

UROX is software designed for the interactive fitting of atomic models into electron-microscopy reconstructions. The main features of the software are presented, along with a few examples.
Keywords: UROX, electron microscopy, model fitting, graphical software, normal modes
Abstract
Electron microscopy of a macromolecular structure can lead to three-dimensional reconstructions with resolutions that are typically in the 30–10 Å range and sometimes even beyond 10 Å. Fitting atomic models of the individual components of the macromolecular structure (e.g. those obtained by X-ray crystallography or nuclear magnetic resonance) into an electron-microscopy map allows the interpretation of the latter at near-atomic resolution, providing insight into the interactions between the components. Graphical software is presented that was designed for the interactive fitting and refinement of atomic models into electron-microscopy reconstructions. Several characteristics enable it to be applied over a wide range of cases and resolutions. Firstly, calculations are performed in reciprocal space, which results in fast algorithms. This allows the entire reconstruction (or at least a sizeable portion of it) to be used by taking into account the symmetry of the reconstruction both in the calculations and in the graphical display. Secondly, atomic models can be placed graphically in the map while the correlation between the model-based electron density and the electron-microscopy reconstruction is computed and displayed in real time. The positions and orientations of the models are refined by a least-squares minimization. Thirdly, normal-mode calculations can be used to simulate conformational changes between the atomic model of an individual component and its corresponding density within a macromolecular complex determined by electron microscopy. These features are illustrated using three practical cases with different symmetries and resolutions. The software, together with examples and user instructions, is available free of charge at http://mem.ibs.fr/UROX/.
1. Introduction
The three-dimensional structure of a macromolecular complex provides important information about the intricate interactions between its components. Some macromolecular complexes have been produced in homogeneous form, crystallized and analyzed at high resolution (3–2 Å or better) using X-ray crystallography (XR). However, in many cases they are too large or too unstable to be crystallized and therefore only individual components of such complexes can be analyzed. In contrast, electron microscopy (EM) allows three-dimensional reconstructions of whole macromolecular complexes under close-to-native conditions but is limited to relatively low resolutions. By fitting atomic models of individual components into the EM reconstruction, the latter can be interpreted at a higher than nominal resolution, thereby effectively bridging the different resolution ranges (for recent reviews, see Rossmann et al., 2005 ▶; Volkmann & Hanein, 2003 ▶). The first combination of EM and XR relied simply on visual inspection of the EM map and manual docking of the XR models. Despite the subjectivity inherent to such a procedure, it led to significant results such as the identification of several components of the adenovirus (Stewart et al., 1993 ▶) and its binding footprint (Wang et al., 1992 ▶).
Recent methodological developments have improved the quality of the fitting procedure. A variety of algorithms are currently implemented, including CoAn (Volkmann & Hanein, 1999 ▶), DockEM (Roseman, 2000 ▶), EMfit (Rossmann, 2000 ▶), Foldhunter (Jiang et al., 2001 ▶), Situs (Wriggers et al., 1999 ▶), 3SOM (Ceulemans & Russell, 2004 ▶) and URO (Navaza et al., 2002 ▶). Careful use of such packages enhances the information that can be gained from the fitting compared with a manual docking procedure and allows errors to be estimated using criteria other than the human eye.
However, as far as visual operations are concerned, such as placing the atomic models at initial positions or inspecting putative solutions, most of the above-mentioned packages have to rely on external programs for graphics [e.g. O (Jones et al., 1991 ▶), Coot (Emsley & Cowtan, 2004 ▶), PyMOL (DeLano, 2002 ▶) or VMD (Humphrey et al., 1996 ▶)]. For some algorithms it is also necessary to carve out a piece of the density to reduce the size of the computation problem, which can cause artifacts because some a priori knowledge about the location of the molecule is introduced. The resulting fitting procedure can be cumbersome, especially in difficult cases in which numerical criteria do not unambiguously discern the correct solution. This often occurs when some atomic structures to be fitted into the EM map are not available, leaving unaccounted-for density (Lescar et al., 2001 ▶). In such cases, it is important to be able to graphically position the molecules in the EM map while obtaining rapid (ideally immediate) feedback on the quality of the fitting. The visualization package Chimera (Pettersen et al., 2004 ▶) provides a step in this direction with its real-space fitting module integrated with the graphical display. Unfortunately, it only provides local optimization and will seldom rotate the model by more than 90° or move it more than its diameter (Pettersen et al., 2004 ▶).
