

Summary
Chronic allograft nephropathy, a descriptive term denoting chronic scarring injury of the renal parenchyma and vasculature in allograft kidneys arising from various etiologies including chronic rejection, is the most common cause of late allograft failure, but mediators of this progressive injury largely remain unknown. We hypothesized that platelet-derived growth factor D (PDGF-D) and its specific receptor PDGF-Rβ may be an important mediator in the pathogenesis of chronic allograft nephropathy, and hence sought to identify its expression in this setting. Allograft nephrectomies demonstrating chronic allograft nephropathy, obtained from patients with irreversible transplant kidney failure (n = 15) were compared with renal tissues without prominent histopathological abnormalities (n=18) and a series of renal allograft biopsies demonstrating acute vascular rejection (n=12). Antibodies to PDGF-D and PDGF-Rβ were used for immunohistochemistry. Double and triple immunohistochemistry was used to identify cell types expressing PDGF-D. PDGF-D was widely expressed in most neointimas in arteries exhibiting the chronic arteriopathy of chronic allograft nephropathy, and only weakly expressed in a small proportion of sclerotic arteries in the other two groups. Double and triple immunolabeling demonstrated that the neointimal cells expressing PDGF-D were α-smooth muscle actin expressing cells, but not infiltrating macrophages or endothelial cells. PDGF-Rβ expression evaluated in serial sections was localized to the same sites where neointimal PDGF-D was expressed. PDGF-Rβ was expressed in interstitial cells more abundantly in the chronic allograft nephropathy, group compared with the normal and acute vascular rejection groups, without demonstrable co-localization of PDGF-D. PDGF-D is present in the neointima of the arteriopathy of chronic allograft nephropathy, where it can engage PDGF-Rβ to promote mesenchymal cell migration, proliferation and neointima formation. PDGF-D may engage the PDGF-Rβ to promote interstitial injury in chronic allograft injury, but its sources within the interstitium were unidentified.
Keywords: PDGF-D, transplantation, allograft, nephropathy
1. Introduction
Chronic allograft nephropathy (CAN) is a descriptive term comprising the severe pathologic changes found in failed renal allografts resulting from a multipliticity of etiologies, including, but not limited to, chronic vascular rejection. The pathology of CAN includes glomerular capillary wall thickening with basement membrane duplication, increased mesangial matrix, and focal segmental and focal global glomerulosclerosis (chronic transplant glomerulopathy), tubular atrophy, interstitial fibrosis and an arteriopathy characterized by neointima formation and sclerosis [1–3].
Platelet-derived growth factors (PDGFs) are major mitogens and chemoattractants for many types of mesenchymal cells, including vascular smooth muscle cells, interstitial myofibroblasts, and glomerular mesangial cells [4–6]. There have been reports that PDGFs may play important roles in various human renal diseases. PDGF-A and PDGF-B in particular have been shown to be potentially important mediators of human renal vascular rejection [7].
The PDGF family currently has four known members, which are encoded by four different genes: PDGF-A, -B, and the recently discovered PDGF-C and -D. Both PDGF-A and -B have short N-terminal extensions that undergo intracellular proteolytic processing for activation and are secreted in an active form as disulfide bonded homo- or heterodimers (PDGF-AA, -AB, and -BB), whereas PDGF-C and -D are secreted in a latent form with an N-terminal CUB domain, which is required to be cleaved by an extracellular protease to release the active growth factor domain. Active PDGFs bind to specific tyrosine kinase receptors, PDGF receptor alpha (PDGF-Rα) and receptor beta (PDGF-Rβ). The activated receptors consist of dimers of α and β subunits (PDGF R-αα, -αβ, and -ββ). PDGF-A can bind PDGF R-αα, PDGF-B can bind all the three isoforms, and PDGF-C can bind both PDGF R-αα and PDGF R-αβ, whereas PDGF-D can only bind PDGF-Rβ [8–10].
It is well established that PDGF-A and -B play important roles in both human and experimental renal diseases [7, 9–16], but there is relatively little information on their role in the injuries affecting human transplant kidneys. We have shown previously that expression of both PDGF-A and –B are up-regulated in renal vascular rejection [7, 9, 12]. Others have shown that PDGF-B and PDGF β receptor also are localized to chronically injured arteries in murine models of kidney transplantation [17, 18]. Currently, the potential role of PDGF-D in renal transplants is unexplored. However, there is evidence from other settings indicating that PDGF-D might be a mediator of CAN, including (1) its presence in neointimal hyperplastic injuries in a rat model of vessel injury [19], (2) its expression in human normal glomeruli [20] and in experimental glomerulonephritis [6, 14], and (3) its expression during the interstitial fibrosis that occurs in human and experimental obstructive nephropathy [12]. Its obligate receptor, PDGF-Rβ, is expressed at sites of injury in these human and experimental diseases [12, 20, 21]. Because CAN is an important and deleterious outcome in human kidney transplantation, involving all the lesions mentioned above, we sought to localize PDGF-D and its expression at relevant sites of injury in this disease process.
2. Materials and methods
2.1 Source of tissue
Nephrectomies obtained from patients with irreversibly injured renal allografts and with pathologic features of chronic allograft nephropathy were enrolled in the main study group (n = 15). All of them had chronic transplant glomerulopathy, extensive tubular atrophy, interstitial fibrosis and arteriopathy with neointima formation. Fourteen of these cases also demonstrated superimposed acute vascular rejection (AVR) and cellular rejection, presumably a consequence of discontinued immunosuppression prior to nephrectomy.
Comparison groups included a group of renal tissues (n=18) without prominent histopathological abnormalities, including biopsies of donor kidneys obtained before transplantation (n = 5) and normal protocol biopsies of patients after transplantation (n=13). A second comparison group of tissues (n=12) consisted of renal biopsies with findings of acute vascular rejection (AVR) concurrent with acute cellular rejection.
