

Abstract
Increased myocardial cGMP, achieved by enhancing cyclase activity or impeding cGMP hydrolysis by phosphodiesterase type-5 (PDE5A), suppresses cellular and whole organ hypertrophy. The efficacy of the latter also requires cyclase stimulation and may depend upon co-activation of maladaptive signaling suppressible by cGMP-stimulated kinase (cGK-1). Thus, PDE5A inhibitors could paradoxically be more effective against higher than lower magnitudes of pressure-overload stress. To test this, mice were subjected to severe or moderate trans-aortic constriction (sTAC, mTAC) for 6 weeks ±co-treatment with oral sildenafil (SIL 200 mg/kg/d). LV mass (LVM) rose 130% after 3-wks sTAC and SIL blunted this by 50%. With mTAC, LVM rose 56% at 3wks but was unaffected by SIL, whereas a 90% increase in LVM after 6wks was suppressed by SIL. SIL minimally altered LV function and remodeling with mTAC until later stages that stimulated more hypertrophy and remodeling. SIL stimulated cGK-1 activity similarly at 3 and 6 wks of mTAC. However, pathologic stress signaling (e.g. calcineurin, ERK-MAPkinase) was little activated after 3-wk mTAC, unlike sTAC or later stage mTAC when activity increased and SIL suppressed it. With modest hypertrophy (3-wk mTAC), GSK3β and Akt phosphorylation were unaltered but SIL enhanced it. However, with more severe hypertrophy (6-wk mTAC and 3-wk sTAC), both kinases were highly phosphorylated and SIL treatment reduced it. Thus, PDE5A-inhibition counters cardiac pressure-overload stress remodeling more effectively at higher than lower magnitude stress, coupled to pathologic signaling activation targetable by cGK-1 stimulation. Such regulation could impact responses of varying disease models to PDE5A inhibitors.
Keywords: hypertrophy, phosphodiesterase, cyclic nucleotides, sildenafil, mouse models, Galphaq, PDE5A, remodeling, heart failure, stress response kinase, hemodynamics
INTRODUCTION
Heart failure (HF) is a leading cause of morbidity and mortality affecting an estimated six million patients in the United States alone [1]. Current treatments target excess fluid load and block neurohormonal stimulation at the level of cell surface receptors. However, recent research has uncovered multiple intracellular signaling cascades central to maladaptive HF remodeling, and many are now being explored as new potential therapeutic targets [2,3]. A prominent example is the regulatory system controlled by cyclic guanosine monophosphate (cGMP) and cGMP-stimulated protein kinase (cGK-1). Cardiac cGMP/cGK-1 signaling counters acute and chronic stress responses and protects against cell death [4,5]. Cyclic GMP is generated by soluble and receptor guanylate cyclases stimulated by nitric oxide and natriuretic peptides, respectively, and catabolized by selective members of the phosphodiesterase (PDE) superfamily. Among the latter, PDE-type 5 (PDE5A) remains the best characterized, and its inhibition by drugs such as sildenafil (Viagra®) is widely used to treat erectile dysfunction and pulmonary hypertension [6].
PDE5A was the first cGMP-selective PDE discovered, and while important in platelets and vascular smooth muscle, its role in the heart was long thought to be negligible given low levels of protein expression and activity [7]. Indeed, in un-stressed hearts and myocytes, acute and chronic inhibition of PDE5A has negligible impact [8–10]. However, under conditions of cardiac stress, PDE5A inhibitors have been recently shown to suppress beta-adrenergic stimulation [9–11], and protect hearts against ischemic cardiomyopathy [12], mitochondrial toxicity [13], and chronic pressure-overload [8]. Genetic studies now provide more direct support for PDE5A pro-hypertrophic effects in isolated myocytes, and show its inhibition by either gene silencing or pharmacologic blockers is anti-hypertrophic in a cGK-1 dependent manner [14].
The lack of effects from PDE5A inhibition in the normal resting heart yet substantial impact in hearts exposed to various stressors led investigators to propose that its cardiac modulation acts much like a brake [7]. However, while brakes often work best when countering less force, PDE5A inhibition might actually require a sufficiently high stress to be influential for the following reasons. First, it can only enhance cGK-1 activity if there is sufficient cGMP synthesis produced by the relevant cyclase, with NO-stimulated sGC being identified thus far as the key source [9–11]. Second, PDE5A must itself be active so its inhibition can further stimulate cGK-1. Third, cGK-1 is not a generalized effecter, but suppresses selective cascades involved with maladaptive hypertrophy, and these would have to be activated if PDE5A inhibition was to offset this signaling. Thus, anti-hypertrophic effects from PDE5A-inhibition may only become manifest if the stress meets these conditions.
