

Abstract
Estrogen promotes changes in cytoskeletal architecture not easily attributed to the biological action of estrogen receptors, ERα and ERβ. The Gs protein-coupled transmembrane receptor, GPR30, is linked to specific estrogen binding and rapid estrogen-mediated release of heparin-bound epidermal growth factor. Using marker rescue and dominant interfering mutant strategies, we show that estrogen action via GPR30 promotes fibronectin (FN) matrix assembly by human breast cancer cells. Stimulation with 17β-estradiol or the ER antagonist, ICI 182, 780, results in the recruitment of FN-engaged integrin α5β1 conformers to fibrillar adhesions and the synthesis of FN fibrils. Concurrent with this cellular response, GPR30 promotes the formation of Src-dependent, Shc-integrin α5β1 complexes. Function-blocking antibodies directed against integrin α5β1 or soluble Arg-Gly-Asp peptide fragments derived from FN specifically inhibited GPR30-mediated epidermal growth factor receptor transactivation. Estrogen-mediated FN matrix assembly and epidermal growth factor receptor transactivation were similarly disrupted in integrin β1-deficient GE11 cells, whereas reintroduction of integrin β1 into GE11 cells restored these responses. Mutant Shc (317Y/F) blocked GPR30-induced FN matrix assembly and tyrosyl phosphorylation of erbB1. Interestingly, relative to recombinant wild-type Shc, 317Y/F Shc was more readily retained in GPR30-induced integrin α5β1 complexes, yet this mutant did not prevent endogenous Shc-integrin α5β1 complex formation. Our results suggest that GPR30 coordinates estrogen-mediated FN matrix assembly and growth factor release in human breast cancer cells via a Shc-dependent signaling mechanism that activates integrin α5β1.
GPR30 transactivates the epidermal growth factor receptor in human breast cancer cells as a consequence of integrin α5β1-mediated fibronectin matrix assembly.
Fibronectin (FN) is essential for embryogenesis, blood vessel formation, and wound healing (1,2). FN is a soluble plasma protein that is converted into an insoluble fibrillar structure in the extracellular matrix (ECM) by an orderly process known as FN matrix assembly (3). This process is initiated by the interaction between FN and integrin α5β1 and involves a number of intracellular signaling pathways (4,5,6) that promote cellular adhesion, haptotaxis, and survival. Upon engagement of FN, integrin α5β1 undergoes conformational alterations associated with an increase in receptor affinity (7), ultimately resulting in the mechanical deformation of FN (8,9). FN-occupied integrin α5β1 is then recruited to sites of close cell-matrix contact known as “focal adhesions” that contain transmembrane actin stress fibers where robust anchorage to FN occurs. Ligated α5β1 integrin then translocates centripetally out of focal adhesions generating fibrillar adhesions. This directional movement along the actin cytoskeleton stretches and organizes bound FN into fibrils (10). FN-engaged integrin α5β1 has been readily detected with the conformer-specific monoclonal antibody, SNAKA-51, which marks integrin α5β1 at matrix adhesion sites where emergent fibrils are produced (11).
FN has been implicated in cancer development (12). Non-small-cell lung carcinomas often express elevated levels of FN, and adhesion to FN enhances tumorigenicity and confers resistance to apoptosis by chemotherapeutic drugs (13). In the context of the tumor microenvironment, the interaction of integrin α5β1 with soluble FN has been associated with constitutive invasiveness of human prostate carcinoma cells (14). Moreover, successful implantation of mammary tumor xenografts in immunocompromised mice is facilitated by the introduction of exogenous FN, indicating a survival advantage for tumor cells that interact with FN (15). This observation is supported by studies that have shown that mammary adenocarcinoma cells are capable of converting soluble FN into fibrils (16), resulting in increased responsiveness to growth factors and enhanced anchorage-independent growth (17), the attribute that best predicts metastatic potential of transplanted tumor cells in mice.
Recent work has shown that the addition of exogenous FN negatively impacts acinar differentiation and creates a permissive environment for mammary epithelial cell growth (18), suggesting a role for FN in mammary gland function. Both FN and integrin α5β1 are abundantly expressed in the mammary gland and modulated by ovarian steroid hormones (19). However, the mechanism by which estrogen regulates FN-adhesive function remains unclear. A direct influence of estrogen on breast tumor cell behavior has been shown by studies measuring cytoarchitectural alterations in cells cultured in serum in response to hormone stimulation (20,21). Similar cytostructural changes, including enhanced actin stress fiber formation and the establishment of prominent focal adhesions, have been measured in long-term cultures stimulated with the ER antagonist, tamoxifen (22,23), suggesting that alternative estrogen receptors may influence the interaction of breast tumor cells with their ECM.
The Gs-coupled seven-transmembrane receptor, GPR30/GPER-1, has been linked to specific estrogen binding (24,25), stimulation of adenylyl cyclase (26), and transactivation of the epidermal growth factor receptor (EGFR) (27). Additional studies have shown that GPR30 promotes gene transcription in ovarian cancer cells (28) and proliferative effects in endometrial cancer cells (29). Although the biological role of GPR30 has yet to be established, its expression has been linked to the development of advanced breast cancer (30) and high-grade endometrial tumors (31). The studies outlined here were designed to test the hypothesis that GPR30 may regulate breast tumor cell morphology by altering their ability to adhere to FN and deposit a FN matrix.
Results
Estrogen promotes cellular spreading and mobilization of integrin α5β1 to matrix adhesion sites in human SKBR3 breast cancer cells
To examine the influence of estrogen on the breast cancer cell morphology and FN matrix assembly, human SKBR3 breast cancer cells were plated onto glass coverslips and treated with 17β-estradiol (17β-E2) in the absence or presence of exogenous FN (Fig. 1). In the absence of FN, SKBR3 cells adhered but only partially spread, and this cellular morphology was still apparent 24 h later (Fig. 1A). Stimulation with 17β-E2 resulted in significant cellular spreading by 30 min, and cells were fully spread by 2 h after treatment (Fig. 1B). A trivial amount of FN was detected on the surface of SKBR3 cells that were seeded in the absence of exogenous matrix protein (Fig. 1, C and D). Integrin α5β1, the primary FN receptor expressed by SKBR3 cells (data not shown), colocalized at adhesion sites at the periphery of the cell along with human FN, with slightly more receptor accumulating at these sites in 17β-E2-stimulated cells (Fig. 1, E–J). The addition of exogenous human plasma FN resulted in increased cellular spreading in both untreated and 17β-E2-treated cells (Fig. 1, K and L); the degree of spreading, however, was much more pronounced in 17β-E2-stimulated cells. 17β-E2-treated cells produced detectable FN fibrils relative to untreated cells (Fig. 1, M and N). A dramatic increase was observed in the amount of integrin α5β1 codistributing with FN (Fig. 1, P–U), with emergent FN fibrils originating from points where integrin α5β1 clustered at the cell surface (Fig. 1, N, S, and U).
Figure 1.

17β-E2 stimulation of human SKBR3 breast cancer cells promotes morphological alterations and enhanced FN matrix assembly. SKBR3 cells were allowed to adhere to glass coverslips in FN-reduced serum and then left unstimulated or treated with 17β-E2 (10 nm) for 2 h in the absence (A–J) or presence (K–U) of human FN (hFN) (2 μg/ml) and then fixed. Changes in cellular morphology were visualized by differential interference contrast (DIC) microscopy. Integrin α5β1 was detected using the integrin α5β1 inhibitory mAB, SAM-1 and Alexa 488-coupled antimouse IgG. Surface-associated hFN and emergent fibrils were visualized with hFN-specific rabbit polyclonal antibodies and Alexa 594-coupled antirabbit IgG. Colocalization of hFN and integrin α5β1 and an overlay of hFN and DIC are shown. The white arrow illuminates confluence of hFN and integrin α5β1 in samples receiving no exogenous FN. Bar, 10 μm.
To determine whether integrin α5β1 was directly involved in estrogen-mediated FN matrix assembly, SKBR3 cells were fed exogenous human plasma FN and immunostained with SNAKA-51, a monoclonal antibody (mAB) that specifically detects FN-occupied integrin α5β1 conformers, and has been used to measure FN matrix adhesion sites (11). SNAKA-51 codistributed with FN along the perimeter of both unstimulated and estrogen-stimulated SKBR3 cells (Fig. 2), suggesting that integrin α5β1 was engaging FN at these sites. In stimulated cells, SNAKA-51 was not only concentrated more heavily at the edge of the cell but also accumulated along filopodia and at extracellular sites where FN fibrils were deposited (Fig. 2, D and F). This finding, shown clearly in Fig. 2F, is consistent with the concept that engagement of immobilized ECM proteins by integrin leads to an irreversible state (32) that results in integrin retention at former adhesion sites. Whereas the vast majority of SNAKA-51 colocalized with FN at the edge of the cell (Fig. 2F), it is noteworthy that this was not the pattern observed using the inhibitory integrin α5β1-specific mAB, SAM-1 (supplemental Fig. 1S published as supplemental data on The Endocrine Society’s Journals Online web site at http:/mend.endojournals.org). In this case, codistribution of SAM-1 and FN was found neither at the extreme edge of the cell where FN was deposited, nor at extracellular sites of FN deposition but at a site circumscribed by FN (supplemental Fig. 1S). This difference in staining pattern is consistent with the concept that SAM-1 is unable to detect integrin α5β1 that has engaged FN.
Figure 2.

Concentration of FN-occupied integrin α5β1 at matrix adhesions in response to estrogen stimulation. SKBR3 cells were allowed to adhere to glass coverslips in FN-reduced serum, and then left unstimulated (A, C, E, and G) or treated with 17β-E2 (10 nm) (B, D, F, and H) for 2 h in the presence of hFN (2 μg/ml) and then fixed. Changes in cellular morphology were visualized by differential interference contrast (DIC) microscopy. Conformer-specific integrin α5β1 mAB (SNAKA51) was employed to identify FN-occupied integrin α5β1 and was visualized using Alexa 488-coupled antimouse IgG. FN was detected using hFN-specific rabbit antibodies and Alexa 594-coupled antirabbit IgG. Colocalization of hFN and FN-occupied integrin α5β1 and an overlay of hFN and DIC are shown. Bar, 10 μm.