The original aim of UROX was to provide a graphical tool with real-time interactive fitting between the EM map and model-derived electron density. In practice, when the user moves a molecule on the graphical display with the mouse, a new correlation is computed and displayed for each incremental motion of the mouse. The calculation should be so fast that the correlation appears as if it were continuously changing while the molecule is moved. This real-time interactivity is designed to serve as a guide for determining a suitable starting point for least-squares minimization. Conceptually, this is comparable to currently available tools for model building in crystallography, such as those implemented in O (Jones et al., 1991 ▶) or Coot (Emsley & Cowtan, 2004 ▶), in which the model is refined interactively when a residue is moved in the density. As an alternative to least-squares minimization, exhaustive searches are included in the package, which can be useful when numerical criteria can clearly discern the correct solution.
An additional difficulty arises when an EM reconstruction possesses a particular symmetry which should be taken into account appropriately in the fitting procedure. UROX incorporates the symmetry of the reconstruction in the graphical representation as well as in the calculations.
Mismatches between the EM map and the fitted molecules can point to inaccuracies or plausible modifications of the models, such as those produced by flexible fitting (Suhre et al., 2006 ▶; Hinsen et al., 2005 ▶; Delarue & Dumas, 2004 ▶; Wriggers et al., 1999 ▶). Normal-mode calculations based on NORMA (Suhre et al., 2006 ▶) are available in UROX.
2. Software design
We start with a summary of the reciprocal-space formalism (Navaza et al., 2002 ▶) and then describe how this formalism is integrated with the graphics.
2.1. Reciprocal-space fitting
The fitting problem is formulated in reciprocal space as the minimization of the so-called ‘quadratic misfit’ (Q) between the electron density based on the molecules (including their symmetry mates) and the EM map (Navaza et al., 2002 ▶). In real space, Q is expressed as
where ρem(r) is the electron density of the EM map, ρmod(r) is the electron density derived from the independent molecules and their symmetry mates and λ is the relative scale between these two densities. The integral in (1) is performed over a volume containing the EM map. On the other hand, in reciprocal space Q is expressed as
where F em(s) and F mod(s) are the Fourier transforms of ρem(r) and ρmod(r), respectively. Explicitly, F mod(s) is expressed in terms of the molecular scattering factors fm of the independent molecules as (Navaza et al., 2002 ▶)
where m refers to one of the M independent molecules located at position X m in orientation R m with respect to a reference position (as detailed in Navaza, 2002 ▶), while g refers to the symmetry operator represented by the translation T g and the rotation M g. F mod is thus a function of the positional variables of the independent molecules.
It is worth noting that minimizing (2) amounts to maximizing the correlation coefficient (CC),
where the overline represents the complex conjugate. In practice, integrals are calculated on discrete regularly spaced grids, which amounts to substituting the integrals over the continuous variable s by summations over the discrete variable h. (1) and (2) are strictly equivalent for both continuous and discrete Fourier transforms. This is not a ‘superficial invocation of Parseval’s theorem’, as stated in Fabiola & Chapman (2005 ▶), but its rigorous application.
The reciprocal-space formalism, as implemented in URO, has been successfully applied to more than 20 fitting problems currently deposited in the EMsearch database (http://www.ebi.ac.uk/msd-srv/emsearch/index.html). This formalism has been adapted in UROX to allow interaction with graphics. Its main advantages are as follows. Firstly, it is extremely fast, which allows real-time calculations. Secondly, one can use the entire EM reconstruction, or at least a substantial part of it (containing all the independent molecules and several of their symmetry mates). Thirdly, it incorporates the symmetry of the reconstruction (see equation 3). Fourthly, it is sufficiently general to be used directly with an electron-density map instead of an atomic model, which corresponds to the ‘map on map’ option in UROX. Additionally, it can be used with low-resolution maps derived from experimental sources other than EM (e.g. small-angle X-ray scattering). We also found that the so-called ‘R factor’ widely used in crystallography and specific to reciprocal space,
 |
helps in assessing the resolution of the EM reconstruction.