To test this hypothesis, we contrasted the efficacy of sildenafil treatment in hearts subjected to moderate versus more severe pressure-overload. Based on data showing cGK-1 (and PDE5A inhibition) suppresses Gαq-signaling pathways [8], we examined whether the efficacy of sildenafil corresponds with the occurrence of such stimulation. The results show that PDE5A inhibition has features of a targeted modulator, being more effective with higher magnitudes of hypertrophic stimulation.
MATERIALS AND METHODS
Animal models
Male C57Bl/6 mice (9–12 wks; Jackson Labs, Bar Harbor, ME) were subjected to two models of pressure-overload produced by transverse aortic constriction (TAC) [8], using either a 27-gauge (severe TAC; sTAC) or 25-gauge (moderate TAC; mTAC) needle. Sham-operated control mice underwent the same surgical procedures but without aortic constriction. Sildenafil was prepared fresh every other day by grinding 50 mg Viagra® tablets into a soft diet (Bio-Serv), and administered at 200 mg/kg/day from the onset of TAC and continued for 6 weeks. Controls received the soft diet only. The dose was based on higher catabolism of sildenafil in mice compared to human (~100×), and was increased over that initially reported [8] as the prior dose while effective yielded free plasma sildenafil concentrations of ~10 nM, very close to the in vitro IC50 for PDE5A. The 200 mg/kg/dose resulted in a 55.6±13 nM level (measured after 3 weeks of chronic therapy) similar to that for humans following a 100 mg oral dose [15], and still well below the IC50 for PDE1. The protocol was approved by the Johns Hopkins Medical Institutions Animal Care and Use Committee.
Physiologic studies
Cardiac function was assessed in conscious mice by transthoracic, two-dimensional, guided M-mode echocardiography (Sequoia C256, Siemens, NY) with the 15 MHz linear-array transducer. More comprehensive in vivo cardiac mechanics was determined using a miniature pressure-volume catheter (SPR-839, Millar Instruments, Inc., Houston, TX) inserted via the LV apex in anesthetized, open-chest mice, as described [9].
Protein and Gene Expression
Protein extract was obtained from snap-frozen cardiac tissue. Immunoblots were performed using primary antibodies to phospho-(Ser473)-Akt (1:500, Santa Cruz Biotechnology, Inc., Santa Cruz, CA), Akt, ERK, phospho-(Thr202/Thr204)-ERK, glycogen synthase 3β (GSK3β), phospho-(Ser9)-GSK3β, GAPDH (1:1000, Cell Signaling Technology, Inc., Danvers, MA), and calcineurin (Cn; 1:2000, BD Biosciences, Sparks, MD). Secondary antibodies used were either goat anti-rabbit immunoglobulin G (IgG) or goat anti-mouse IgG2b conjugated with horseradish peroxidase (1:3000, Santa Cruz Biotechnology). Protein bands were detected using GE Amersham chemiluminescence reagent and visualized by exposure to X-ray film. Protein band intensity was quantified by laser scanner and results expressed as arbitrary units. Equal loading was confirmed by Ponceau (Sigma-Aldrich, Louis, MO) method.
Total RNA was extracted from homogenized mouse heart tissue with TRIzol (Invitrogen). Messenger RNA expression was determined by quantitative rtPCR after converting to cDNA using either SYBR green method or Taqman probe method. PCR reactions were performed, recorded, and analyzed by using the ABI PRISM Detection System (Applied Biosystems, Foster City, CA). The following primers were used: Mouse ANP 5′-TCGTCTTGGCCTTTTGGCT-3′ (forward) and 5′TCCAGGTGGTCTAGCAGGTTCT-3′ (reverse); Mouse BNP 5′-AAGTCCTAGCCAGTCTCCAGA-3′ (forward) and 5′-GAGCTGTCTCTGGGCCATTTC-3′ (reverse); Mouse β-MHC 5′-ATGTGCCGGACCTTGGAAG-3′ (forward) 5′-CCTCGGGTTAGCTGAGAGATCA-3′ (reverse). PCR samples were run in duplicate and normalized to GAPDH. PCR conditions were 15 min at 95°C and 40 cycles of 95°C for 30 s, 57°C for 15 s, and 72°C for 15 s. Specificity of the SYBR green assays was confirmed by dissociation curve analysis. Regulator of Calcineurin-1 (RCAN-1) expression was used to index calcineurin activation (Applied Biosystems: MCIP1, Mm01213407_m1). PCR samples (25ng) were run in duplicate and normalized to GAPDH. Reactions were performed with 900 nM of the specific primer pairs and 250 nM of specific probe. PCR conditions used were 15 min at 95°C and 40 cycles of 95°C for 15 s and 60°C for 60 s.