Because SKBR3 breast cancer cells lack ERα and ERβ, mRNA as demonstrated by RT-PCR (Ref. 32 and supplemental Fig. 1S) and protein (27), these findings indicate that 17β-E2 acts in an ER-independent manner to induce significant changes in cellular morphology and promote the mobilization of integrin α5β1 to FN matrix adhesion sites associated with fibrillogenesis.
Estrogen-mediated FN matrix assembly occurs via GPR30 and requires Src-like kinases and matrix metalloproteinases (MMPs) but not the release of pro-HB-EGF or the activation of EGFRs
Conversion of soluble FN into a detergent-insoluble form has been used as a measure of FN matrix assembly by fibroblasts (33). Here we employed this methodology to determine the requirements for enhanced FN fibril formation by estrogen (Fig. 3A). For this purpose, quiescent SKBR3 cells were fed exogenous rat FN in the presence of either 17β-E2, the estrogen receptor (ER) antagonist, ICI 182,780, or no stimulus for 18 h, after which cells were solubilized in deoxycholate (DOC). Cellular proteins were partitioned into DOC-soluble and -insoluble fractions, and equivalent amounts of each fraction were resolved on SDS-polyacrylamide gels and immunoblotted with the rat-specific FN mAB, IC3. The representative data shown in Fig. 3A show that 17β-E2-stimulated cells had a mean increase of 5.2-fold (P < 0.001) in their capacity to assemble a FN matrix as compared with untreated cells. Similar increases were observed in SKBR3 cells that were treated with the ER antagonist, ICI 182,780 (Fig. 3A), consistent with prior data that ICI 182,780 acts as a GPR30 agonist (25,26,27,28,29). Estrogen-mediated FN matrix assembly was inhibited in cells that were pretreated with the Src inhibitor, PP1, which blocks GPR30-mediated EGFR transactivation (27), indicating a requirement for Src-like kinases in estrogen-mediated FN matrix assembly. Similarly, SKBR3 cells pretreated with the pan-MMP inhibitor, GM6001 (Ilomastat), were unable to synthesize insoluble FN, suggesting a requirement for MMPs, in estrogen-stimulated FN matrix assembly. In contrast, prior exposure to the diphtheria toxin mutant, CRM-197, which binds and sequesters proHB-EGF and blocks GPR30-mediated EGFR transactivation (27), had no impact on estrogen-mediated FN matrix assembly, implying that release of pro-HB-EGF is not necessary for FN matrix assembly. CRM-197 did not affect FN matrix assembly by ICI 182, 780-treated SKBR3 cells, despite the fact that this treatment inhibits ICI 182,780-mediated EGFR transactivation by these cells (data not shown). To further discern the order of events in GPR30-mediated estrogen signaling, AG1478 was employed to block EGFR function and ascertain the role of growth factor receptor activation in FN matrix assembly. As shown in Fig. 3B, AG1478 does not inhibit FN matrix assembly induced by 17β-estradiol, ICI 182,780, and angiotensin II (ATII). In addition, EGF and HB-EGF are unable to promote FN matrix assembly under any of the conditions tested. In contrast, AG1478 effectively blocked E2, EGF, and HB-EGF induced activation of EGFR/erbB1 (Fig. 3C). These data suggest that estrogen-mediated FN matrix assembly is not likely manifested by ER and may be associated with GPR30-dependent action. Further, these results indicate that estrogen-mediated FN matrices form independently of growth factor release and subsequent downstream activation of erbB1.
Figure 3.

GPR30-mediated FN matrix assembly occurs independently of EGFR transactivation in SKBR3 breast cancer cells. A, SKBR3 cells were fed rat FN (rFN) (25 μg/ml) in the presence of either 17β-E2 (1 nm) or ICI 182,780 (1 μm) or no agonist (untreated, UN) for 18 h. Alternatively, cells were pretreated with the pan MMP-inhibitor, GM6001 (10 μm for 15 min), Src-like kinase inhibitor, PP1 (50 nm for 15 min), or CRM-197 (200 ng/ml for 1 h) before receiving rFN in the presence of 17β-E2. Equivalent amounts of DOC-soluble or -insoluble protein were blotted with rFN-specific mAB IC3. The immunoblot shown is a representative of four independent experiments. Band intensities from individual experiments were measured using Scion Image software. Mean integrated optical densities (±sem) were measured for unstimulated (28.6 ± 11.5) and 17β-E2-stimulated (148.1 ± 15.7) cells. B, SKBR3 cells were pretreated with either the erbB1-specific inhibitor, tyrphostin AG1478 (1 nm) or vehicle (DMSO) for 15 min before receiving rFN in the presence of 17β-E2 (1 nm), ICI 182, 780 (1 μm), EGF (10 ng/ml), HB-EGF (10 ng/ml), or no agonist. FN matrix assembly was measured after 18 h of treatment as described above. C, SKBR3 cells treated with AG1478 or vehicle as described in panel B in the absence of exogenous rFN. Cells were then lysed and erbB1 tyrosyl phosphorylation was assessed in erbB1 immunoprecipitates that were immunoblotted with phosphotyrosine-specific 4G10 mAB. ErbB1 recovery was determined by reprobing the same filter with sheep EGFR antibodies. DMSO, Dimethylsulfoxide.
We have previously shown that forced expression of GPR30 into GPR30-deficient human MDA-MB-231 breast cancer cells is required for estrogen-mediated activation of adenylyl cyclase (26) or erbB1 (27). Notably, forced GPR30 expression in MDA-MB-231 cells has no effect on the basal level of ERα mRNA (supplemental Fig. 1) or the protein level of ERα and ERβ as reported previously (27). Restoration of GPR30 expression in MDA-MB-231 cells is required to promote FN matrix assembly, albeit, the increase in FN matrix assembly in response to estrogen stimulation was not as substantial as measured by immunofluorescence or immunoblotting in SKBR3 cells (data not shown). Therefore, to address the potential influence of endogenous GPR30 protein expressed in SKBR3 cells on estrogen-mediated FN fibril formation, a dominant-negative strategy was employed. Because truncated versions of seven-transmembrane receptors (7TMRs) have been widely used as negative regulators of their WT receptor counterparts (34,35,36,37), a carboxyl terminally truncated, hemagglutinin-tagged mutant of GPR30, termed “GPR30Δ154” was engineered and stably expressed in SKBR3 cells. Expression of GPR30Δ154 selectively inhibited 17β-E2-enhanced FN matrix assembly with no impact on ATII-mediated FN matrix assembly (Fig. 4A). In contrast, 17β-E2-enhanced FN matrix assembly was maintained in SKBR3 cells that were stably transfected with pcDNA3.1 Zeo (+) vector DNA. The stereoisomer, 17α-E2, which neither competes for GPR30 binding activity (25,26) nor promotes erbB1 activation (27), does not facilitate FN matrix assembly by vector-transfected SKBR3 cells (Fig. 4A). Approximately equivalent amounts of DOC-soluble rat FN were associated with vector- or GPR30Δ154-transfected SKBR3 cells indicating that the relative differences in DOC-insoluble material were not associated with a difference in the capacity of these cells to bind soluble rat FN. Notably, the expression of the dominant-negative acting GPR30 mutant GPR30 (Δ154) had no impact on the expression of endogenous GPR30 mRNA as demonstrated by RT-PCR (supplemental Fig. 2). This result is not surprising based on the fact that dominant-negative mutants of G protein-coupled receptors (GPCRs) often inhibit signaling through ligand saturation or the formation of heterogenic signaling molecules with endogenous receptors, and this has been shown by both FRET and BRET analyses (reviewed in Refs. 36 and 37).
Figure 4.

Expression of truncated GPR30 blocks FN matrix assembly and EGFR transactivation in SKBR3 breast cancer cells. A, SKBR3 (GPR30Δ154) or (vector) cells were fed exogenous rFN in the absence of stimulus (untreated, UN) or in the presence of 17β-E2, 17α-E2, or (ATII) for the indicated periods of time (hours). DOC-soluble or -insoluble proteins were immunoblotted with the rFN-specific mAB, IC3. B, SKBR3 (GPR30Δ154) or (vector) cells were left untreated or exposed to 17β-E2 (1 nm), ICI 182,780 (1 μm), ATII (1 μm), or EGF (1 ng/ml) for 15 min and then lysed in detergent. ErbB1 was immunoprecipitated from equivalent amounts of total cellular protein. Tyrosyl phosphorylation was measured by blotting with the phosphotyrosine-specific mAB 4G10. ErbB1 recovery was determined by reprobing the same filter with sheep EGFR antibodies. The data are representative of three independent experiments. Integrated optical densities from the experiment shown are plotted below. Data are presented as a fold-increase in EGFR tyrosyl phosphorylation relative to unstimulated cells and are normalized for erbB1 recovery.
The fact that GPR30Δ154 selectively blocked estrogen-dependent FN matrix assembly in SKBR3 cells that express endogenous GPR30 protein provided the rationale to utilize this mutant in studies designed to examine GPR30-dependent erbB1 transactivation (Fig. 4B). Treatment of vector-transfected SKBR3 cells with 17β-E2, ICI 182,780, or EGF resulted in increased tyrosyl phosphorylation of erbB1 (8.7-, 4.9-, and 9.8-fold, respectively). In contrast, GPR30Δ154-expressing SKBR3 cells were selectively impaired with regard to their capacity to promote erbB1 tyrosyl phosphorylation in response to stimulation by either 17β-E2 or ICI 182,780. However, SKBR3 (GPR30Δ154) cells remained responsive to EGF (9.3-fold increase), demonstrating that they retain competent erbB1 (Fig. 4B). Likewise, ATII stimulation of SKBR3 (GPR30Δ154) cells also activated erbB1 (4.9-fold increase), further suggesting that the GPR30-interfering mutant had no effect on EGFR transactivation mediated via another G protein-coupled receptor in these cells. Although not the focus of this paper, it is interesting to note that ATII-mediated FN assembly and transactivation of the EGFR are both blocked by inhibitors of Src (data not shown). These data suggest that a common pathway may be used by both ATII receptor and GPR30 with regard to the initiation of FN matrix assembly and downstream activation of EGFRs, despite the fact that they are coupled to different G proteins.
