriyasak P, Demchenko AV. Tetrahedron: Asymm. 2005;16:433–439. [Google Scholar]
- 94.De Meo C, Parker O. Tetrahedron Asymm. 2005;16:303–307. [Google Scholar]
- 95.Cacciapaglia R, Van Doorn AR, Mandolini L, Reinhoudt DN, Verboom W. J Am Chem Soc. 1992;114:2611–2617. [Google Scholar]
- 96.Lu T, Yoo HK, Zhang H, Bott S, Atwood JL, Echegoyen L, Gokel GW. J Org Chem. 1990;55:2269–2270. [Google Scholar]
- 97.Illuminati G, Mandolini L, Masci BJ. Am Chem Soc. 1983;105:555–563. [Google Scholar]
- 98.Yoo HK, Davis DM, Chen Z, Echegoyen L, Gokel GW. Tetrahedron Lett. 1990;31:55–58. [Google Scholar]
- 99.(a) Cacciapaglia R, Mandolini L. Chem Soc Rev. 1993;22:221–231. [Google Scholar]; (b) Cacciapaglia R, Mandolini L. Pure App Chem. 1993;65:533–538. [Google Scholar]
- 100.Cacciapaglia R, Mandolini L, Romolo FS. J Phys Org Chem. 1992;5:457–460. [Google Scholar]
- 101.Ercolani G, Mandolini L. J Am Chem Soc. 1990;112:423–427. [Google Scholar]
- 102.Cacciapaglia R, Lucente S, Mandolini L, Van Doorn AR, Reinhoudt DN, Verboom W. Tetrahedron. 1989;45:5293–5304. [Google Scholar]
- 103.Cacciapaglia R, Mandolini L, Reinhoudt DN, Verboom W. J Phys Org Chem. 1992;5:663–669. [Google Scholar]
- 104.Kraft D, Cacciapaglia R, Boehmer V, Abu El-Fadl A, Harkema S, Mandolini L, Reinhoudt DN, Verboom W, Vogt W. J Org Chem. 1992;57:826–834. [Google Scholar]
- 105.Brandt K, Siwy M, Porwolik-Czomperlik I, Stilberring J. J Org Chem. 2001;66:5701–5712. doi: 10.1021/jo001725o. [DOI] [PubMed] [Google Scholar]
- 106.Hanessian S, Kagotani M, Komaglou K. Heterocycles. 1989;28:1115–1120. [Google Scholar]
- 107.Hanessian S, Johnstone S. J Org Chem. 1999;71:3285–3286. [Google Scholar]
- 108.Gobbi A, Landini D, Maia A, Secci D. J Org Chem. 1995;60:5954–5957. [Google Scholar]
- 109.Gobbi A, Landini D, Maia A, Penso M. J Chem Soc Perk Trans 2: Phy Org Chem. 1996;11:2505–2509. [Google Scholar]
- 110.Albanese D, Landini D, Maia A. J Org Chem. 2001;66:3249–3252. doi: 10.1021/jo0056388. [DOI] [PubMed] [Google Scholar]
- 111.Lepore SD, Bhunia AK, Cohn PC. J Org Chem. 2005;70:8117–8121. doi: 10.1021/jo051241y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lepore, S.D.; Mondal, D. Unpublished results.
- 113.Kozikowski AP, Lee J. Tet Lett. 1988;29:3053–3056. and references cited therein. [Google Scholar]
- 114.Lepore SD, Bhunia AK, Mondal D, Cohn PC, Lefkowitz C. J Org Chem. 2006;71:3285–3286. doi: 10.1021/jo052333q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sinnott ML, Storesund HJ, Whiting MC. J Chem Soc D: Chem Commun. 1969:1000–1001. [Google Scholar]
- 116.Lepore, S.D.; Mondal, D.; Li, S. Unpublished results.