cGK-1 Activity
cGMP protein kinase activity was assessed by colorimetric analysis (CycLex, Nagano, Japan) following manufacturer’s instructions, and utilizing the same protein extracts used for immunoblot analyses. Protein concentration was determined by the BCA method (Pierce), and 30 μg of whole heart lysate from each sample used to assess cGK-1 activity.
Statistical Analysis
Data are presented as mean ± SEM. Differences between multiple groups were compared by analysis of variance (ANOVA, one- or two-way as appropriate) followed by a Tukey’s multiple comparisons test. Serial studies were tested by repeated measures ANOVA. P values of <0.05 were considered significant.
RESULTS
Sildenafil more effectively suppresses hypertrophy in severe versus moderate TAC
Figure 1 shows example echocardiographic M-mode tracings (1A) and corresponding summary data (1B) for these experiments. Hearts subjected to sTAC developed >100% increase in estimated left ventricular mass after 3 weeks, which was reduced by half in animals co-treated with sildenafil. Cardiac remodeling/dilation from sTAC was reflected by chamber dilation (LV end-diastolic and end-systolic dimension) and a decline in fractional shortening. All of these changes were ameliorated by sildenafil treatment. In contrast, mice subjected to mTAC developed about half the extent of LV hypertrophy after 3 weeks (+56%), and LV end-diastolic volumes declined, consistent with concentric compensated hypertrophy. At this time point, sildenafil treatment had no impact on any mechanical or remodeling parameters (Figure 1B). After 6 weeks of mTAC, non-treated mice now developed chamber dilation, with reduced fractional shortening (FS), and increased hypertrophy (~100% increase). However, this remodeling was now significantly blunted in mice receiving sildenafil therapy. These differential responses were confirmed at post-mortem analysis (heart weight/tibia length; 76.9±1.1 vs 82.0±3.2 mg/mm 3-wks mTAC; 92.2±2.7 vs 79.9±4.2 mg/mm 6-wks mTAC; vehicle vs SIL, respectively, p<0.01 by 2-way ANOVA).
Figure 1.

A) Example M-mode echocardiographic images at baseline (sham operated), and vehicle versus sildenafil treated animals for 3-wks severe TAC, and 3-wks, 6-wks moderate TAC models. Chamber end-diastolic dimension is highlighted (arrows) and shows reduction at 3-wks sTAC, but no change until 6-wks mTAC. B) Summary echocardiographic data for LV diastolic dimension (LVDd), LV systolic dimension (LVDs), fractional shortening (FS), and LV mass (N=5–10). After 3-wks sTAC, SIL reduced chamber dilation and dysfunction, and suppressed hypertrophy. With mTAC, SIL did not alter heart function at 3-wks where there was moderate LVH (about half the extent with sTAC), but did blunt hypertrophy and remodeling at a later stage (6-wks).
PDE5 inhibition enhances function in mTAC hearts exposed to more sustained stress
To examine the effect of sildenafil treatment on mTAC induced changes in cardiac function in greater detail, we performed pressure-volume loop analysis. Sildenafil has been previously shown to enhance left ventricular systolic function in hearts exposed to sTAC for 3-wks [8] (e.g. increase in dP/dtmax/IP from 192±6.9 to 256±8.4 sec−1, reduce end-systolic volumes from 23.3±3.3 to 6.4±1.7 μL; sTAC±SIL respectively, both p<0.01; increase end-systolic elastance by 90%, p<0.05; details in supplemental table in ref [8]). These changes occurred with no alteration in LV afterload. In contrast, sildenafil had negligible effects after 3-wks of mTAC (Fig. 2A, B). Chamber volumes and end-systolic elastance (upper left corners of the loops) was unaltered, though there was a slight rise in dP/dtmax/IP. After more prolonged mTAC exposure and greater hypertrophic stimulation, LV chambers dilated and displayed a rightward shift of the end-systolic pressure-volume relation (ESPVR). Mice receiving SIL treatment now displayed less remodeling and improved systolic function (both slope of ESPVR, Ees, and and dP/dtmax/IP increased; Figure 2B).