Truncated GPCRs are commonly used to inhibit GPCR signaling, and we found that truncated GPR30 similarly inhibited its signaling capacity. Targeting of GPCR expression through antisense oligonucleotide-mediated reduction of GPR30 also yielded similar results. SKBR3 cells pretreated with antisense phosphothioate oligonucleotides showed a dramatic decrease in GPR30 mRNA when compared with cells treated with a sense control (supplemental Fig. 3). Antisense-induced reduction in GPR30 RNA expression from SKBR3 cells correlated with a deficiency in 17β-estradiol-mediated FN matrix assembly. These cells remained competent in their capacity to produce a FN matrix after stimulation with ATII that acts through its cognate receptor. In contrast, cells treated with control GPR30 sense oligonucleotides were unaffected and formed matrices in the presence of either ATII or E2. These data mimic the results seen with the GPR30 dominant-negative mutant (Δ154) and further support the role of GPR30 as a stand-alone receptor with regard to rapid estrogen signaling.
These findings imply that estrogen signaling through endogenous GPR30 not only facilitates transactivation of erbB1 but also enhances integrin α5β1-mediated FN matrix assembly.
Engagement of integrin α5β1 is required for GPR30-dependent transactivation of erbB1 in human breast cancer cells
In some instances, ECM engagement by integrin β1 is required for 7TMR signaling (38,39). Therefore, we examined whether activation of integrin α5β1 and FN matrix assembly by GPR30 were associated with erbB1 transactivation. To test this possibility, tyrosyl phosphorylation of erbB1 was measured in GPR30-expressing breast cancer cells that were treated with inhibitory antibodies that interfere with the interaction of integrin α5β1 with FN and block FN matrix assembly (Fig. 5A). Pretreatment of MDA-MB-231 (GPR30) cells with the integrin α5β1-inhibitory mAB, P1D6, abrogated 17β-E2-mediated erbB1 tyrosyl phosphorylation. In contrast, estrogen-mediated erbB1 tyrosyl phosphorylation was unaffected in cells that were exposed to integrin α2β1-inhibitory antibody, AK-7. Interesting to note is that although the AK-7 antibody is unable to block EGFR activation in MDA-MB-231 (GPR30) cells it has profound inhibitory effects on the ability of these cells to adhere to collagen but not FN (data not shown). A requirement for integrin α5β1 engagement of FN in GPR30-mediated EGFR transactivation was also shown in experiments using soluble peptide fragments of FN containing the RGD (arginine-glycine-aspartic acid) recognition sequence. Interaction of integrin α5β1 with the RGD recognition sequence with the type III repeats within FN is required for FN matrix assembly (33). Although pretreatment of MDA-MB-231 (GPR30) cells with soluble RGD peptide inhibited 17β-E2-mediated erbB1 tyrosyl phosphorylation, control RGE (arginine-glycine-glutamic acid) peptide did not (Fig. 5B). Neither peptide had a significant effect on EGF-induced erbB1 activation. Similar results were obtained in SKBR3 cells (data not shown), and neither SKBR3 cells nor MDA-MB-231 cells expressed significant amounts of integrin αvβ3 (data not shown), which is also capable of binding FN via the RGD recognition sequence (40).
Figure 5.
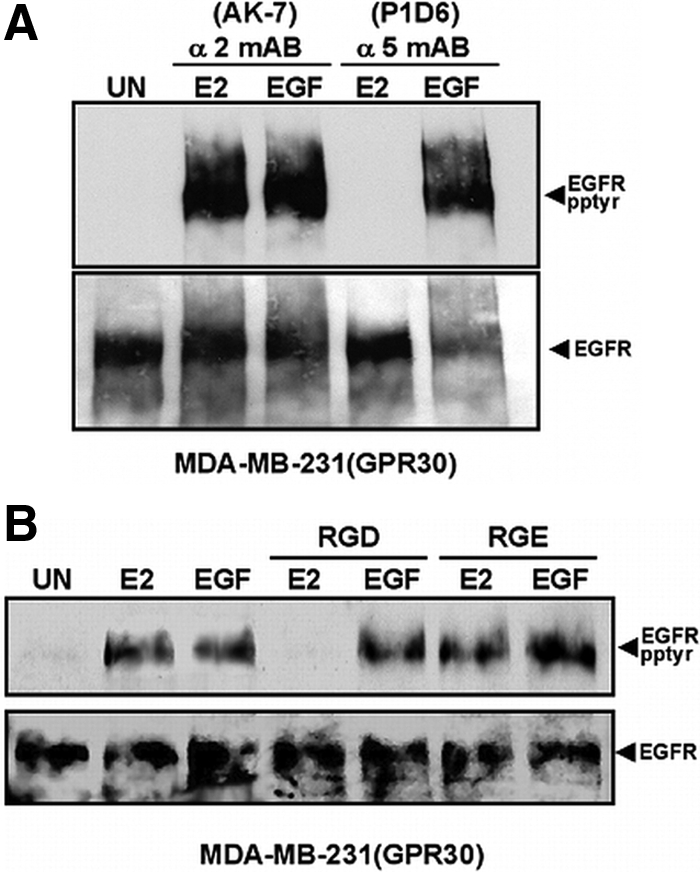
Integrin α5β1 is a necessary signaling intermediary for GPR30-dependent EGFR transactivation. ErbB1 tyrosyl phosphorylation was measured in MDA-MB-231 (GPR30) cells that were left untreated (UN) or pretreated with: (A) function blocking integrin-specific mABs for α5β1 (mAB P1D6; 10 μg/ml) or α2β1 (mAB AK-7; 20 μg/ml) before stimulation or (B) soluble RGD or RGE peptides (1 mm) and then stimulated with E2 or EGF. Tyrosyl phosphorylation of immunoprecipitated erbB1 was determined by immunoblotting with phosphotyrosine-specific mAB, 4G10. Reprobing with sheep EGFR antibodies assessed ErbB1 recovery.
To further demonstrate the role of integrin α5β1 in estrogen-mediated EGFR activation and FN matrix assembly, these estrogen-dependent signaling events were examined in GE11 epithelial cells derived from β1-null mice (41), and in GE11 (β1) cells in which integrin β1 subunit protein expression was restored by transfection (Fig. 6). Neither 17β-E2 nor ICI 182,780 was capable of promoting erb1 activation in GE11 cells despite the fact that these cells express high levels of GPR30 (supplemental Fig. 2) and erbB1 activation was readily measured in response to EGF stimulation (Fig. 6A). Upon expression of β1 subunit protein in GE11 cells, the resultant GE11 (β1) cell population acquired the capacity to transactivate erbB1 after exposure to either 17β-E2 or ICI 182,780. A similar dependency on β1-integrin expression was observed for estrogen-mediated FN matrix assembly in the GE11 cell background (Fig. 6B).
Figure 6.

Restoration of integrin β1 expression in integrin β1-null mouse epithelial cells is necessary for GPR30-induced erbB1 tyrosyl phosphorylation and FN fibril formation. Mouse GE11 (integrin β1 knockout) epithelial cells or GE11 cells stably transfected with integrin β1 subunit protein (GE11β1) were assessed for their capacity to promote estrogen-mediated (A) erbB1 tyrosyl phosphorylation or (B) FN matrix assembly. Cells were made quiescent and then left untreated (Un) or stimulated with 17β-E2 (1 nm), ICI 182,780 (1 μm), or EGF (10 ng/ml). FN matrix assembly (18 h) and EGFR transactivation (15 min) were measured as described in Fig. 3. C, GE11β1 cells were stably transfected with HA-GPR30Δ154 Hygro (B, D, F, and H) or vector, pcDNA3.1(+) Hygro (A, C, E, and G) and then treated with 17α-E2, 17β-E2, ICI 182,780, or ATII for 18 h in the presence of hFN (2 μg/ml). Fibril formation was assessed as in Fig. 2. Insets depict expanded views of fibrillogenesis. Bar, 10 μm.
To determine whether the estrogen-mediated responses that were measured in GE11 (β1) cells occurred via GPR30, the truncation mutant GPR30Δ154, or a control vector was stably introduced into GE11 (β1) cells, and FN fibril formation was examined. Vector-transfected GE11 (β1) cells produced FN fibrils in response to either 17β-E2 or ICI 182,780 (Fig. 6C). Similarly, ATII facilitated the production of FN fibrils, whereas 17α-E2 remained incapable of promoting FN matrix assembly in vector-transfected GE11 (β1) cells. Expression of GPR30Δ154 selectively inhibited estrogen and ICI 182,780-mediated FN fibril formation but had no impact on ATII-induced fibrillogenesis. Interestingly, as shown (in supplemental Fig. 2), endogenous mouse GPR30 expression is unaffected by expression of GPR30Δ154, whereas FN matrix assembly is markedly reduced.
These data in mouse GE11 (β1) cells mimic the results observed in human breast cancer cells that express GPR30 and further indicate that α5β1 is required for GPR30-mediated estrogen-induced erbB1 tyrosyl phosphorylation and enhanced FN-matrix assembly. Together, these data suggest that FN matrix engagement by integrin α5β1 is a necessary intermediary signaling component for GPR30-mediated erbB1 transactivation.