These studies have received human subject approval by an institutional IRB review panel (UW 96-1826-A-08). Tissues were fixed in methyl Carnoy’s (methacarn) solution (60% methanol, 30% chloroform, and 10% acetic acid), processed and embedded in paraffin according to conventional techniques. The pathologic features resulting in a diagnosis of rejection were those formulated by the Cooperative Clinical Trials in Transplantation working group (Class I, acute cellular rejection and Class II, acute vascular rejection concurrent with acute cellular rejection) [22] and consistent with the general categories (Class IIA/IIB with arteritis) of the most recently published Banff (Banff 97) classification [23, 24].
2.2 Antibodies
PDGF-D
Rabbit polyclonal antibody (lot number E3812, provided by ZymoGenetics Inc., Seattle, USA) is an affinity-purified antibody raised against the middle component of the PDGF-D molecule and is similar to antibody lot E0259 previously used and described [20]. It recognizes the full-length human PDGF-D peptide and appropriately sized bands in homogenates of human kidneys by Western blotting as previously described [20].
PDGF-Rβ
Murine monoclonal antibody (PR7212, R & D system Inc., Minneapolis, MN, USA) has been demonstrated to react specifically with human PDGF-Rβ and to detect patterns of PDGF-Rβ localization in tissues. The specificity of this antibody was demonstrated by absorption of the antibodies with PDGF-Rβ peptide, which abolished positive staining patterns in both Western blots and immunohistochemical analysis[20].
Alpha-smooth muscle actin
Murine monoclonal antibody (clone 1A4, DAKO Corp., Carpinteria, CA, USA) recognizes alpha-smooth muscle actin and was used to identify vascular smooth muscle cells, activated glomerular mesangial cells, and interstitial myofibroblasts [25].
Ulex europaeus agglutinin I
This lectin glycoprotein (lot no. 91129, Vector Laboratories, Burlingame, CA, USA) is an established marker of glomerular and vascular endothelial cells[26].
CD 68
Murine monoclonal antibody (clone PG-M1, DAKO Corp., Carpinteria, CA, USA) detects the CD68 epitope of human monocytes and macrophages [27].
Epithelial membrane antigen (EMA)
Murine monoclonal antibody (clone E29, DAKO Corp., Carpinteria, CA, USA) detects epithelial membrane antigen of human distal tubules and collecting ducts [28].
2.3 Immunohistochemistry
Immunohistochemistry was performed on four-micron sections of methyl Carnoy’s fixed, paraffin embedded tissues. Sections were deparaffinized in xylene and rehydrated with graded ethanol. Endogenous peroxidase was blocked with 3% hydrogen for 5 minutes. Sections were incubated in 10% normal goat serum (Vector Laboratories) for 20 minutes. The sections were then incubated with rabbit anti-human PDGF-D polyclonal antibody for 1 hour in room temperature. After washing in PBS, the sections were incubated with anti-rabbit Ig ImmPRESS reagent (MP-7402, Vector Laboratories) at room temperature for 30 minutes. After washing in PBS, 3, 3′-diaminobenzidine (DAB) used as the chromogen and sections were counterstained with hematoxylin, dehydrated, and coverslipped. To evaluate the immunohistochemical staining of anti-PDGF-Rβ, the next sequential histologic sections were incubated with murine monoclonal anti-human PDGF-Rβ at room temperature for 2 hours, washed in PBS, and incubated with anti-mouse Ig ImmPRESS reagent (MP-7401, Vector Laboratories) for 30 minutes. The subsequent steps are as described above.
For all samples, a negative antibody control consisted of substitution of the primary antibody with normal rabbit or mouse IgG and performance of all the subsequent steps of the immunohistochemical procedures on serial histological sections to establish the specificity of the antisera and staining patterns for PDGF-D and PDGF-Rβ.
2.4 Double immunohistochemistry
Double immunolabeling was utilized to identify the cell types expressing PDGF-D. After the sections were deparaffinized and reacted with 3% hydrogen peroxide to block endogenous peroxidase, they were first incubated with the rabbit anti-human PDGF-D polyclonal antibody using the single labeling described above, and positive staining was visualized by a brown label using DAB. For the second immunohistochemical label, the sections were then blocked with 3% hydrogen peroxide, and then incubated either with murine monoclonal anti-alpha-smooth muscle actin antibody, anti-CD68 antibody, or anti-EMA antibody. Then, the sections were incubated with anti-mouse Ig ImmPRESS reagent, and positive label identified as a blue-gray signal, utilizing the Vector SG substrate kit for peroxidase (SK-4700, Vector Laboratories). For endothelial cell labeling, the sections were first incubated with anti-PDGF-D antibody and then were incubated sequentially with Ulex europaeus agglutinin I, biotinylated anti-Ulex, followed by the ABC-alkaline phosphatase reagent (Vector Laboratories) and finally developed using Vector Blue Alkaline Phosphatase Substrate Kit III (Vector Laboratories) to yield a blue color in the areas of positive signal. The slides were then dehydrated and coverslipped.
2.5 Triple immunohistochemistry
The sections that were already doubly immunostained with PDGF-D and alpha-smooth muscle actin were sequentially blocked with 3% hydrogen peroxide, incubated with monoclonal anti-CD68 antibody and then anti-mouse Ig ImmPRESS reagent, and then followed by vector VIP substrate kit for peroxidase (SK-4600, Vector Laboratories), yielding a purple color signal in positively stained areas. These slides were dehydrated and coverslipped.