2.2. Interaction with the graphics
The main characteristic of UROX that distinguishes it from other fitting packages is the close connection between the graphics and the computations via graphical libraries from the Visual ToolKit (VTK; http://www.vtk.org).
The Python language is used to wrap together Fortran computation subroutines and VTK graphics (Fig. 1 ▶).
Figure 1.

The design of UROX. Core calculations (Fortran77) and graphical libraries (Visualization Toolkit; VTK) are wrapped together with the Python language, using its TKinter module for the graphical user interface.
The core of the interaction between the calculations and the graphics is as follows. The positions and orientations of all molecules are extracted by graphical subroutines each time a molecule is moved by the user. This information is passed to a subroutine that computes a correlation coefficient (4), which is then returned to the display. This computation is extremely fast: 10−7 s per Fourier coefficient and per symmetry operation on a single-processor (2.2 GHz) machine, which makes it possible to compute the CC in real time for an entire EM map.
Moreover, if the map is sizeable, a ‘BoxWidget’ tool from the VTK libraries can be manipulated interactively to inspect local portions of the EM map (see Fig. 3, right). This box can be used to conveniently reduce the field of view and speed up the graphics, but is not used in computations.
A graphical user interface (GUI) is also provided, with a modular architecture so that the user can add or modify components as necessary. Fig. 2 ▶ presents a general overview of the UROX interface. All figures except Fig. 8 are snapshots produced using the ‘take snapshot’ option from the interface’s menu.
Figure 2.

Snapshot of the UROX interface for an icosahedral case (DLP, described in §3). The real-time correlation coefficient and R factor are shown on the left. Further options are available from the menu above the display, including a wizard to facilitate most basic operations.
2.3. Symmetry
Several built-in symmetries are available: icosahedral, tetrahedral, octahedral, helicoidal, dihedral (D n) and cyclic (C n), including of course the case of no symmetry (called C 1 or P 1). These symmetries have been chosen to cover most of the practical cases in EM, but the user has the possibility of adding another one if it is not in the set provided. The symmetry is included in the calculation of (3), with the option of defining a different set of operators for each molecule. This is useful, for example, in the case of a trimeric protein lying on a threefold symmetry axis, as for the icosahedral rotavirus described in §3.
The symmetry is also taken into account in the display: when the user moves one independent molecule in the map, the symmetry mates move as well in real time.
2.4. Strategy
The real-time correlation coefficient is typically used as a guide to place the models in the EM map, which is followed by a least-squares refinement. The latter uses a fast algorithm which has been proven to have a large convergence radius (Castellano et al., 1992 ▶). Alternatively, exhaustive searches can be performed (see below). The choice between these strategies (least-squares minimization or exhaustive searches) depends on the nature of the problem, as explained below.
The radius of convergence of the least-squares minimization is roughly proportional to the resolution of the data. For example, data up to 20 Å typically lead to a radius of convergence of about 30 Å (Navaza et al., 2002 ▶). As the resolution becomes higher, the convergence radius becomes smaller, enhancing the dependence on the initial positioning of the molecules. Therefore, the usual strategy is to perform several cycles of minimization, starting at low resolution and moving to high resolution, while monitoring the CC as well as the positions and orientations of the molecules in real time. In the reciprocal-space formalism, changing resolution is straightforward because the Fourier transforms of the EM map and of the models (F em and F mod, respectively) are calculated only once at the resolution of the EM reconstruction. Overall, the procedure leading to a converged fitting solution should only take a few minutes of CPU time on a single-processor computer (one cycle of least-squares minimization effectively takes about 2.5 × 10−7 s per reflection, per symmetry operator and per molecule).