Shc is required for GPR30-mediated erbB1 transactivation and FN fibril formation
The adaptor protein Shc associates with the EGFR after GPR30 stimulation (27), consistent with its role in facilitating the recruitment of protein kinase signaling modules that are activated downstream of EGFR autophosphorylation, such as erk-1/-2. However, surprisingly, Shc317Y/F, a point mutant that lacks its primary tyrosyl phosphorylation site, selectively blocks activation of erk-1/-2 by 17β-E2, but not EGF, despite the fact that intracellular signaling by these stimuli converge at the EGFR (27). Because integrin α5β1 is an upstream requirement for GPR30-mediated EGFR transactivation and Shc is commonly associated with integrin activation (42,43,44,45,46), the hypothesis that GPR30 may employ Shc upstream of erbB1 was tested. Tyrosine phosphorylation of erbB1 was measured in detergent lysates prepared from SKBR3 cells expressing glutathione-S-transferase (GST) fusion proteins containing either WT or mutant (317Y/F) Shc (Fig. 7). Cells transfected with either recombinant Shc protein demonstrated approximately equivalent amounts of both endogenous Shc isoforms (p52 and p46) and exogenous GSTShc (apparent molecular mass of 78 kDa) (Fig. 7A). [As reported previously, SKBR3 cells fail to express the p66Shc isoform (47)]. Tyrosyl phosphorylation of erbB1 was selectively inhibited in 317Y/FShc cells treated with 17β-E2, but not EGF (Fig. 7B). In contrast, erbB1 activation by either 17β-E2 or EGF was unimpaired in WTShc cells. These results indicate a novel position for Shc protein in EGFR signaling, acting upstream of the EGFR in GPR30-mediated transactivation.
Figure 7.

Expression of 317Y/F Shc inhibits GPR30-mediated erbB1 transactivation and FN matrix assembly. A, Equivalent amounts of total cellular protein prepared from SKBR3 breast cancer cells transfected with vector, GST-WT-Shc, or GST-317Y/F-Shc were immunoblotted with the pan-Shc mAB PG797. B, ErbB1 tyrosyl phosphorylation (panel B) or FN matrix assembly (panel C) was measured in SKBR3 cells transfected with GST-WT-Shc or GST-317Y/F-Shc and left untreated (Un) or stimulated with 17β-E2 or EGF for the times indicated. Vec, Vector.
Similarly, an inhibitory effect by 317Y/FShc was observed on GPR30-mediated FN matrix assembly by SKBR3 cells (Fig. 7C). Whereas WTShc-expressing SKBR3 cells produced insoluble FN in response to 17β-E2, expression of 317Y/F Shc completely blocked estrogen-induced FN matrix assembly by SKBR3 cells. Conversion of soluble FN into an insoluble form in response to ATII was also abrogated in 317Y/F Shc cells (supplemental Fig. 4), indicating a general role for Shc in FN matrix assembly. Thus, the data presented in Figs. 5–7 suggest that Shc and integrin α5β1 are necessary intermediaries for GPR30-mediated FN matrix assembly as well as erbB1 transactivation.
GPR30 promotes the formation of Src-dependent, Shc-integrin α5β1 complexes
Shc is known to associate with β1 and β3 integrin subunit proteins after their engagement of ECM proteins and has been used as a measure of outside-in integrin activation (42,44,46), a process associated with activation of Src-like kinases. Because GPR30-dependent activation of the EGFR involves Src-like kinases (27), we tested whether integrin α5β1-Shc complexes formed in response to GPR30 stimulation in either MDA-MB-231 or SKBR3 cell backgrounds (Figs. 8 and 9). Triton-soluble detergent extracts were prepared from vector- or GPR30-transfected MDA-MB-231 cells that were treated with 17β-E2 or EGF. Integrin α5β1 or integrin α2β1 was immunoprecipitated with antibodies specific for integrin α5 or α2 subunit proteins, and the resulting immunoprecipitates were then assayed for associated Shc proteins. Unlike SKBR3, MDA-MB-231 breast cancer cells express all three isoforms of endogenous Shc. Cells treated with 17β-E2, but not EGF, promoted the association of p66, p52, and p46 Shc isoforms with α5β1 protein in MDA-MB-231 (GPR30) cells (Fig. 8). Pretreatment of MDA-MB-231 (GPR30) cells with the Src family kinase inhibitor, PP1, abolished the formation of 17β-E2-induced integrin α5β1-Shc complexes, implying that Src-like kinase activity is required for these complexes to form. Integrin α5β1-Shc complexes were not detectable in 17β-E2-stimulated, MDA-MB-231 (vector) cells. In contrast, integrin α2β1-Shc complexes were readily detected in untreated MDA-MB-231 (GPR30) cells, and a marginal increase in the formation of this complex was observed after treatment with either 17β-E2 or EGF. These data suggest that Src-like kinases are required for GPR30 recruitment of Shc to integrin α5β1, a finding that is consistent with the fact that Src is necessary for FN matrix assembly by fibroblasts (4) and SKBR3 cells treated with E2 (Fig. 3A). In contrast, integrin α2β1 forms stable complexes with Shc under conditions of quiescence, indicating that further stimulation is not required for integrin α2β1-Shc complex formation.
Figure 8.

GPR30 signaling promotes the formation of Src-dependent, Shc-integrin α5β1 protein complexes. Triton X-100 extracts prepared from MDA-MB-231 cells stably expressing vector or GPR30 were left untreated (UN) or stimulated with 17β-E2, EGF, or pretreated with the Src family kinase inhibitor, PP1 (50 nm; 15 min) before stimulation with 17β-E2. Integrin α5β1 or α2β1 was immunoprecipitated with rabbit antibodies specific for the integrin α5 or α2 subunit cytoplasmic tails, respectively. Associated Shc proteins were detected with Shc mAB, PG797. Recovery of integrin α5β1 or α2β1 was determined by blotting with integrin α5 or α2 subunit protein antibodies. Relative positions of endogenous p66, p52, and p46 Shc are designated.
Figure 9.

Engagement of integrin α5β1 is required for GPR30-mediated, Shc-integrin α5β1 protein complexes. SKBR3 cells transfected with mutant GST-317Y/F-Shc, GST-WT-Shc, or GPR30Δ154 received no peptide, RGD peptide, or control RGE peptide and then were left untreated or stimulated with 17β-E2 or EGF for 15 min and lysed in Triton X-100. Shc-integrin α5β1 complexes were determined as described above. The relative amount of integrin α5β1-associated recombinant Shc protein was computed from densitometric tracings from two experiments and normalized relative to the amount of total integrin α5β1.
FN engagement is required for GPR30-induced integrin α5β1-Shc complex formation but tyrosyl phosphorylation of Shc residue 317 is not
To evaluate the requirement of FN engagement on integrin α5β1-Shc complex formation, SKBR3 cells expressing either recombinant WT or mutant (317Y/F) Shc protein were pretreated with monomeric soluble RGD or RGE peptide before stimulation by 17β-E2 (Fig. 9). After hormone stimulation, detergent lysates were prepared, and integrin α5β1-Shc complexes were assessed as described above. Analysis of recombinant and endogenous Shc proteins immunoprecipitated with integrin α5β1 from these samples showed that 17β-E2 promoted the association of p52- and p46Shc isoforms with integrin α5β1 in either WT- or 317Y/F Shc cells (Fig. 9). Interestingly, both WT and mutant Shc GST-fusion proteins associated with integrin α5β1 after stimulation by 17β-E2. However, despite the fact that equivalent amounts of total GSTShc fusion proteins were expressed in these cells, as shown in the representative experiment in Fig. 7A, recombinant mutant Shc protein accumulated in significant excess relative to recombinant WT Shc protein with integrin α5β1 (∼5-fold). Association of integrin α5β1 with either recombinant or endogenous Shc isoforms was not influenced by RGE peptide, whereas RGD peptide prevented all isoforms from complexing with this integrin. Although endogenous Shc isoforms did not complex with integrin α5β1 isolated from 17β-E2-stimulated SKBR3Δ154 cells (Fig. 9), p46 and p52 Shc proteins did couple to integrin α5β1 after stimulation with ATII, suggesting that the recruitment of Shc to integrin α5β1 requires GPCR signaling (data not shown). These findings imply that, although tyrosyl phosphorylation of Shc at residue 317 is required for GPR30-mediated EGFR transactivation and FN matrix assembly (Fig. 7), the ability of Shc to complex with integrin α5β1 is independent of tyrosyl phosphorylation at this site (Fig. 9). Moreover, our data indicate that the ability of 317Y/F Shc to block GPR30 mediated-EGFR transactivation and FN matrix assembly is not due to the result of a failure of the endogenous p52- and p46 Shc isoforms to complex with integrin α5β1 in the presence of this mutant.
Discussion
In this study, we report that estrogen action via the G protein-coupled receptor, GPR30, results in activation of integrin α5β1, resulting in enhanced FN matrix assembly in breast cancer cells. As summarized in the model in Fig. 10, we further show that integrin α5β1 and the signaling adapter Shc are necessary signaling intermediaries required for GPR30-mediated EGFR transactivation. These data are particularly interesting in light of the fact that β1 integrin and Shc are required for mammary gland homeostasis. Namely, transgenic mice conditionally expressing an interfering mutant of β1-integrin exhibit defects in mammary gland development and function as well as Shc signaling (48).
Figure 10.

Model depicting the role of integrin α5β1 and Shc in FN matrix assembly and EGFR transactivation. Estrogen action via the 7TMR GPR30 results in Gβγ-subunit protein-dependent activation of integrin α5β1 as measured by Shc protein coupling and FN matrix assembly. Activated integrin α5β1, in turn, promotes erbB1 transactivation by release of membrane-tethered HB-EGF, resulting in EGFR tyrosyl phosphorylation (PY). Data provided here indicate that activation of integrin α5β1 is a prerequisite event for EGFR transactivation by GPR30 and that sequestration of pro-HB-EGF or inhibition of the tyrosine kinase activity of erbB1 does not interfere with GPR30-mediated FN matrix assembly.