2.6 Morphometric analysis
For analysis of neointimal sclerosis, which occurs in relatively larger arteries, we utilized a criterion for artery inclusion as two layers of smooth muscle cells in the medial portion of the arterial wall. All of the arteries meeting this criterion that were present in one histologic section per case were counted with every artery with neointimal sclerosis enumerated and their PDGF-D expression analyzed.
Morphometry was performed on slides immunostained for PDGF-Rβ by an observer blinded to their origin. For each case, 10 consecutive glomerular cross-sections and first 5 cortical tubulointerstitial fields were photographed using Olympus DP11 digital camera (OlympusAmerica, Melville, NY), and the images were analyzed by Image-pro Plus software (Media Cybernetics, Silver Spring, MD). The parameters of PDGF-Rβ expression were determined as the percentage of positively stained area per glomerular tuft cross-sectional area (not including the urinary space) and percentage of positively stained area compared to the total tubulointerstitial area examined.
3. Results
3.1 PDGF-D and PDGF-Rβ expression in arteries
Neointimal sclerosis was much more severe in the CAN group. It was present in all 15 cases, and was characterized by much thicker intimal layers and higher prevalence of arterial involvement (339/368 of total counted arteries, 92.1%) compared to the normal group, where it was present in 6/18 cases (33.3%) and involved 11/74 (14.9%) of total counted arteries. The CAN group also demonstrated more widespread arteriopathy, compared with the AVR group, in which it was present in 5/12 cases (41.7%) and involved 5/31 (16.1%) of total counted arteries.
PDGF-D was expressed uniformly in the cytoplasm of medial smooth muscle cells of the arteries and variably by some adventitial cells in a similar pattern in all three groups (Table 1). It was also expressed widely in the neointima of the arteriopathy, but most prominently in the CAN group. Double and triple labeling immunohistochemistry identified neointimal cells expressing PDGF-D as medial smooth muscle cells or myofibroblasts by virtue of co-expressed α-smooth muscle actin and the absence of co-expressed markers of infiltrating macrophages (CD68, Fig. 1A, C, and E) or endothelial cells (Ulex lectin).
Table 1.
PDGF-D expression in human renal tissue.
| Glomeruli | Tubules | Vasculature | Interstitial Myofibroblasts | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MC | VEC | PEC | Distal | Proximal | CD | Media SMC | Int SMC | Endo | ||
| Normal, n=18 | − | +++ | Focal + | Focal + | − | ++ | +++ | − | − | − |
| AVR, n=12 | − | +++ | Focal + | Focal + | − | ++ | +++ | ++ | − | Focal ++ (n=1) |
| CAN, n=15 | − | +++ | Focal + | Focal + | − | ++ | +++ | +++ | − | Focal ++ (n=2) |
MC, mesangial cell; VEC, visceral epithelial cell; PEC, parietal epithelial cell; CD, collecting duct; SMC, smooth muscle cell; Int, intimal; Endo, endothelium.
Scale is negative (−), weak (+), moderate (++) and strong (+++) immunohistochemical signal.
Fig. 1.
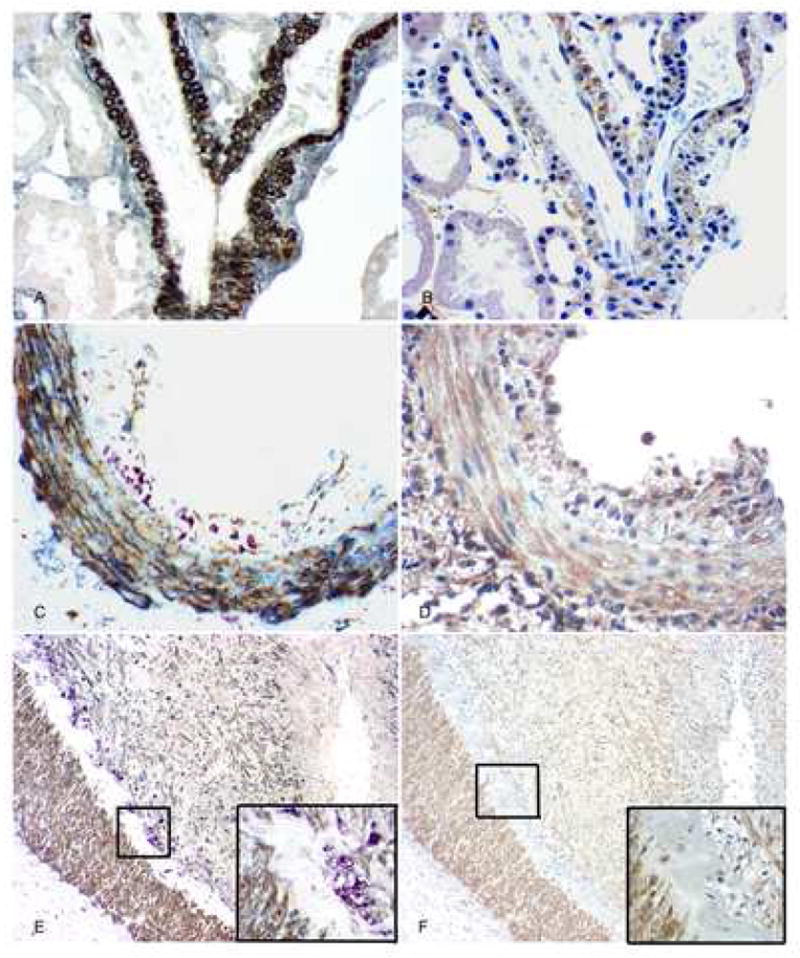
PDGF-D and PDGF-Rβ expression in arteries. A: In normal group, triple immunolabeling shows that PDGF-D (brown) is expressed in arterial medial smooth muscle cells that also express alpha-smooth muscle actin (blue-gray); no monocyte/macrophages (purple) are found. There is no expression of PDGF-D seen in the interstitium. B: In serial section of A, PDGF-Rβ (brown) is expressed in the similar distribution of PDGF-D in arterial wall, and is also expressed mildly in interstitium. C: In kidneys with AVR, triple immunolabeling shows that PDGF-D (brown) is expressed in arterial medial smooth muscle cells, some adventitial cells and some neointimal cells that also have alpha-smooth muscle actin (blue-gray) expression; It is not expressed by the subendothelial infiltrating monocyte/macrophages (purple). D: In serial section of C, PDGF-Rβ (brown) is expressed in the similar distribution of PDGF-D in the arterial wall. E: Part of an artery in kidneys with CAN, triple immunolabeling shows that PDGF-D (brown) is expressed in arterial medial smooth muscle cells and the more prominent neointimal layers compared with the other two groups, which also have alpha-smooth muscle actin (blue-gray) expression; It is not expressed by the subendothelial infiltrating monocyte/macrophages and foam cells (purple). The inset shows the framed area with high power. F and its inset: In serial section of D, PDGF-Rβ (brown) is expressed in a similar distribution of PDGF-D in the artery. Original magnification, AD, × 400, E and F, × 100.