Exhaustive searches (translational and/or rotational) can also be performed in a user-defined region of the map. A full six-dimensional search (three rotations and three translations) is generally fairly time-consuming and can be avoided, since the positions of the molecules can already be found quite accurately by the least-squares algorithm, especially when symmetry is present. However, we found that including all data up to high resolution in the early stages of refinement can lead to molecules being trapped in false positions corresponding to a local maximum of CC (4). In such a case, rotational exhaustive sampling can prove quite useful.
To accelerate the rotational sampling a Burdina–Lattman parameterization is used (Burdina, 1971 ▶; Lattman, 1972 ▶), taking into account the moments of inertia of the molecule. Indeed, the mean-square shift of the atomic positions {r o} when we move from a rotation and a translation (R, T) to (R + δR, T + δT) is
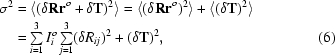 |
where Iio are the model’s principal moments of inertia. Imposing the mean-square shift σ to be of the order of the resolution leads to the shift for the Euler angles δα, δβ and δγ being inversely proportional to the square roots of (Iyy + Izz), (Ixx + Izz) and (Ixx + Iyy), respectively.
2.5. Additional features for speedup
When the number of Fourier coefficients multiplied by the number of symmetry operators becomes greater than 105 (e.g. the rotavirus example below), the computations are too slow to allow real-time interactions if all coefficients are used. In this case, the following additional procedures can be used to speed up the calculations by limiting the number of coefficients, thereby allowing real-time computations.
(i) Perform the calculations at a lower resolution.
(ii) Extract a subset of Fourier coefficients using a decimation procedure.
(iii) Select only those coefficients belonging to the asymmetric unit in reciprocal space.
The rationale behind this is as follows: if N independent molecules have to be placed in the map, only 6N parameters have to be determined, corresponding to the positions and orientations of the molecules. Even though all the Fourier coefficients are not independent of one another, the fitting problem is widely over-determined.
The first two options are applicable in a general case, while the last is particularly useful in the case of high point-group symmetry (e.g. icosahedral symmetry). Note that the loss of high resolution is not critical for the real-time computation, since its goal is to provide a suitable starting point for a subsequent least-squares minimization procedure, which then uses the whole resolution range of the data.
Moreover, the display can also be accelerated either by decimating the EM map, by using the above-mentioned ‘BoxWidget’ and/or by taking advantage of the symmetry of the reconstruction.
2.6. Optimization and flexible fitting
Several parameters can be optimized after a fitting solution is obtained. Firstly, because EM map magnification can have errors of as much as 5%, the absolute scale of the reconstruction is determined by automatically performing least-squares refinement at several magnifications. Secondly, the overall isotropic temperature factors (B factors) of the molecules can also be refined. Thirdly, if the absolute handedness of the EM map is unknown, fitting can be performed with left- and right-handed maps and the correlation coefficient can be used to discriminate between them.
Moreover, after a rigid-body solution has been determined using the procedure described above, the remaining mismatches between the EM map and the fitted molecules can point to inaccuracies or plausible modifications of the models, such as those produced by flexible fitting (Suhre et al., 2006 ▶; Hinsen et al., 2005 ▶; Delarue & Dumas, 2004 ▶; Wriggers et al., 1999 ▶). Normal-mode calculations were included in version 2.0 of UROX in the following two complementary ways.
(i) Each molecule can be perturbed along any normal mode. The perturbation is visualized immediately on the display and a new correlation is computed corresponding to the perturbed molecule. This serves as an estimate of whether a given normal mode is likely to improve the fitting solution.
(ii) A group of normal modes can be selected (after visual inspection as described above) and a downhill simplex algorithm (Press, 1992 ▶) is applied to select the combination of normal-mode amplitudes that maximize the correlation. This procedure is similar to that used in NORMA (Suhre et al., 2006 ▶) but it has been adapted to allow efficient interaction with the graphics. The result is represented in Fig. 3 ▶.
Figure 3.