17β-Estradiol action via GPR30 promotes the spreading of human breast cancer cells and FN fibril production (Fig. 1) associated with the localization of FN-engaged integrin α5β1 in fibrillar adhesions (Fig. 2). In our experiments, exogenous FN (2 μg/ml) was required to readily measure FN fibril formation stimulated by 17β-E2 (Fig. 1). This concentration of FN did not modulate FN receptor affinity because equivalent SNAKA-51 staining was observed in unstimulated vs. 17β-E2-stimulated cells (Fig. 2). These findings indicate that FN engagement was not sufficient to drive integrin α5β1 to fibrillar adhesions but that additional signaling from GPR30 was required for fibrillogenesis to occur. FN fibrils continued to mature and grow in SKBR3 cells that were stimulated with 17β-E2 up to 30 h, but after this time FN fibrils were no longer contiguous and showed evidence of degradation (Quinn, J. A., and E. J. Filardo, unpublished results). In this context, it is interesting to note that fragments of FN activate matrix-degrading proteases that facilitate the release of EGF-like sequences from matrix proteins (49).
For several reasons, integrin α5β1 serves as a likely conduit to promote transmembrane signaling between GPR30 and the EGFR. First, Src-like kinase activity is generally required for GPCR-mediated EGFR transactivation (27), and Src family members play an important role in integrin function, including the following: their recruitment to focal adhesions (50), FN matrix assembly (4), and focal adhesion kinase (FAK) activation at cell-matrix interaction sites (51). Second, Shc interacts directly with integrin and focal adhesion kinase, FAK, and other proteins that become tyrosyl phosphorylated in focal adhesion plaques (50,51). Third, GPR30 (52) like other GPCRs, employ membrane-associated proteases for the purpose of releasing membrane-tethered EGF-like ligands. In support of this concept, integrin αvβ3 has been reported to associate directly with MMP-2 (53), and ADAMs (a Disintegrin and a Metalloproteinase) family members have integrin interaction domains (54,55). Thus, integrins may provide a general mechanism for localizing and concentrating these matrix-degrading proteases at the cell surface, which, in turn, may trigger the release of membrane-tethered EGF-like ligands. It is noteworthy that the engagement of native or partially proteolyzed FN by integrin is associated with increased MMP activity (56,57,58). Similarly, the interaction of soluble FN with hepatocellular carcinoma cells induces ADAM-mediated proliferation and invasion that are abrogated by gefitinib, a small molecule EGFR inhibitor (59). Collectively, these observations further support the case that integrin engagement by FN is not only a prerequisite step in FN matrix assembly but may lead to EGFR activation.
Integrins are bidirectional transmembrane signaling effectors (60,61) that regulate their ability to recognize their cognate ECM proteins. Affinity modulation of leukocyte or platelet integrins by a process referred to as “inside-out” signaling, is triggered by vasoactive substances or chemokines, respectively. These agonists share the common property that they generally promote their intracellular actions via 7TMRs. 7TMR agonists also induce integrin clustering, the formation of focal adhesions, and downstream signaling events. Conversely, “outside-in” signaling is triggered by integrin engagement of matrix protein and results in increased integrin affinity. Inhibition of GPR30-induced EGFR tyrosine phosphorylation by monomeric soluble RGD peptides (Fig. 5B) implies that this process requires integrin engagement. Prior studies have similarly shown that ECM engagement by integrin β1 is required for downstream signaling by other GPCRs. Namely; ECM engagement is required for muscarinic acid receptor-mediated activation of FAK and paxillin (39) as well as bombesin-induced induced activation of pro-MMP-9 (40). Tyrosyl phosphorylation of FAK and paxillin are associated with outside-in integrin signaling (50,51); however, we failed to detect differences in FAK phosphorylation at its autoactivation site after GPR30 stimulation (Quinn, J. A., and E. J. Filardo, unpublished results). It has been argued that FAK activation occurs as a direct result of ligand occupancy or tethering (62) and is proportional to the number of integrin α5β1-FN bonds (63). One possible explanation for our inability to measure FAK autophosphorylation is that GPR30 stimulation in human breast carcinoma cells does not generate a number of integrin α5β1-FN bonds sufficient to exceed a signaling threshold that permits FAK activation.
Ectopic expression of GPR30 restores estrogen-induced release of pro-HB-EGF from the surface of breast cancer cells (27). Here we show that a carboxyl-terminal deletion mutant of GPR30 (GPR30Δ154), blocks estrogen-induced EGFR transactivation and FN matrix assembly in cells that express endogenous GPR30 (Fig. 4, A and B). These results are consistent with work that has shown that carboxyl-terminal truncated versions of the heptahelical receptors for vasopressin and dopamine have a negative regulatory effect on signaling (34,35). Truncated versions of these receptors function as dominant-negative receptors by interfering with the surface expression of WT counterparts. This is consistent with a growing body of evidence suggesting that GPCRs function as multimeric molecules on the cell surface (36,37). Our present findings provide additional evidence indicating that estrogen-induced EGFR tyrosine phosphorylation is ER independent and occurs via GPR30. Support for this concept is offered by the fact that the ER antagonist, ICI 182,780, activates the EGFR in vector-transfected SKBR3 cells that lack ER, and expression of truncated GPR30 blocks EGFR transactivation by this same antagonist (Fig. 4B).
Our data indicate that Shc acts in an unfamiliar position in EGFR signaling, upstream of the EGFR. This conclusion is supported by the observation that expression of a mutant Shc protein lacking its primary tyrosine phosphorylation site at residue 317, blocks 17β-E2-induced, but not EGF-induced, EGFR tyrosine phosphorylation (Fig. 7B). Prior work has shown that tyrosyl phosphorylation of Shc is not necessary for its association with integrin αllbβ3 (42). Consonant with this observation, Shc317Y/F associates with integrin after GPR30 stimulation, indicating that phosphorylation of this tyrosine is not necessary for Shc to associate with integrin α5β1 (Fig. 9). In fact, mutant GST-317Y/F Shc is more readily detected on integrin α5β1 than GST-WTShc or endogenous Shc isoforms, even though equivalent amounts of both recombinant and endogenous Shc proteins are expressed in these SKBR3 transfectants (Fig. 7A). The results presented here do not explain the mechanism by which GST-317Y/F Shc interferes with GPR30-dependent FN matrix assembly or EGFR transactivation. The observation that this mutant is more readily detected with integrin α5β1 suggests that phosphorylation at 317 may result in Shc disengagement from integrin. Conversely, dephosphorylation of this tyrosyl residue may cause Shc-integrin disassembly. It is tempting to speculate that GST-317Y/F interferes with integrin clustering by preventing Shc from interacting with cytoskeletal-associated proteins containing SH2-binding domains, and that this, in turn, prevents enhanced FN matrix assembly and downstream activation of the EGFR. However, our data reported here does not investigate the status of Shc Y239/240 phosphorylation (64,65), or how this may vary between WT and 317Y/F Shc proteins, or whether phosphorylation of Y239/240, or the phosphotyrosine-binding domain of Shc affects Shc-integrin α5β1 interaction.
Estrogen mediates integrin-dependent reordering of the actin cytoskeleton. SKBR3 cells that lack ER proteins, yet express endogenous GPR30, recruit FN-engaged integrin α5β1 to fibrillar adhesions in response to estrogen stimulation (Fig. 2), suggesting that this action is ER independent. The fact that integrin α5β1 participates in GPR30-dependent EGFR transactivation is consistent with the idea that FN is a common component of provisional matrices that must be remodeled during glandular development. Thus, as summarized in the schematic model in Fig. 10, GPR30 may serve a dual purpose in that it may regulate integrin α5β1-dependent FN matrix remodeling and also promote release of EGF-like growth factors to ensure cellular survival during estrus cycle-dependent remodeling of the mammary gland. Breast tumors that maintain GPR30 may employ this integrin-dependent autocrine loop to their advantage. The fact that ER antagonists also act through GPR30 further suggests a mechanism by which ER antagonists may actually facilitate tumor growth and invasion.
Materials and Methods
Cell culture
SKBR3 (ERα−, ERβ−, GPR30+) breast carcinoma cells were obtained from the American Type Culture Collection (Manassas, VA). SKBR3 variants expressing WT Shc, dominant-negative Shc, or dominant-negative GPR30 were generated as described below. Human MDA-MB-231 (ERα−, ERβ+, GPR30−) breast cancer cells expressing vector or full-length GPR30 have been described previously (27). GE11 epithelial cells were generated by immortalization and in vitro selection of β1-null knockout embryonic stem (ES) cells as previously detailed (41). In short, ES cells were propagated in foster mice, harvested, and immortalized in vitro using a transducing retrovirus expressing simian virus-40 large T antigen and lac Z. Drug-resistant ES cells were cloned by limiting dilution, and the GE11 subclone was isolated based on its polarized epithelial morphology and expression of high levels of lac Z protein. GEII cells were recloned and confirmed to lack the expression of functional β1 integrin protein. GE11β1 epithelial cells were produced by stable transfection of β1 integrin cDNA into GE11 (β1 null) cells (41). Stable expression of dominant-negative GPR30 in mouse GE11β1 cells was accomplished through methods described below. All cultures were grown in phenol red-free (PRF) DMEM/Ham’s F12 media (1:1) (DMEM-F12) supplemented with 5% fetal bovine serum and 50 μg/ml gentamicin.
cDNAs and dominant-negative constructs
Carboxyl-terminal truncated GPR30 was generated by altering a cDNA encoding full-length human GPR30 protein (66). To accomplish this task, a PCR product encoding the first 153 amino acids of GPR30 was synthesized using forward 5′-GGTCTTGAATTCT TTCG-GCAAATCTTGAAAGCTGCA-3′ and reverse 5′-AACTCGAGTCTAGAAGCTC-ATCCAGGTGAGGAA-3′ oligonucleotide primers. The PCR-amplified fragment, termed “GPR30Δ154,” was subcloned into EcoRI and XbaI sites of pcDNA3.1 Zeo (+) or pcDNA3.1 Hygro (+) (Invitrogen, Carlsbad, CA). Molecular clones encoding GST fused to either mouse WT or mutant (317Y/F) Shc were a kind gift from Dr. K. Ravichandran (67).