PDGF-Rβ was also expressed uniformly by medial smooth muscle cells of arteries and by some adventitial cells of arteries in all groups. There was weak expression of PDGF-Rβ by arterial neointimal cells in the normal and AVR groups and stronger expression in the CAN group (Fig. 1B, D, and F). In serial sections, PDGF-D and PDGF-Rβ were co-localized in a similar anatomic distribution in the walls of arteries showing chronic allograft arteriopathy (Fig. 2).
Fig 2.
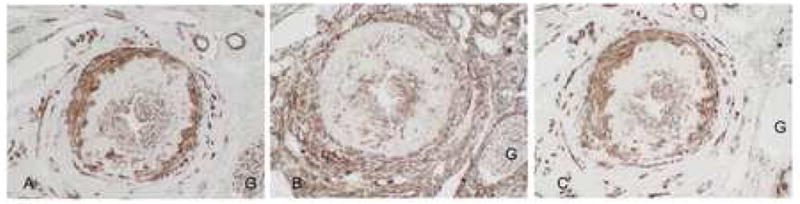
PDGF-D, PDGF-Rβ and smooth muscle actin in transplant arteriopathy. A: PDGF-D is expressed by medial smooth muscle cells, neointimal smooth muscle cells and some adventitial cells. B: In a serial section, PDGF-Rβ is expressed in a similar pattern by medial smooth muscle cells and neointimal smooth muscle cells. In addition, extravascular interstitial and adventitial cells strongly express PDGF-Rβ. C: Alpha smooth muscle actin immunolabeling of a serial section demonstrates a pattern of expression similar to PDGF-D, with strong staining of medial smooth muscle cells, neointimal smooth muscle cells and some adventitial cells and indicates co-localization of PDGF-D to these cells. Expression by interstitial myofibroblasts is also seen (*), which lack PDGF-D co-localization. G: glomerulus Original magnification A-C, ×200.
3.2 PDGF-D and PDGF-Rβ expression in tubulointerstitial areas
PDGF-D and PDGF-Rβ were expressed in each of the three groups, but with different patterns. PDGF-D was expressed focally by a few tubules, without detectable differences between groups (Fig. 3A, B and C). Double immunohistochemistry with the anti-EMA antibody localized PDGF-D expression to the epithelial cytoplasm of some, but not all distal tubules and collecting ducts. Proximal tubular expression was not identified. PDGF-Rβ was extensively expressed in the interstitial area in each of the groups (Fig. 3D, E and F). Morphometric analysis revealed that PDGF-Rβ was constitutively expressed in the thin interstitial space in the normal group (3.6%±2.4% of total tubulointerstitial area), but not significantly increased in the group with AVR (5.4%%±3.6% of total tubulointerstitial area, compared with the normal group, P>0.05, Student’s t test). PDGF-Rβ expression became more prominent in the group with CAN (10.1%±3.9% of total tubulointerstitial area, compared with the normal group, P<0.001, and compared with the AVR group, P<0.01, Student’s t test).
Fig. 3.
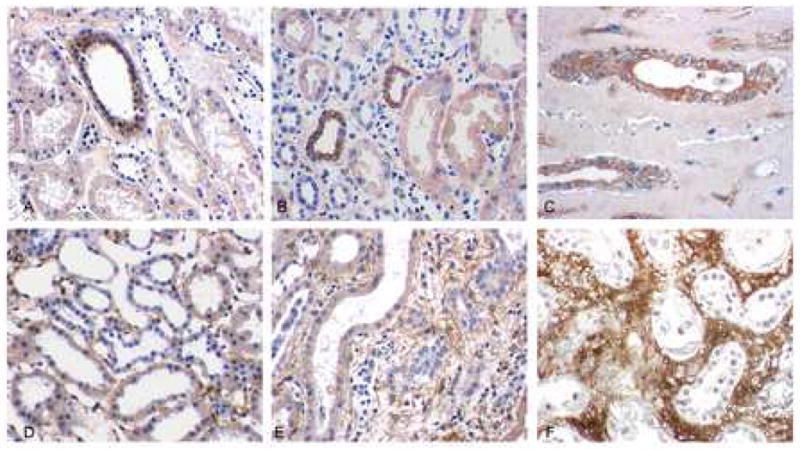
PDGF-D and PDGF-Rβ expression in tubulointerstitial areas. PDGF-D is expressed uniformly in a few tubuli in A: normal kidneys, B: kidneys with acute vascular rejection (AVR), and C: kidneys with chronic allograft nephropathy (CAN). PDGF-Rβ is expressed in interstitial areas in normal kidneys (D) and in kidneys with AVR (E), and more extensively in kidneys with CAN (F). Original magnification A-E, ×400.