Normal modes in UROX: snapshot of user interface (left) and result (right). The initial solution (red) along with the normal-mode perturbed solution (blue) are shown on the right. This image also illustrates the BoxWidget tool for truncating the field of view, using the white spheres as handles to reduce or enhance the box that limits the view.
2.7. Error estimates
The fitting algorithm purposefully does not prevent clashes between different molecules placed in the EM map, again with the idea of using only experimental data to avoid bias. The amount of overlap between different molecules can therefore serve as a rough indication of the quality of the fit. Indeed, a model may not exactly fit the EM map because the molecule has undergone modifications, some of which may be taken into account by normal-mode analysis. In addition to CC, other figures of merit such as the R factor (5) are optionally computed to help assess the quality of the solution.
2.8. Additional features
Anisotropy can exist in the EM data, as in the case of tomographic data with a missing wedge region. This can be taken into account by detecting the Fourier coefficients falling in the missing wedge region and excluding them from the summation in (2) or (4). This would not be possible in the real-space formulation (1). A tool to visualize the Fourier coefficients corresponding to an EM map is provided (Fig. 4 ▶). The missing wedge regions can be eliminated by adjusting the threshold.
Figure 4.

Visualization of the Fourier transform of an EM map (GroEL; see §3) to which an artificially created missing wedge was applied. The coefficients are coloured according to their moduli.
Another feature concerns the case of partial occupancy of some molecules. In this case, it is necessary to assign a different occupancy to them by accordingly weighing its contribution to the term F h mod in the discrete form of (2).
3. Applications
3.1. Simple ‘benchmark’: GroEL
To illustrate the difficulties encountered in practice, several of the packages mentioned in §1 were tested on a common case, GroEL. We fitted the crystal structure of GroEL (PDB code 1oel; Braig et al., 1995 ▶) into a cryo negative-stain EM reconstruction of GroEL (DeCarlo et al., 2002 ▶). It is considered to be a ‘simple’ case because there is only one independent molecule to be placed in the map, which contains in total of 14 molecules related by D 7 symmetry, and all the density in the EM map can be accounted for by the models.
It is important to note that the goal of this ‘benchmark’ is not to perform a thorough evaluation of each individual package, but rather to point out the difficulties encountered in practice.
Fig. 5 ▶(a) presents a wrong solution commonly reached by several packages (the correct solution is shown in Fig. 5 ▶ b), which has the model placed at the intersection between the two sevenfold-symmetric rings. This illustrates the importance of taking into account the symmetry of the reconstruction, as in UROX.
Figure 5.

Fitting the solution of the crystal structure of GroEL into a cryo negative-stain EM reconstruction. (a) Wrong solution, (b) correct solution.
Alternatively, one could extract the portion of the map corresponding to one independent molecule, in which case most packages converge to a similar solution. The problem with the latter approach is that it introduces a priori knowledge about the location of the molecule and thus bias.
3.2. Rotavirus capsid proteins
The X-ray crystal structure of VP6, the major capsid protein of rotavirus (PDB code 1qhd; Mathieu et al., 2001 ▶) was fitted into EM reconstructions corresponding to assemblages of different symmetries.
3.2.1. Helical VP6 assemblies
The helical high-pH VP6 assembly (referred to as ‘small tubes’; Lepault et al., 2001 ▶) was reconstructed to a resolution of 20 Å. This reconstruction was chosen to illustrate the difficulty in carving out a volume of density around one molecule (Fig. 6 ▶). Indeed, although several VP6 trimers can be distinguished by eye, the density is continuous between them and it would be difficult to decide where to delineate the contour of a monomer. This problem is circumvented through the reciprocal-space formulation by using an EM map containing several symmetry-related molecules (44 VP6 monomers, more than 20 000 Fourier coefficients to 20 Å). After optimization of the scale factor corresponding to the magnification of the EM map, we obtained a correlation of 94.1% and an R factor of 33.4% (Fig. 7 ▶), which are in agreement with the previously obtained result (Navaza et al., 2002 ▶).
Figure 6.