Figure 2.

A) Example pressure-volume loops for sham control, and moderate TAC ±SIL treatment at 3 and 6-wks timepoints. Increased cardiac loading is demonstrated by the higher systolic pressures, but at 3-wks there was negligible functional impact of SIL, whereas at 6-wks, untreated hearts dilated and had reduced function compared with SIL treated hearts. B) Summary results from PV analysis for LV end-diastolic and end-systolic volumes (LV-EDV, LV-ESV), maximal LV pressure (LVPmx), end-systolic elastance (Ees), and maximal rate of pressure rise normalized to instantaneous developed pressure (dP/dtmax/IP) (N=5–7).
PKG is equally potentiated by sildenafil at 3 and 6 weeks of mTAC
To test whether late but not early efficacy of PDE5-inhibition with mTAC reflected an inadequate stimulation of cGK-1 at the earlier time point, perhaps due to insufficient cyclase activity, we determining cGK-1 activity by in vitro assay (Figure 3A). cGK-1 activation increased after 3 and 6-wks of mTAC in a gradual manner, with the latter reaching a similar 2-fold increase as previously observed with untreated 3-wks sTAC [8]. SIL treatment further increased cGK-1 activity to ~4–5 times above sham controls at both time points. This level is also similar to that previously reported after 3-wks of sTAC+SIL [8] (also shown in Fig. 3A). Thus, the difference in SIL modulation of cardiac function and remodeling on 3- versus 6-wk mTAC could not be explained based on differential potentiation of cGK-1 activity.
Figure 3.

A) cGK-1 activity measured in sham and after 3 or 6-wks mTAC±SIL (N=4). Rest (vehicle treated) activity increased at each time point, but was further and similarly enhanced (~4-fold over sham) with SIL treatment. Augmentation of activity after 3-wks sTAC ±SIL co-treatment is shown for comparison. * p<0.02 versus sham control; †p<0.01 versus vehicle treated at same time point. B) Influence of mTAC±SIL on ERK1/2 phosphorylation and calcineurin (Cn) protein expression (N=3). SIL had minimal impact at 3 wks, but significantly depressed expression levels when treated out to 6-wks of mTAC. Comparison expression levels after 3-wks of sTAC (previously shown to be blunted by SIL treatment [8]) were similar to those observed after 6-wks mTAC. C) Gene expression for RCAN-1, ANP, and β-MHC (N=4). * p<0.05 versus sham control; † p<0.05 versus 6-wks mTAC vehicle treated.
PDE5A Inhibition is Effective when Hearts Display Activation of Targeted Signaling
Given similar cGK-1 activation with SIL treatment at both mTAC time points, we next hypothesized that the efficacy of SIL to suppress hypertrophy may depend upon the activation of specific kinase targets that contribute to the stress response. Figure 3B shows results for two major enzymes that contribute to pathologic hypertrophy and have been previously shown to be targeted by PDE5A-inhibitor/cGK-1 signaling, extracellular response kinase (ERK1/2) and calcineurin (Cn) [8]. ERK1/2 phosphorylation modestly increased after 3-wks of mTAC but was unaffected by SIL co-treatment, and even less stimulation of Cn was observed in either condition. However, after 6-wks of mTAC, ERK1/2 activation and Cn expression were markedly upregulated, at levels similar to those observed after 3-wks sTAC. In this condition, SIL significantly suppressed both responses (previously reported with 3-wks sTAC [8,16]). Cn activity was further assayed by RCAN-1 gene expression (Fig. 3C). RCAN-1 expression was negligibly changed after 3 weeks (±SIL), but increased by 6-wks mTAC. Importantly, at the higher level of activation, SIL now reduced expression to control levels. RCAN-1 expression at 3-wks sTAC is shown by way of comparison, and prior studies have demonstrated SIL suppressed this response [16].