In one case of AVR and in 3 cases of CAN, there were small sharply defined areas of marked scarring with extensive fibrosis, tubular dropout, and the presence of prominent spindled mesenchymal cells having the appearance of fibroblasts and/or myofibroblasts strongly expressing PDGF-D and PDGF-Rβ in an identical distribution. Double and triple immunohistochemistry proved that they were all alpha-smooth muscle actin expressing cells, and not CD 68 + monocyte/macrophages (Fig. 4).
Fig. 4.
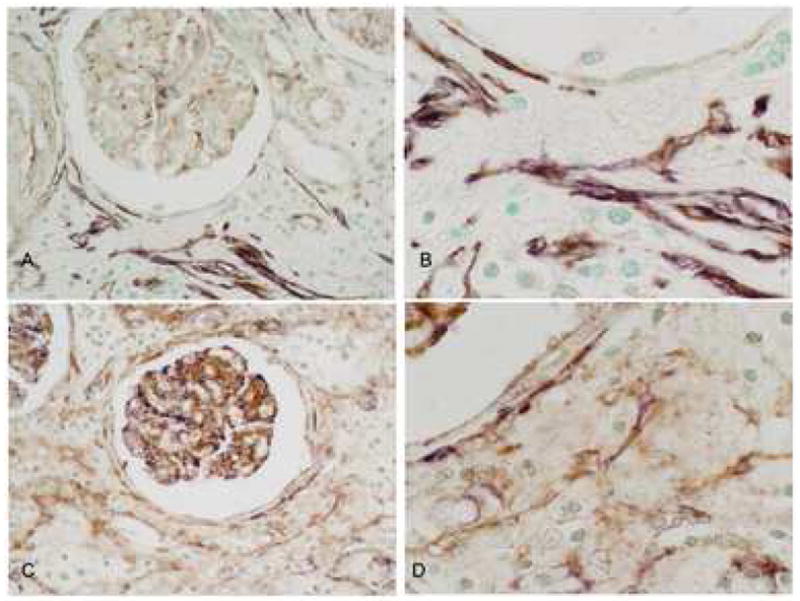
PDGF-D colocalization with smooth muscle actin and PDGF-Rβ in the interstitium. A and B: Double IHC showing PDGF-D (brown) and α-smooth muscle actin (purple). PDGF-D is expressed by glomerular podocytes and a subset of interstitial cells (A). High power view demonstrates colocalization of PDGF-D and α-smooth muscle actin by interstitial myofibroblasts.
C and D. Double IHC showing PDGF-D (purple) and PDGFR-beta (brown). PDGF-D is expressed by glomerular podocytes and a subset of interstitial cells (C). High power view shows PDGF-D colocalized PDGFR-beta in a subset of interstitial myofibroblasts (D). Original magnification A and C ×400, B and D ×1000.
3.3 PDGF-D and PDGF-Rβ expression in glomeruli
PDGF-D was extensively and uniformly expressed in the cytoplasm of podocytes in each of the three groups, with no differences noted between the three groups. No identifiable expression was detected in mesangial or endothelial cells (Fig. 5A, B, and C, Table 1). PDGF-Rβ was also expressed in all groups, with expression confined to mesangial areas (Fig. 1D, E, and F). Morphometric analysis of PDGF-Rβ expression showed it to be increased prominently in the CAN group (10.2%±7.7% per glomerular tuft area, p< 0.05, Student’s t test) compared with the normal group (2.9%±2.8% per glomerular tuft area) and AVR group (2.6%±3.2% per glomerular tuft area). There was no significant difference between the latter two groups. There was similar PDGF-D and PDGF-Rβ expression in a few parietal epithelial cells in all three groups.
Fig. 5.
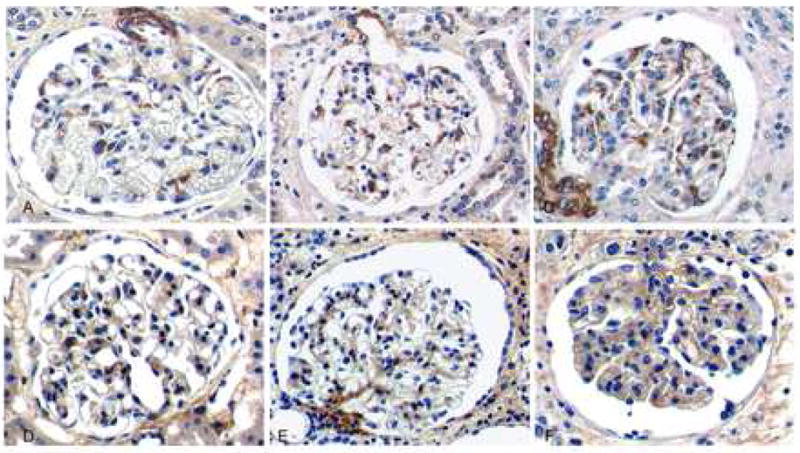
PDGF-D and PDGF-Rβ expression in glomeruli. PDGF-D is uniformly expressed in visceral epithelial cells and arteriolar wall in A: normal kidneys, B: kidneys with AVR, and C: kidneys with CAN. PDGF-Rβ is expressed weakly in mesangial areas in D: normal kidneys, E: kidneys with AVR, and more prominently in mesangial areas in F: kidneys with CAN. It is also expressed in some parietal epithelial cells and periglomerular areas in all three groups. Original magnification, × 400.