Transfections and selection of stable cell lines
SKBR3 cells were transfected with either pcDNA3.1 Zeo (+) or GPR30Δ154 using Lipofectamine (GIBCO-BRL). Three days after transfection, 200 μg/ ml of Zeocin (Invitrogen) was added to the growth medium. The resulting uncloned population of Zeocin-resistant cells was propagated to generate a cell line that was used for further study. SKBR3 cells expressing GSTShc proteins were generated as follows. SKBR3 cells were cotransfected with pNeoNut and a 4-fold molar excess of either pEBG WT-Shc or pEBG 317Y/F-Shc in serum-free media. Transfectants were first selected in media supplemented with 500 μg/ml G418 (GIBCO-BRL). Well-isolated colonies were harvested with the aid of cloning cylinders and expanded individually. As assessed by immunoblotting with rabbit Shc antibodies, clones that expressed approximately equivalent levels of GSTShc protein were selected for this study. Expression of GPR30Δ154 in GE11β1 cells was accomplished using identical methods as described for the SKBR3 cell line, although in this case the cDNA was expressed using pcDNA3.1 Hygro (+).
Phosphothioate antisense and RT-PCR amplification
All custom-made phosphothioate-containing and standard oligonucleotides were purchased from Sigma-Genosys (Woodlands, TX). For antisense experiments, SKBR3 cells were treated with 15 nm of phosphothioate antisense 5′-TTGGGAAGTCACAT-CCAT-3′ or sense oligonucleotides 5′-ATGGAT-GTGACTTCCCAA-3′ in the presence of Lipofectamine for 6 h before agonist stimulation or measurement of estrogen receptor mRNA expression by RT-PCR.
RT-PCR was performed on total RNA template extracted from cells using the High Purity RNA Isolation Kit (Roche, Mannheim, Germany). cDNA was amplified from total RNA using the Titan One Tube RT-PCR Kit (Roche), and specific oligonucleotide primers for ERα or GPR30 according to the manufacturer’s recommendations. The following primers sets were used:
human GPR30: forward, 5′-ATACTAGAATTCCTGTCTAC-ACGGCACGCTGAACCT-3′; and reverse, 5′-GATGTCCGGGAGGTGCAGTGG-3′.
human ERα: forward, 5′-TCCGGGATGGCCCTACTGC-ATCAG-3′; and reverse, 5′-TCTCTT-GAAGAAGGCCTTGCAGCC-3′.
mouse GPR30: forward, 5′-GGAGCTGTCACATAAAACAG-C-3′; and reverse, 5′-GCTAGGTGTGTGCA-AGTCCT-3′.
Reverse transcription was performed for 30 min at 45 C (human GPR30 or ERα) or at 50 C (murine GPR30). After first-strand synthesis, RNA-cDNA heteroduplexes were denatured at 94 C for 2 min. Conditions for PCR amplification varied slightly. Human GPR30 cDNA was synthesized using a 40-cycle program consisting of the following thermal steps: denaturation (30 sec at 94 C); annealing (30 sec at 55 C), elongation (2 min at 68 C), and a final prolonged elongation step (10 min at 68 C). Human ERα cDNA was amplified using the same temperature increments but for 30 thermal cycles. Mouse GPR30 cDNA was amplified using a 40-cycle program consisting of: denaturation (60 sec at 94 C), annealing (60 sec at 52 C), and elongation (2 min at 72 C). This was followed by a single prolonged elongation step of 10 min at 72 C.
Growth factors, estrogens, antiestrogens, inhibitors, and matrix proteins
Recombinant human EGF was purchased from Upstate Biotechnology, Inc. (Lake Placid, NY). Water-soluble 17β-estradiol, 17α-estradiol, and ATII were purchased from Sigma (St. Louis, MO). ICI 182,780 was obtained from Tocris Bioscience (Ellisville, MO). Src-family tyrosine kinase inhibitor, PP1, was purchased from AG Scientific Inc. (San Diego, CA). RGD and RGE peptides were obtained from American Peptide Co. (Sunnyvale, CA). The broad-spectrum MMP inhibitor, GM6001 (Ilomastat) was purchased from Chemicon (Temecula, CA). Diphtheria toxin mutant, CRM-197, was acquired from Berna Products (Coral Gables, FL). The EGFR inhibitor AG1478 was obtained from BIOMOL Research Laboratories, Inc. (Plymouth Meeting, PA).
Antibodies
mAB IC3 specific for rat FN has been previously described (33). Rabbit polyclonal antibodies specific for human FN were obtained from Rockland Immunochemicals (Philadelphia, PA). The SNAKA51 antibody was a gift from Martin Humphries (University of Manchester, UK) (11). The following antibodies were purchased as indicated: sheep polyclonal EGFR antibody from Millipore Corp. (Milford, MA); erbB1-specific mAB, Ab-1, from EMD Biosciences (San Diego, CA); phosphotyrosine-specific mAB, 4G10, from Upstate Biotechnology, Inc.; inhibitory integrin α5β1 mAB SAM-1 from Chemicon; rabbit polyclonal antibodies (AB1949) specific for the cytoplasmic tail of integrin α5 subunit protein from Chemicon; mAB clone 2, against the N terminus of integrin α2 (611017) and mAB clone 1, against the C terminus of integrin α5 (610633) from BD Transduction Laboratories (Lexington, KY); function-blocking integrin mABs specific for integrins α2β1 (AK-7) and integrin α5β1 (P1D6) from EMD Biosciences. Shc was detected using mAB, PG797, raised against the SH2 domain, and specific for the p66, p52, and p46 isoforms (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) or rabbit antibodies (06-203; Millipore) raised against the same region. Alexa fluor dye-conjugated secondary antibodies were obtained from Molecular Probes, Inc. (Eugene, OR)/Invitrogen. Horseradish peroxidase (HRP)-conjugated secondary antibodies were obtained from GE Healthcare (Piscataway, NJ).
Cellular stimulation and protein extraction
Conditions for quiescence, cell stimulation, and protein extraction were noted previously (27).
Analysis of EGFR tyrosine phosphorylation
Tyrosine phosphorylation of the EGFR was measured as previously described (27). A requirement for integrin engagement in EGFR activation was determined in cells that were pretreated for function-blocking integrin-specific mABs for α5β1 (P1D6; 10 μg/ml) or α2β1 (AK-7; 20 μg/ml) or soluble peptides (1 mm) containing either RGD or RGE for 15 min before agonist stimulation.
Immunofluorescence
The localization of integrin α5β1 and human FN was visualized in SKBR3 cells that were seeded onto glass coverslips in PRF-DMEM/F12 medium containing FN-reduced serum and allowed to adhere overnight at 37 C. After adhesion, serum was removed by washing 3× PRF-DMEM/F12, and the cells were then cultured in the same media in the absence of serum for an additional 30 h. Serum-starved cells were fed human plasma FN (2 μg/ml) or no exogenous FN in the absence or presence of hormone for 2 h. Cells were then washed, fixed for 5 min in 4% paraformaldehyde, and blocked in 5% BSA/PBS for 30 min. Cells were incubated with 10 μg/ml of α5β1-specific mAB, SAM-1, and 4 μg/ml of human FN-specific polyclonal antibody delivered in PRF-DMEM/F12 containing 0.2% BSA for 60 min. Coverslips were washed in PRF-DMEM/F12, and cell-associated antibodies were detected using Alexa 594-conjugated antirabbit IgG or Alexa 488-conjugated antimouse IgG diluted at 1:800 and delivered in PRF-DMEM/F12 containing 0.2% BSA for 30 min. After staining, coverslips were washed and mounted on glass slides in Vectashield/4′6-diamidino-2-phenylindole (Vector Laboratories, Inc., Burlingame, CA). Experiments examining SNAKA51 staining in SKBR3 cells were done as above except 2 μg/ml of SNAKA51 mAB was substituted for SAM-1 mAB, and all treatments were carried out in the presence of exogenous FN.
The formation of FN fibrils was examined in GE11β1 and GE11β1Δ154 mouse epithelial cells seeded onto glass coverslips in PRF-DMEM/F12 medium containing FN-reduced serum. Starved cells were fed rat plasma FN (25 μg/ml) in PRF-DMEM/F12 medium in the presence of ligand (10 nm E2α, 10 nm E2β, 1 μm ICI, or 10 μm ATII) for 18 h and then fixed and prepared for immunostaining as above. Fixed cells were stained with IC3 ascites diluted 1:1000 and delivered in PBS containing 1% BSA for 60 min. IC3 mAB was detected by staining with Alexa 594 conjugated antimouse IgG (1:800) and processed for microscopy as above. All immunofluorescent images were visualized with a Nikon Eclipse 80i microscope (Nikon, Inc., Melville, NY) equipped with a Nikon Plan Fluor 100× 0.5–1.3 Oil Iris with differential interference contrast and epifluorescent capabilities. Digital images were captured using a QImaging Retiga 2000R digital camera and Nikon imaging software (Elements-Basic Research 3.0).
Detection of DOC-insoluble FN
Cells were seeded in FN-depleted serum at 1 × 106 cells per 60-mm culture dish. Cells were allowed to adhere overnight at 37 C and then serum starved for 30 h. After starvation, cells were incubated with 25 μg/ml of rat FN alone or in the presence of 10 nm 17α-E2, 10 nm 17β-E2, 1 μm ICI 182,780, or 1 μm ATII for the indicated amount of time. In some experiments, before stimulation, cells were pretreated with inhibitors as follows: 10 μm GM6001 for 15 min; 50 nm PP1 for 15 min; 1 nm AG1478 for 15 min; or 200 ng/ml CRM-197 for 2 h. DOC-soluble and -insoluble fractions were isolated and analyzed as described (33). Equivalent amounts of total protein were electrophoresed through 6% reducing SDS polyacrylamide gels and transferred to nitrocellulose for immunodetection using a 1:5000 dilution of IC3 antirat FN ascites. Immobilized FN antibodies were visualized using HRP-conjugated antimouse IgG secondary antibodies and enhanced chemiluminescence.