Fetal gene re-expression is also a marker of pathologic hypertrophy. At 3-wks mTAC, ANP expression increased significantly, but not markedly, and was unaltered by SIL treatment, whereas much higher expression was observed at 6-wks mTAC and was blunted by SIL. β-MHC expression remained at control levels till 6-wks of mTAC, and was unaltered by SIL. Reference expression change with 3-wks sTAC are shown for these genes as well, and the impact of SIL is consistent with prior reports [8,16].
Sildenafil enhances Akt and GSK3β activation early but reduces it later with mTAC
The serine/threonine kinase Akt and its downstream target glycogen synthase kinase 3β (GSK3β) are potent modulators of cardiac hypertrophy. Their upregulation is thought to contribute to physiologic hypertrophic responses, but if enhanced at sufficiently high levels, they can contribute to pathologic remodeling. We reported that sTAC activates both Akt and GSK3β, and that this can be reversed by SIL treatment [8]. Here we examined how these signaling cascades were activated by mTAC and modulated by SIL. At 3-wks of mTAC, neither kinase was activated over sham controls, but interestingly, SIL treatment resulted in a significant increase in phosphorylation. The opposite pattern was observed after 6-wk mTAC, where phosphorylation of both proteins was significantly increased over baseline, and now SIL depressed this response. Thus, the impact of SIL was bimodal, and depended upon the severity of the hypertrophic stimulus and activation level of Akt/GSK3β kinases.
DISCUSSION
The current findings reveal an intriguing and important phenomenon regarding the ability of PDE5A inhibition to suppress hypertrophic responses induced by pressure-overload. Specifically, this strategy is more effective when stresses are of a higher magnitude and activate targeted pathologic signaling cascades, whereas moderate hypertrophy stimulation is less impacted. Prior studies reporting efficacy of sildenafil when treatment was initiated at the start [8] or delayed until more established disease developed [16] have employed a severe TAC model that induces a marked cardiac stress response. Here we showed far less initial impact of PDE5A inhibition when confronting mTAC, yet over time as the hypertrophic stimulus reached a sufficient magnitude and was accompanied by activation of targeted stress kinases, (e.g. ERK1/2 and calcineurin), SIL treatment became effective. This behavior differs somewhat from approaches that directly inhibit a single enzyme or block transcriptional regulation, where modest hypertrophy is often more readily suppressed than severe disease.
Despite the recognition that the cGMP-cGK-1 pathway has anti-hypertrophic effects in the heart, therapeutic strategies to effectively harness this have faced limitations. In particular, stimulation of upstream cGMP synthesis either by providing nitric oxide donors or natriuretic peptides have not translated into effective anti-hypertrophic treatments. Both have prominent effects on peripheral vascular tone, and particularly for NO donors and organo-nitrates this dominates the pharmacology. Secondly, chronic nitric oxide stimulation results in tolerance, in part from S-nitrosylation of sGC itself [17], and in part from up-regulation of PDEs such as PDE1c [18] and PDE5A [19,20]. Lastly, administering compounds exogenously does not necessarily yield the same intracellular signal, and this appears particularly important for cyclic nucleotides which are tightly regulated in microdomains within the cell [21]. This results in different functional pools of cGMP produced by sGC versus rGC [11,22] (i.e. natriuretic peptide stimulated), and corresponding regulation. In contrast, inhibition of a specific phospho- diesterase provides more local control of cGMP and in the cells of interest. For PDE5A, vasodilation targets the pulmonary vasculature and corpus cavernosum, and in a sufficiently stressed myocardium, there appears substantial impact as shown by augmented cGK-1 activity.
Even genetic strategies that modify guanylate cyclase have only been reported to suppress rather modest hypertrophy [23,24], whereas PDE5A inhibition was shown to counter a far more potent hypertrophic stimulus [8,16]. This may be due to the greater impact of blocking cGMP hydrolysis as a means of stimulating cGK-1 activity, whereas stimulating cGMP synthesis without concomitant blockade can be offset by feedback activation of its hydrolysis [7]. However, for PDE5A inhibition to be effective there must also be sufficient activation of the underlying cyclase, as well as activation of maladaptive enzymes targeted by cGK-1. The finding that cGK-1 activity is enhanced similarly at both 3 and 6wks of mTAC, at levels similar to those after 3wks of sTAC, indicates that sufficient cyclase activation occurred even when SIL did not exhibit anti-hypertrophic effects. Thus, the primary factor would appear to be activation cGK-1 targetable proteins. This insight may be relevant to studies exploring the efficacy of a PDE5A strategy in other forms of hypertrophic disease (e.g. related to genetic mutations) where such signaling may not be involved, or conditions with less severe disease.