4. Discussion
Chronic allograft nephropathy is a descriptive term that encompasses chronic fibrosing injury in renal allografts regardless of underlying primary etiology, since such injuries are often multifactorial in etiology and/or the primary etiology may be inapparent. Chronic allograft arteriopathy is one of the main pathological manifestations of CAN, which is characterized by neointima formation by mesenchymal cells with accumulation of sclerotic matrix in arteries [1, 3]. This arteriopathy is often a consequence of rejection, as recently detailed in studies of a non human primate model of transplant rejection [29], and it is likely most of the cases of CAN included in the present study are a consequence of chronic vascular rejection. Current leading opinions about the origin of the mesenchymal neointimal cells in this arteriopathy is that they are derived either from smooth muscle cells that have migrated from the vessel media as a result of stimulation by growth factors and cytokines released during vascular injuries, or that they are derived from myofibroblasts migrating from the vascular adventitia. Cell culture experiments, balloon models of arterial injury, and models of transplant ateriopathy have each provided evidence that smooth muscle cell migration is regulated by PDGF-A and –B, acting through α or β receptors [30, 31].
Therefore, we asked whether the recently discovered PDGF-D might also be present in CAN and be in a position to promote the lesions of CAN, particularly arteriopathy, by acting through PDGF-Rβ. This study clearly establishes that this is the case. Our previous studies of acute and chronic vascular rejection in human renal allografts demonstrated that the other principal ligand of PDGF-Rβ, PDGF-B, is expressed by infiltrating monocytes/macrophages within the arterial walls [7]. PDGF-Rα is expressed by the intimal smooth muscle cells in a distribution similar to that found for PDGF-Rβ in the present study [13]. Furthermore, the principal ligand for PDGF-Rα, PDGF-A, has been found to be expressed in the arterial endothelial cells and neointimal smooth muscle cells continuously in the evolution of acute to chronic vascular rejection [7, 13]. Taken together, these studies provide an integrated view of how the PDGF ligand/receptor system may play a major role in promoting the neointimal formation that characterizes CAN arteriopathy. The evidence suggests that the injury process involves at least three main participants: endothelial cells which produce PDGF-A, infiltrating monocytes/macrophages which produce PDGF-B, and migrating smooth muscle cells and/or myofibroblasts which express PDGF-A, -B, -D, and their receptors α and β, and which synthesize the extracellular matrix that leads to intimal sclerosis. It is likely that these three cell types have interactions within the intima of rejecting arteries that involve autocrine and paracrine signaling activities. One possibility suggested by previous and current data is that in the early stage of arterial injury in renal transplants, PDGF-A induced by endothelial injury and PDGF-B produced by inflammatory infiltrates in arterial walls have chemoattractant activity for medial smooth muscle cells or adventitial myofibroblasts. These and other factors may then lead to medial smooth muscle cell or myofibroblast activation, further upregulation of PDGF-Rβ by these cells, and induction of migration to the subendothelial space. In this site these cells then may proliferate and synthesize extracellular matrix to form neointima [29]. PDGF-D may play a pivotal role in the sclerosing stages of arteriopathy. Here we have shown that PDGF-D and its receptor PDGF-Rβ expression are detectable in a similar distribution in only some of the sclerotic arteries in protocol renal biopsies and in biopsies with AVR, but that their expression becomes extensive and prominent in the chronic arterial injury of CAN with end stage transplant function. This finding suggests that PDGF-D, together with PDGF-A and –B, participates in the pathogenensis of transplant arteriopathy in an organized sequence of activities. This proposed scenario is consistent with the evolution of morphologic changes recently described in a non-human primate model of chronic vascular rejection which also suggest that many if not all of the cases of CAN in our series are indeed the result of chronic rejection [29]. Our findings do not identify what may be the functional role of PDGF-D that appears to be constitutively expressed by vascular medial smooth muscle cells.
Interstitial fibrosis is also an important component of CAN. Previous studies demonstrated a key role for PDGF-Rβ expressing fibroblasts and myofibroblasts in mediating renal interstitial fibrotic processes [12]. In particular, PDGF-B has been demonstrated to drive migration and proliferation of these cells and mediate the transformation of fibroblasts to an active myofibroblast phenotype [32–34]. In a previous study of the murine unilateral ureteral obstruction (UUO) model and human obstructive nephropathy, we showed that PDGF-D, as well as PDGF-B, was present in myofibroblastic cells in sites of interstitial fibrosis, where they co-localized with the PDGF-Rβ [12]. In the present study of human CAN, we observed an important difference from the findings in the UUO model and human obstructive nephropathy, in that, although PDGF-Rβ was expressed prominently, no identifiable PDGF-D expression was found in the extensively fibrotic interstitium except for limited areas of scarring present in only a small proportion of AVR or CAN cases. This result indicates that PDGF-B but not PDGF-D might have a principal role in mediating the interstitial fibrosis of CAN, although a role for PDGF-D produced from paracrine or exogenous systemic sources is not excluded.
Finally, this study found that in glomeruli of patients with CAN, PDGF-D expression was limited to podocytes and was unchanged in pattern or intensity from that observed in kidneys undergoing protocol biopsy and kidneys with AVR. In contrast, PDGF-Rβ demonstrated increased expression in mesangial areas in kidneys with CAN compared with the other two groups. It is yet to be determined whether there can be cross-talk between podocytes and mesangial cells, whereby podocyte produced PDGF-D can be secreted and gain access to the mesanguim where it might then bind to PDGF-Rβ. Alternately, or concurrently, this upregulated PDGF-Rβ may be active principally through engagement by PDGF-B or PDGF-AB. We cannot exclude the additional possibility that PDGF-D in the circulation can participate in glomerular diseases, as assays to detect circulating active forms of PDGF-D in humans are not currently available to test this hypothesis.