Electron-density map of part of a helical VP6 assembly (‘small tubes’) contoured at 1.5σ. The enlargement of a VP6 dimer reveals the contiguous density between VP6 monomers.
Figure 7.

Electron-density map of part of a helical VP6 assembly (‘small tubes’), contoured at 1.5σ, with the result of the fit after refinement (CC = 94.1%, R = 33.4%). There are two independent VP6 trimers, coloured in cyan and in blue; molecules related by symmetry are shown in the same colour.
3.2.2. Icosahedral VP6 assemblies
We fitted the atomic model of VP6 into double- and triple-layer assemblies (DLP and TLP, respectively; Libersou et al., 2008 ▶). Both DLP and TLP are icosahedral [with a triangulation number (Caspar & Klug, 1962 ▶) T = 13 for the VP6 layer (Ludert et al., 1986 ▶; Roseto et al., 1979 ▶)] and contain five independent VP6 molecules (four trimers and a monomer). The Fourier transform of each EM map leads to more than 650 000 coefficients at 20 Å resolution. We used only about 11 000 coefficients belonging to the asymmetric unit of the icosahedron to reduce the computational cost to 1 s per refinement cycle. The resulting fit is shown in Fig. 8 ▶. As described elsewhere (Libersou et al., 2008 ▶), we fitted the VP6 atomic model in six reconstructions of viral particles containing different layers of capsid proteins from the rotavirus. The handedness of each reconstruction was checked by fitting into a left-handed and in a right-handed map. The EM magnification was estimated by fitting into a series of reconstructions with different scales (e.g. from 0.9 to 1.1). This example illustrates that the speed of the algorithm is instrumental, considering the number of fits to be performed.
Figure 8.

Electron-density map and results of the fitting of VP6 into double-layer (DLP; left) and triple-layer (TLP; right) assemblies. The VP6 molecules related by symmetry are shown in the same colour. This figure was prepared with PyMOL (DeLano, 2002 ▶).
4. Conclusion
UROX is an interactive software package for fitting atomic models into electron-microscopy reconstructions. It is based on a reciprocal-space formulation adapted for interactive positioning of the molecules in the EM map, with real-time calculation and display of the correlation between them. The symmetry of the EM reconstruction is used both in the calculations and in the graphics.
A user-friendly graphical interface is provided, with a variety of options. The fastest strategy to obtain a fitting solution is based on least-squares refinement, but exhaustive searches are also available. Version 2.0 of UROX now includes normal-mode flexible fitting based on the NORMA package. It is also possible to fit two electron-density maps together, as well as to exclude Fourier coefficients according to a threshold on their moduli. The latter can be used in tomographic applications to exclude missing wedge regions.
As the main programs for the graphical interface are written in a modular way using Python, additional user scripts can easily be incorporated. The UROX software package is available at http://mem.ibs.fr/UROX. At present a compiled version is only available for Linux, but sources can be provided upon request for compilation on other platforms. This site also provides detailed installation instructions including a user manual and several solved examples.
Acknowledgments
We would like to thank Jean Lepault for providing the VP6 reconstructions and Karsten Suhre for his help with NORMA. XS was supported by a Marie Curie International Reintegration Grant (IRG-021715).
References
- Braig, K., Adams, P. & Brünger, A. (1995). Nature Struct. Biol.2, 1083–1094. [DOI] [PubMed]
- Burdina, V. I. (1971). Soviet Phys. Crystallogr.15, 545–550.
- Caspar, D. L. & Klug, A. (1962). Cold Spring Harb. Symp. Quant. Biol.27, 1–24. [DOI] [PubMed]
- Castellano, E. E., Oliva, G. & Navaza, J. (1992). J. Appl. Cryst.25, 281–284.
- Ceulemans, H. & Russell, R. B. (2004). J. Mol. Biol.338, 783–793. [DOI] [PubMed]
- De Carlo, S., El-Bez, C., Alvarez-Rua, C., Borge, J. & Dubochet, J. (2002). J. Struct. Biol.138, 216–226. [DOI] [PubMed]
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, USA. http://www.pymol.org.