The lack of SIL effects on mild hypertrophy generated after 3-wks of mTAC was not simply due to the lack of any pressure-overload response to begin with. Though reduced in magnitude, LV mass rose significantly (confirmed by post-mortem analysis) to levels previously generated and inhibited by upregulating GC-A [24]. Secondly, SIL treatment induced a marked rise in cGK-1 activity at the 3-wk mTAC time point. This indicates stimulation of resting cyclase activity, a response that is not observed in un-stressed (sham operated) controls treated with SIL for the same duration [8]. Third, we found increased in ERK1/2 phosphorylation and ANP expression (nearly 2-times increased) at this time point, further supporting activation of a pathologic stress response, albeit at a lower level. These data show that modest but compensated hypertrophic responses were induced in the early phase mTAC, yet were not suppressed by SIL.
cGK-1 inhibition of Cn-activated NFAT transcriptional regulation was first revealed by Fiedler et al in a study of neonatal myocytes [25]. SIL similarly suppresses NFAT stimulation induced by Gq-coupled α-adrenergic receptor agonists in isolated myocytes [8], and in the intact heart, reduces Cn expression and activity reflected by RCAN-1 expression [16]. More recently, we have found that an important mechanism of SIL suppression of early-phase hypertrophic remodeling is its interaction and activation of regulator of G-protein coupled signaling-2 (RGS2) [26]. This GTPase particularly targets Gαq-coupled signaling, restoring the hetero-trimeric G-protein complex. In vascular smooth muscle, activated cGK-1 binds to and phosphorylates RGS2, and then co-migrates to the sarcolemmal membrane where it suppresses angiotensin-stimulated vasoconstriction [27]. Though RGS proteins target many G-protein species and coupled receptors, in cardiomyocytes, RGS2 now appears to particularly regulate Gαq/11 signaling [28]. We have observed that mice lacking RGS2 develop profound early hypertrophy, heart failure, and mortality after TAC, with increased Gαq-coupled signaling (including ERK1/2 and Cn), and SIL cannot suppress this response [29]. Yet, chronic swimming exercise, which does not trigger these pathologic signaling cascades, was performed perfectly well by RGS2−/− mice [26]. SIL treatment also does not influence physiologic hypertrophy induced by swimming exercise [30]. Thus, an important early target of SIL via cGK-1 activation appears to be negative regulation of Gαq. The present data further support the notion that sufficient amplitude of pathologic stress signaling, likely involving Gαq activation, is important for PDE5-inhibition to effectively blunt hypertrophy.
GSK3β is a negative regulator of cardiac hypertrophy and positive regulator of cell survival. Thus, phosphorylation inactivating the enzyme stimulates hypertrophy [31] while de-phosphorylation (activation) provides cardiomyocyte protection against apoptosis [32]. Both GSK3β and Akt, an upstream kinase that can modulate GSK3β activity, were phosphorylated by SIL therapy after 3-wk mTAC. Yet after 6-wks, when both proteins displayed enhanced resting phosphorylation, SIL treatment now reduced it. In contrast, with sTAC [8], at 1-wk (compensated hypertrophy), pAkt and pGSK3β both increased, and SIL depresses pAkt but did not impact pGSK3β whilein late-stage disease (9-wk), enhanced phosphorylation of both kinases was suppressed by SIL as found here for mTAC after 6 wks.
The current results support a positive impact of a cGK-1 signaling (if not cGK-1 itself) to enhance GSK3β phosphorylation so long as it is not already pathologically inactivated. This is supported by a recent study by Das and Kukreja [33], who showed that SIL-induced cardio-protection against simulated ischemia and re-oxygenation in adult ventricular myocytes depended on cGK-1 activation, and was accompanied by GSK3β, ERK1/2, and Akt phosphorylation. Akt phosphorylation in this cellular model was not found to be cGK-1 dependent, however, suggesting an alternative activation pathway.