In summary, we demonstrate a potential role for PDGF-D in the arteriopathy of human CAN, but were largely unable to localize its presence in sites of established interstitial fibrosis encountered as part of this injury process despite the presence of its receptor at appropriate sites of injury. These findings should be useful to consider in the context of identifying potential therapeutic targets for amelioration of sclerosing renal injury.
Acknowledgments
Funding Sources: This study was supported in part by an NIH funded O’Brien Kidney Center of Research Excellence (DK 47659), in part by grant DK69912 from the National Institutes of Health and by a grant and generous provision of reagents from ZymoGenetics, Inc.
Dr. Gang Liu and Dr. Siribha Changsirikulchai are recipients of International Society of Nephrology Fellowship Award.
Abbreviations
- CAN
Chronic allograft nephropathy
- PDGF
Platelet derived growth factor
- PDGF-R
Platelet derived growth factor receptor
- AVR
acute vascular rejection
- DAB
3,3′-diaminobenzidine
- EMA
epithelial membrane antigen
Footnotes
Conflict of interest statement: There are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Paul LC. Chronic allograft nephropathy: An update. Kidney Int. 1999;56 (3):783–93. doi: 10.1046/j.1523-1755.1999.00611.x. [DOI] [PubMed] [Google Scholar]
- 2.Seron D. Early diagnosis of chronic allograft nephropathy by means of protocol biopsies. Transplant Proc. 2004;36 (3):763–4. doi: 10.1016/j.transproceed.2004.03.037. [DOI] [PubMed] [Google Scholar]
- 3.Aull MJ. Chronic allograft nephropathy: pathogenesis and management of an important posttransplant complication. Prog Transplant. 2004;14 (2):82–8. doi: 10.1177/152692480401400202. [DOI] [PubMed] [Google Scholar]
- 4.Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15 (4):255–73. doi: 10.1016/j.cytogfr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 5.Betsholtz C. Insight into the physiological functions of PDGF through genetic studies in mice. Cytokine Growth Factor Rev. 2004;15 (4):215–28. doi: 10.1016/j.cytogfr.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 6.Hudkins KL, Gilbertson DG, Carling MD, et al. Exogenous PDGF-D is a potent mesangial cell mitogen and causes a severe mesangial proliferative glomerulopathy. J Am Soc Nephrol. 2004;15:286–98. doi: 10.1097/01.asn.0000108522.79652.63. [DOI] [PubMed] [Google Scholar]
- 7.Alpers CE, Davis CL, Barr D, Marsh CL, Hudkins KL. Identification of platelet-derived growth factor A and B chains in human renal vascular rejection. Am J Pathol. 1996;148 (2):439–51. [PMC free article] [PubMed] [Google Scholar]
- 8.Fredriksson L, Li H, Eriksson U. The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004;15 (4):197–204. doi: 10.1016/j.cytogfr.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Floege J, Eitner F, Van Roeyen C, Ostendorf T. PDGF-D and Renal Disease: Yet Another One of Those Growth Factors? J Am Soc Nephrol. 2003;14 (10):2690–1. doi: 10.1097/01.asn.0000090831.40856.69. [DOI] [PubMed] [Google Scholar]
- 10.Reigstad LJ, Varhaug JE, Lillehaug JR. Structural and functional specificities of PDGF-C and PDGF-D, the novel members of the platelet-derived growth factors family. Febs J. 2005;272 (22):5723–41. doi: 10.1111/j.1742-4658.2005.04989.x. [DOI] [PubMed] [Google Scholar]
- 11.Uehara G, Suzuki D, Toyoda M, Umezono T, Sakai H. Glomerular expression of platelet-derived growth factor (PDGF)-A, -B chain and PDGF receptor-alpha, -beta in human diabetic nephropathy. Clin Exp Nephrol. 2004;8 (1):36–42. doi: 10.1007/s10157-003-0265-8. [DOI] [PubMed] [Google Scholar]
- 12.Taneda S, Hudkins KL, Topouzis S, et al. Obstructive Uropathy in Mice and Humans: Potential Role for PDGF-D in the Progression of Tubulointerstitial Injury. J Am Soc Nephrol. 2003;14 (10):2544–55. doi: 10.1097/01.asn.0000089828.73014.c8. [DOI] [PubMed] [Google Scholar]
- 13.Floege J, Hudkins KL, Davis CL, Schwartz SM, Alpers CE. Expression of PDGF alpha-receptor in renal arteriosclerosis and rejecting renal transplants. J Am Soc Nephrol. 1998;9 (2):211–23. doi: 10.1681/ASN.V92211. [DOI] [PubMed] [Google Scholar]
- 14.Ostendorf T, van Roeyen CR, Peterson JD, et al. A fully human monoclonal antibody (CR002) identifies PDGF-D as a novel mediator of mesangioproliferative glomerulonephritis. J Am Soc Nephrol. 2003;14 (9):2237–47. doi: 10.1097/01.asn.0000083393.00959.02. [DOI] [PubMed] [Google Scholar]
- 15.Lutz J, Yao Y, Song E, et al. Inhibition of matrix metalloproteinases during chronic allograft nephropathy in rats. Transplantation. 2005;79 (6):655–61. doi: 10.1097/01.tp.0000151644.85832.b5. [DOI] [PubMed] [Google Scholar]