- Delarue, M. & Dumas, P. (2004). Proc. Natl Acad. Sci. USA, 101, 6957–6962. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fabiola, F. & Chapman, M. S. (2005). Structure, 13, 389–400. [DOI] [PubMed]
- Hinsen, K., Reuter, N., Navaza, J., Stokes, D. L. & Lacapere, J. J. (2005). Biophys. J.88, 818–827. [DOI] [PMC free article] [PubMed]
- Humphrey, W., Dalke, A. & Schulten, K. (1996). J. Mol. Graph.14, 33–38. [DOI] [PubMed]
- Jiang, W., Baker, M. L., Ludtke, S. J. & Chiu, W. (2001). J. Mol. Biol.308, 1033–1044. [DOI] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Lattman, E. E. (1972). Acta Cryst. B28, 1065–1068.
- Lepault, J., Petitpas, I., Erk, I., Navaza, J., Bigot, D., Dona, M., Vachette, P., Cohen, J. & Rey, F. A. (2001). EMBO J.20, 1498–1507. [DOI] [PMC free article] [PubMed]
- Lescar, J., Roussel, A., Wien, M. W., Navaza, J., Fuller, S. D., Wengler, G., Wengler, G. & Rey, F. A. (2001). Cell, 105, 137–148. [DOI] [PubMed]
- Libersou, S., Siebert, X., Ouldali, M., Estrozi, L. F., Navaza, J., Charpilienne, A., Garnier, P., Poncet, D. & Lepault, J. (2008). J. Virol.82, 2844–2852. [DOI] [PMC free article] [PubMed]
- Ludert, J. E., Gil, F., Liprandi, F. & Esparza, J. (1986). J. Gen. Virol.67, 1721–1725. [DOI] [PubMed]
- Mathieu, M., Petitpas, I., Navaza, J., Lepault, J., Kohli, E., Pothier, P., Prasad, B. V., Cohen, J. & Rey, F. A. (2001). EMBO J.20, 1485–1497. [DOI] [PMC free article] [PubMed]
- Navaza, J. (2002). Acta Cryst. A58, 568–573. [DOI] [PubMed]
- Navaza, J., Lepault, J., Rey, F. A., Álvarez-Rúa, C. & Borge, J. (2002). Acta Cryst. D58, 1820–1825. [DOI] [PubMed]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004). J. Comput. Chem.25, 1605–1612. [DOI] [PubMed]
- Press, W. H. (1992). Numerical Recipes in FORTRAN, 2nd ed. Cambridge University Press.
- Roseman, A. M. (2000). Acta Cryst. D56, 1332–1340. [DOI] [PubMed]
- Roseto, A., Escaig, J., Delain, E., Cohen, J. & Scherrer, R. (1979). Virology, 98, 471–475. [DOI] [PubMed]
- Rossmann, M. G. (2000). Acta Cryst. D56, 1341–1349. [DOI] [PubMed]
- Rossmann, M. G., Morais, M. C., Leiman, P. G. & Zhang, W. (2005). Structure, 13, 355–362. [DOI] [PMC free article] [PubMed]
- Stewart, P. L., Fuller, S. D. & Burnett, R. M. (1993). EMBO J.12, 2589–2599. [DOI] [PMC free article] [PubMed]
- Suhre, K., Navaza, J. & Sanejouand, Y.-H. (2006). Acta Cryst. D62, 1098–1100. [DOI] [PubMed]
- Volkmann, N. & Hanein, D. (1999). J. Struct. Biol.125, 176–184. [DOI] [PubMed]
- Volkmann, N. & Hanein, D. (2003). Methods Enzymol.374, 204–225. [DOI] [PubMed]
- Wang, G. J., Porta, C., Chen, Z. G., Baker, T. S. & Johnson, J. E. (1992). Nature (London), 355, 275–278. [DOI] [PMC free article] [PubMed]
- Wriggers, W., Milligan, R. A. & McCammon, J. A. (1999). J. Struct. Biol.125, 185–195. [DOI] [PubMed]