Detection of integrin-Shc complexes
After treatment, cellular proteins were extracted in Triton-buffered detergent (125 mm NaCl; 50 mm Tris, pH 8.0; 1% Triton X-100; 10% glycerol; 1 mm MgCl2; and 1 mm CaCl2 plus inhibitors) and solubilized on ice for 10 min. Crude lysates were clarified by centrifugation, and protein concentrations were determined by the bichinchoninic acid method and analyzed directly. Triton-soluble protein (500 μg) was incubated with protein G-agarose beads for 1 h at 4 C to preclear proteins that nonspecifically adhere. Beads were collected by centrifugation, and the cleared lysate was incubated for 3 h at 4 C with 2 μg/sample of antibody. Samples were treated with either mAB clone 2 (611017) specific for integrin α2 or rabbit polyclonal antibodies (AB1949) specific for integrin α5 subunit protein antibodies. Protein G beads were used to capture protein complexes overnight at 4 C. Immunoabsorbed proteins were washed in Triton lysis buffer, and the beads were then heated at 95 C for 5 min in 50 μl of 2× Laemmli sample buffer containing 700 mm β-mercaptoethanol. Samples were centrifuged, and the resulting supernatant was collected by pin-hole elution. Heat-denatured, reduced samples were electrophoresed through 8% SDS-polyacrylamide gels and transferred to nitrocellulose. Membranes were cut into sections based on protein of interest and blocked in Tris-buffered saline (TBS) plus 0.5% Tween 20 (TBS-T) and 5% milk for mABs or PBS and 3% milk for rabbit polyclonal antibody. Shc proteins were detected by probing the nitrocellulose filter with 1 μg/ml of mouse Shc antibody, PG797, or rabbit Shc antibody, 06-203. Integrin α5β1 or integrin α2β1 protein recovery was measured using integrin α5 subunit protein antibody (mAB clone 1) or integrin α2 subunit protein antibody (mAB clone 2) diluted 1:500 and delivered in TBS-T containing 5% milk. Antibody-antigen complexes were detected using HRP-conjugated secondary antibodies diluted 1:5000 and enhanced chemiluminescence.
Supplementary Material
Footnotes
This work was supported, in part, by a Research Scholar Award (to E.J.F.) from the American Cancer Society (RSG-02-194-01) and by National Institutes of Health Award CA119165-01A2 (to E.J.F.).
Disclosure Summary: J.A.Q., C.T.G., A.R.F., M.K., and J.E.S. have nothing to disclose. E.J.F. received an honorarium from the Mayo Clinic.
First Published Online April 2, 2009
Abbreviations: ATII, Angiotensin II; DOC, deoxycholate; E2, estradiol; ECM, extracellular matrix; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; ER, estrogen receptor; ES, embryonic stem; FAK, focal adhesion kinase; FN, fibronectin; GPCR, G protein-coupled receptor; GST, glutathione-S-transferase; HB-EGF, heparin-bound epidermal growth factor; HRP, horseradish peroxidase; mAB, monoclonal antibody; MMP, matrix metalloproteinase; PRF, phenol red free; RGD, arg-gly-asp peptide; RGE, arg-gly-glu peptide; SDS, sodium dodecyl sulfate; Shc, src homologous collagen (homology); TBS, Tris-buffered saline; 7-TMR, seven-transmembrane receptor. WT, wild type.
References
- George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO 1993 Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development 119:1079–1091 [DOI] [PubMed] [Google Scholar]
- Francis SE, Goh KL, Hodivala-Dilke K, Bader BL, Stark M, Davidson D, Hynes RO 2002 Central roles of α5β1 integrin and fibronectin in vascular development in mouse embryos and embryoid bodies. Arterioscler Thromb Vasc Biol 22:927–933 [DOI] [PubMed] [Google Scholar]
- Mao Y, Schwarzbauer JE 2005 Stimulatory effects of a three-dimensional microenvironment on cell-mediated fibronectin fibrillogenesis. J Cell Sci 118:4427–4436 [DOI] [PubMed] [Google Scholar]
- Wierzbicka-Patynowski I, Schwarzbauer JE 2002 Regulatory role for SRC and phosphatidylinositol 3-kinase in initiation of fibronectin matrix assembly. J Biol Chem 277:19703–19708 [DOI] [PubMed] [Google Scholar]
- Yang RS, Tang CH, Ling QD, Liu SH, Fu WM 2002 Regulation of fibronectin fibrillogenesis by protein kinases in cultured rat osteoblasts. Mol Pharmacol 61:1163–1173 [DOI] [PubMed] [Google Scholar]
- Somanath PR, Kandel ES, Hay N, Byzova TV 2007 Akt1 signaling regulates integrin activation, matrix recognition, and fibronectin assembly. J Biol Chem 282:22964–22976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faull RJ, Kovach NL, Harlan JM, Ginsberg MH 1993 Affinity modulation of integrin α5β1: regulation of the functional response by soluble fibronectin. J Cell Biol 121:155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong C, Chrzanowska-Wodnicka M, Brown J, Shaub A, Belkin AM, Burridge K 1998 Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly. J Cell Biol 141:539–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi T, Kiehart DP, Erickson HP 2002 Dual labeling of the fibronectin matrix and actin cytoskeleton with green fluorescent protein variants. J Cell Sci 115:1221–1229 [DOI] [PubMed] [Google Scholar]
- Pankov R, Cukierman E, Katz BZ, Matsumoto K, Lin DC, Lin S, Hahn C, Yamada KM 2000 Integrin dynamics and matrix assembly: tensin-dependent translocation of α(5)β(1) integrins promotes early fibronectin fibrillogenesis. J Cell Biol 148:1075–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K, Pankov R, Travis MA, Askari JA, Mould AP, Craig SE, Newham P, Yamada KM, Humphries MJ 2005 A specific α5β1-integrin conformation promotes directional integrin translocation and fibronectin matrix formation. J Cell Sci 118:291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti E 1999 Fibronectin and its integrin receptors in cancer. Adv Cancer Res 76:1–20 [DOI] [PubMed] [Google Scholar]
- Han S, Khuri FR, Roman J 2006 Fibronectin stimulates non-small cell lung carcinoma cell growth through activation of Akt/mammalian target of rapamycin/S6 kinase and inactivation of LKB1/AMP-activated protein kinase signal pathways. Cancer Res 66:315–323 [DOI] [PubMed] [Google Scholar]
- Jia Y, Zeng ZZ, Markwart SM, Rockwood KF, Ignatoski KM, Ethier SP, Livant DL 2004 Integrin fibronectin receptors in matrix metalloproteinase-1-dependent invasion by breast cancer and mammary epithelial cells. Cancer Res 64:8674–8681 [DOI] [PubMed] [Google Scholar]
- Price JE 1996 Metastasis from human breast cancer cell lines. Br Canc Res Treat 39:93–102 [DOI] [PubMed] [Google Scholar]
- Saulnier R, Bhardwaj B, Klassen J, Leopold D, Rahimi N, Tremblay E, Mosher D, Elliott B 1996 Fibronectin fibrils and growth factors stimulate anchorage-independent growth of a murine mammary carcinoma. Exp Cell Res 222:360–369 [DOI] [PubMed] [Google Scholar]
- Qiao H, Saulnier R, Patryzkat A, Rahimi N, Raptis L, Rossiter J, Tremblay E, Elliott B 2000 Cooperative effect of hepatocyte growth factor and fibronectin in anchorage-independent survival of mammary carcinoma cells: requirement for phosphatidylinositol 3-kinase activity. Cell Growth Differ 11:123–133 [PubMed] [Google Scholar]
- Williams CM, Engler AJ, Slone RD, Galante LL, Schwarzbauer JE 2008 Fibronectin expression modulates mammary epithelial cell proliferation during acinar differentiation. Cancer Res 68:3185–3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward TL, Mienaltowski AS, Modi RR, Bennett JM, Haslam SZ 2001 Fibronectin and the α(5)β(1) integrin are under developmental and ovarian steroid regulation in the normal mouse mammary gland. Endocrinology 142:3214–3222 [DOI] [PubMed] [Google Scholar]
- Acconcia F, Barnes CJ, Kumar R 2006 Estrogen and tamoxifen induce cytoskeletal remodeling and migration in endometrial cancer cells. Endocrinology 147:1203–1212 [DOI] [PubMed] [Google Scholar]
- Azios NG, Krishnamoorthy L, Harris M, Cubano LA, Cammer M, Dharmawardhane SF 2007 Estrogen and resveratrol regulate Rac and Cdc42 signaling to the actin cytoskeleton of metastatic breast cancer cells. Neoplasia 9:147–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapino A, Pietribiasi F, Bussolati G, Marchisio PC 1986 Estrogen- and tamoxifen-induced rearrangement of cytoskeletal and adhesion structures in breast cancer MCF-7 cells. Cancer Res 46:2526–2531 [PubMed] [Google Scholar]
- Ehlers EM, Schubert C 1999 Differences in morphology and cytoskeleton of MCF-7 and MX-1 cells after therapy with OH-tamoxifen and the pure estrogen antagonist ZM 182780. An immunofluorescence and scanning electron microscopic study. Ann Anat 181:231–236 [DOI] [PubMed] [Google Scholar]
- Thomas P, Pang Y, Filardo EJ, Dong J 2005 Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 146:624–632 [DOI] [PubMed] [Google Scholar]
- Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER 2005 A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307:1625–1630 [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Frackelton Jr AR, Bland KI 2002 Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol 16:70–84 [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Bland KI, Frackelton Jr AR 2000 Estrogen-induced activation of Erk-1 and Erk-2 requires the G-protein coupled receptor homolog, GPR30, and occurs via transactivation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol 14:1649–1660 [DOI] [PubMed] [Google Scholar]
- Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, Oprea Tl, Prossnitz ER, Musti AM, Andò S, Maggliolini M 2007 G coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17β-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res 67:1859–1866 [DOI] [PubMed] [Google Scholar]
- Vivacqua A, Bonofiglio D, Recchia AG, Musti AM, Picard D, Andò S, Maggiolini M 2006 The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17β-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol Endocrinol 20:631–646 [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Graeber CT, Quinn JA, Resnick MB, Giri D, DeLellis RA, Steinhoff MM, Sabo E 2006 Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin Cancer Res 12:6359–6366 [DOI] [PubMed] [Google Scholar]
- Smith HO, Leslie KK, Singh M, Qualls CR, Revankar CM, Joste NE, Prossnitz ER 2007 GPR30: a novel indicator of poor survival for endometrial carcinoma. Am J Obstet Gynecol 196:386.e1–e9 (discussion e9–e11) [DOI] [PubMed] [Google Scholar]
- Vladusic EA, Hornby AE, Guerra-Vladusic FK, Lakins J, Lupu R 2000 Expresssion and regulation of estrogen receptor β in human breast tumors and cell lines. Oncol Rep 7:157–167 [DOI] [PubMed] [Google Scholar]
- Sechler JL, Takada Y, Schwarzbauer JE 1996 Altered rate of fibronectin matrix assembly by deletion of the first type III repeats. J Cell Biol 134:573–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Wess J 1998 Truncated V2 vasopressin receptors as negative regulators of wild-type V2 receptor function. Biochemistry 37:15773–15784 [DOI] [PubMed] [Google Scholar]
- Karpa KD, Lin R, Kabbani N, Levenson R 2000 The dopamine D3 receptor interacts with itself and the truncated D3 splice variant d3nf: D3–D3nf interaction causes mislocalization of D3 receptors. Mol Pharmacol 58:677–683 [DOI] [PubMed] [Google Scholar]
- Rios CD, Jordan BA, Gomes I, Devi LA 2001 G-protein-coupled receptor dimerization: modulation of receptor function. Pharmacol Ther 92:71–87 [DOI] [PubMed] [Google Scholar]
- Angers S, Salahpour A, Bouvier M 2002 Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu Rev Pharm Toxicol 42:409–435 [DOI] [PubMed] [Google Scholar]
- Slack BE 1998 Tyrosine phosphorylation of paxillin and focal adhesion kinase by activation of muscarinic m3 receptors is dependent on integrin engagement by the extracellular matrix. Proc Natl Acad Sci USA 95:7281–7286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festuccia C, Angelucci A, Gravina G, Eleuterio E, Vicentini C, Bologna M 2002 Bombesin-dependent pro-MMP-9 activation in prostatic cancer cells requires β1 integrin engagement. Exp Cell Res 280:1–11 [DOI] [PubMed] [Google Scholar]
- Adair BD, Xiong JP, Maddock C, Goodman SL, Arnaout MA, Yeager M 2005 Three-dimensional EM structure of the ectodomain of integrin αvβ3 in a complex with fibronectin. J Cell Biol 168:1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimond C, van Der Flier A, van Delft S, Brakebusch C, Kuikman I, Collard JG, Fässler R, Sonnenberg A 1999 Induction of cell scattering by expression of β1 integrins in β1-deficient epithelial cells requires activation of members of the rho family of GTPases and downregulation of cadherin and catenin function. J Cell Biol 147:1325–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan KJ, Law DA, Phillips DR 2000 Identification of shc as the primary protein binding to the tyrosine-phosphorylated β 3 subunit of αIIbβ3 during outside-in integrin platelet signaling. J Biol Chem 275:36423–36429 [DOI] [PubMed] [Google Scholar]
- Dans M, Gagnoux-Palacios L, Blaikie P, Klein S, Mariotti A, Giancotti FG 2001 Tyrosine phosphorylation of the β 4 integrin cytoplasmic domain mediates Shc signaling to extracellular signal-regulated kinase and antagonizes formation of hemidesmosomes. J Biol Chem 276:1494–1502 [DOI] [PubMed] [Google Scholar]
- Weyts FA, Li YS, van Leeuwen J, Weinans H, Chien S 2002 ERK activation and α v β 3 integrin signaling through Shc recruitment in response to mechanical stimulation in human osteoblasts. J Cell Biochem 87:85–92 [DOI] [PubMed] [Google Scholar]
- Niu S, Xie H, Marcantonio EE 2003 Integrin-mediated tyrosine phosphorylation of Shc in T cells is regulated by protein kinase C-dependent phosphorylations of Lck. Mol Biol Cell 14:349–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernig F, Mayr M, Xu Q 2003 Mechanical stretch-induced apoptosis in smooth muscle cells is mediated by β1-integrin signaling pathways. Hypertension 41:903–911 [DOI] [PubMed] [Google Scholar]
- Stevenson LE, Frackelton Jr AR 1998 Constitutively tyrosine phosphorylated p52 Shc in breast cancer cells: correlation with ErbB2 and p66 Shc expression. Br Cancer Res Treat 49:119–128 [DOI] [PubMed] [Google Scholar]
- Faraldo MM, Deugnier MA, Thiery JP, Glukhova MA 2001 Growth defects induced by perturbation of β1-integrin function in the mammary gland epithelium result from a lack of MAPK activation via the Shc and Akt pathways. EMBO Rep 2:431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schedin P, Strange R, Mitrenga T, Wolfe P, Kaeck M 2000 Fibronectin fragments induce MMP activity in mouse mammary epithelial cells: evidence for a role in mammary tissue remodeling. J Cell Sci 113:795–806 [DOI] [PubMed] [Google Scholar]
- Katz BZ, Romer L, Miyamoto S, Volberg T, Matsumoto K, Cukierman E, Geiger B, Yamada KM 2003 Targeting membrane-localized focal adhesion kinase to focal adhesions: roles of tyrosine phosphorylation and SRC family kinases. J Biol Chem 278:29115–29120 [DOI] [PubMed] [Google Scholar]
- Ren XD, Kiosses WB, Sieg DJ, Otey CA, Schlaepfer DD, Schwartz MA 2000 Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J Cell Sci 113:3673–3678 [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Graeber CT 2003 Evidence supporting a role for GPR30, an orphan member of the G-protein-coupled receptor superfamily, in rapid estrogen signaling. In: Watson CM, ed. Identification of membrane-associated steroid hormone receptors. New York: Kluwer Press [Google Scholar]
- Brooks PC, Strömblad S, Sanders LC, von Schalscha TL, Aimes RT, Stetler-Stevenson WG, Quigley JP, Cheresh DA 1996 Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin αvβ3. Cell 85:683–693 [DOI] [PubMed] [Google Scholar]
- Bridges LC, Bowditch RD 2005 ADAM-integrin interactions: potential integrin regulated ectodomain-shedding activity. Curr Pharm Des 11:837–847 [DOI] [PubMed] [Google Scholar]
- Takeda S, Igarashi T, Mori H, Araki S 2006 Crystal structures of VAP1 reveal ADAMs’ MDC domain architecture and its unique C-shaped scaffold. EMBO J 25:2388–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S, Yamaji N, Yasunaga K, Saito T, Matsumoto S, Katoh M, Kobayashi S, Masuho Y 1999 The fibronectin extra domain A activates matrix metalloproteinase gene expression by an interleukin-1-dependent mechanism. J Biol Chem 274:30756–30763 [DOI] [PubMed] [Google Scholar]
- Mitra A, Chakrabarti J, Chatterjee A 2003 Binding of α5 monoclonal antibody to cell surface α5β1 integrin modulates MMP-2 and MMP-7 activity in B16F10 melanoma cells. J Environ Pathol Toxicol Oncol 22:167–178 [DOI] [PubMed] [Google Scholar]
- Das S, Banerji A, Frei E, Chatterjee A 2008 Rapid expression and activation of MMP-2 and MMP-9 upon exposure of human breast cancer cells (MCF-7) to fibronectin in serum free medium. Life Sci 82:467–476 [DOI] [PubMed] [Google Scholar]
- Matsuo M, Sakurai H, Ueno Y, Ohtani O, Saiki I 2006 Activation of MEK/ERK and PI3K/Akt pathways by fibronectin requires integrin αv-mediated ADAM activity in hepatocellular carcinoma: a novel functional target for gefitinib. Cancer Sci 97:155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg MH, Partridge A, Shattil SJ 2005 Integrin regulation. Curr Opin Cell Biol 17:509–516 [DOI] [PubMed] [Google Scholar]
- Luo BH, Carman CV, Springer TA 2007 Structural basis of integrin regulation and signaling. Annu Rev Immunol 25:619–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Boettiger D 2003 A novel mode for integrin-mediated signaling: tethering is required for phosphorylation of FAK Y397. Mol Biol Cell 14:4306–4315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta A, Shi Q, Boettiger DE 2001 Transformation of chicken embryo fibroblasts by v-src uncouples β1 integrin-mediated outside-in but not inside-out signaling. Mol Cell Biol 21:7295–7306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Geer P, Wiley S, Gish GD, Pawson T 1996 The Shc adaptor protein is highly phosphorylated at conserved, twin tyrosine residues (Y239/240) that mediate protein-protein interactions. Curr Biol 6:1435–1444 [DOI] [PubMed] [Google Scholar]
- Ricketts WA, Brown JH, Olefsky JM 1999 Pertussis toxin-sensitive and -insensitive thrombin stimulation of Shc phosphorylation and mitogenesis are mediated through distinct pathways. Mol Endocrinol 13: 1988–2001 [DOI] [PubMed] [Google Scholar]
- Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ 1997 Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 45:607–617 [DOI] [PubMed] [Google Scholar]
- Pratt JC, Weiss M, Sieff CA, Shoelson SE, Burakoff SJ, Ravichandran KS 1996 Evidence for a physical association between the Shc-PTB and the β c chain of the granulocyte-macrophage colony-stimulating factor receptor. J Biol Chem 271:12137–12140 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.