- 16.Savikko J, Taskinen E, Von Willebrand E. Chronic allograft nephropathy is prevented by inhibition of platelet-derived growth factor receptor: tyrosine kinase inhibitors as a potential therapy. Transplantation. 2003;75 (8):1147–53. doi: 10.1097/01.TP.0000062836.93496.CE. [DOI] [PubMed] [Google Scholar]
- 17.Schindler R, Tullius SG, Tanriver Y, et al. Hypertension increases expression of growth factors and MHC II in chronic allograft nephropathy. Kidney Int. 2003;63 (6):2302–8. doi: 10.1046/j.1523-1755.2003.00034.x. [DOI] [PubMed] [Google Scholar]
- 18.Akyurek LM, Paul LC, Funa K, Larsson E, Fellstrom BC. Smooth muscle cell migration into intima and adventitia during development of transplant vasculopathy. Transplantation. 1996;62 (10):1526–9. doi: 10.1097/00007890-199611270-00029. [DOI] [PubMed] [Google Scholar]
- 19.Chen J, Han Y, Lin C, et al. PDGF-D contributes to neointimal hyperplasia in rat model of vessel injury. Biochem Biophys Res Commun. 2005;329 (3):976–83. doi: 10.1016/j.bbrc.2005.02.062. [DOI] [PubMed] [Google Scholar]
- 20.Changsirikulchai S, Hudkins KL, Goodpaster TA, et al. Platelet-derived growth factor-D expression in developing and mature human kidneys. Kidney Int. 2002;62 (6):2043–54. doi: 10.1046/j.1523-1755.2002.00662.x. [DOI] [PubMed] [Google Scholar]
- 21.Alpers CE, Seifert RA, Hudkins KL, Johnson RJ, Bowen-Pope DF. PDGF-receptor localizes to mesangial, parietal epithelial, and interstitial cells in human and primate kidneys. Kidney Int. 1993;43 (2):286–94. doi: 10.1038/ki.1993.45. [DOI] [PubMed] [Google Scholar]
- 22.Colvin RB, Cohen AH, Saiontz C, et al. Evaluation of pathologic criteria for acute renal allograft rejection: reproducibility, sensitivity, and clinical correlation. J Am Soc Nephrol. 1997;8 (12):1930–41. doi: 10.1681/ASN.V8121930. [DOI] [PubMed] [Google Scholar]
- 23.Racusen LC, Colvin RB, Solez K, et al. Antibody-mediated rejection criteria -an addition to the Banff 97 classification of renal allograft rejection. Am J Transplant. 2003;3 (6):708–14. doi: 10.1034/j.1600-6143.2003.00072.x. [DOI] [PubMed] [Google Scholar]
- 24.Racusen LC, Solez K, Colvin RB, et al. The Banff 97 working classification of renal allograft pathology. Kidney Int. 1999;55 (2):713–23. doi: 10.1046/j.1523-1755.1999.00299.x. [DOI] [PubMed] [Google Scholar]
- 25.Skalli O, Ropraz P, Trzeciak A, Benzonana G, Gillessen D, Gabbiani G. A monoclonal antibody against alpha-smooth muscle actin: a new probe for smooth muscle differentiation. J Cell Biol. 1986;103 (6 Pt 2):2787–96. doi: 10.1083/jcb.103.6.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holthofer H, Virtanen I, Kariniemi AL, Hormia M, Linder E, Miettinen A. Ulex europaeus I lectin as a marker for vascular endothelium in human tissues. Lab Invest. 1982;47 (1):60–6. [PubMed] [Google Scholar]
- 27.Falini B, Flenghi L, Pileri S, et al. PG-M1: a new monoclonal antibody directed against a fixative-resistant epitope on the macrophage-restricted form of the CD68 molecule. Am J Pathol. 1993;142 (5):1359–72. [PMC free article] [PubMed] [Google Scholar]
- 28.Hudkins KL, Giachelli CM, Cui Y, Couser WG, Johnson RJ, Alpers CE. Osteopontin expression in fetal and mature human kidney. J Am Soc Nephrol. 1999;10 (3):444–57. doi: 10.1681/ASN.V103444. [DOI] [PubMed] [Google Scholar]
- 29.Wieczorek G, Bigaud M, Menninger K, et al. Acute and chronic vascular rejection in nonhuman primate kidney transplantation. Am J Transplant. 2006;6 (6):1285–96. doi: 10.1111/j.1600-6143.2006.01307.x. [DOI] [PubMed] [Google Scholar]
- 30.Palumbo R, Gaetano C, Antonini A, et al. Different effects of high and low shear stress on platelet-derived growth factor isoform release by endothelial cells: consequences for smooth muscle cell migration. Arterioscler Thromb Vasc Biol. 2002;22 (3):405–11. doi: 10.1161/hq0302.104528. [DOI] [PubMed] [Google Scholar]
- 31.Raines EW. PDGF and cardiovascular disease. Cytokine Growth Factor Rev. 2004;15 (4):237–54. doi: 10.1016/j.cytogfr.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 32.Tang WW, Ulich TR, Lacey DL, et al. Platelet-derived growth factor-BB induces renal tubulointerstitial myofibroblast formation and tubulointerstitial fibrosis. Am J Pathol. 1996;148 (4):1169–80. [PMC free article] [PubMed] [Google Scholar]
- 33.Yu J, Moon A, Kim HR. Both platelet-derived growth factor receptor (PDGFR)-alpha and PDGFR-beta promote murine fibroblast cell migration. Biochem Biophys Res Commun. 2001;282 (3):697–700. doi: 10.1006/bbrc.2001.4622. [DOI] [PubMed] [Google Scholar]
- 34.Oh SJ, Kurz H, Christ B, Wilting J. Platelet-derived growth factor-B induces transformation of fibrocytes into spindle-shaped myofibroblasts in vivo. Histochem Cell Biol. 1998;109 (4):349–57. doi: 10.1007/s004180050235. [DOI] [PubMed] [Google Scholar]