Antihypertrophic effects of activated GSK3β via reduced phosphorylation or expression 14 of phospho-deficient mutants has been demonstrated by a number of genetic in vivo models. Mice overexpressing either wild-type GSK3β or a (S9A) mutant form (phospho-deficient) display reduced hormone or stress stimulated hypertrophy [31], and induction of the S9A mutant in hearts of mice with established hypertrophy induced by TAC also showed regression of LV mass [34]. However, the signaling from GSK3β is complex, as other data from the laboratory of Junichi Sadoshima showed that mice expressing a dominant negative GSK3β developed basal hypertrophy but had improved contractility, and less fibrosis or apoptosis after TAC exposure [35]. Thus, SIL-induced inactivation of GSK3β with modest hypertrophy (early stages of mTAC) may benefit the heart by assisting systolic function (i.e. rise in dP/dtmax/IP even at this time).
The results of this study should help clarify studies examining cardiac remodeling effects of PDE5A inhibition in various models, and may also have clinical implications. In the United States, The National Institutes of Heath has recently initiated a multicenter trial of sildenafil for the treatment of heart failure with a preserved ejection fraction, also termed diastolic heart failure (RELAX; http://clinicaltrials.gov/ct2/show/NCT00763867). These patients commonly display hypertension and substantial ventricular hypertrophy [36,37], but individual responsiveness to therapy may display variability due in part to the magnitude of cGK-1 targeted signaling cascades involved with a given patient’s disease. Another potential implication relates to experimental tests in models in which cGK-1 targets are either not sufficiently activated or not involved with the disease process. While evidence of anti-hypertrophic efficacy has been generated in models of increased hormone stimulation (isoproterenol, angiotensin II) and TAC, other models particularly generated by selective gene mutations may not stimulate these cascades and thus be unresponsive to PDE5A manipulation.
Several study limitations should be considered. First, while we continue to suspect that the major pathway responsible for the effects described following PDE5A inhibition is related to cGK-1 activation, it remains unproven in vivo. We have reported evidence in vitro using both pharmacologic and targeted PDE5A gene silencing that anti-hypertrophic effects in cardiac myocytes are cGK-1 dependent. However, pharmacologic inhibitors used in cell studies are not applicable in intact animals, and myocyte targeted gene deletion or loss-of-function mutation models have not yet been reported. These are under development. The study is also limited to studying one dose of SIL, albeit one achieving a therapeutic plasma level, and one model of hypertrophy. Whether the same findings would apply to low versus higher levels of alternative stresses such as adrenergic or angiotensin II stimulation is unknown.
In summary, we provide new and important insights into the dependence of SIL efficacy on the magnitude and/or duration of pressure-overload stress. Rather than being more effective with less stress, PDE5A inhibition effects strengthen as the stress and critical signaling are more activated. Thus, while a model of the cGMP/cGK-1 system as a car brake may still be apt, it is a targeted one that perhaps works best at higher speeds.
Figure 4.
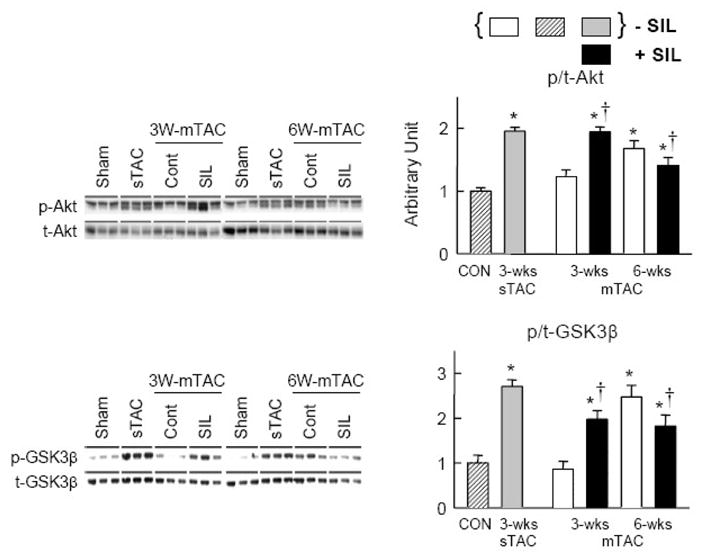
Influence of mTAC±SIL on phosphorylation of Akt and GSK3β varies depending upon the time course of moderate hypertrophic stimulation (N=3). At 3-wks, phosphorylation was little altered from sham controls in vehicle treated hearts, but was significantly enhanced by SIL. However, at 6-wks, with more sustained overload and hypertrophic response, phosphorylation of both kinases was increased over control, and now SIL depressed this response. * p<0.05 versus sham control. † p<0.05 versus vehicle treated at corresponding time point. Basal (non-SIL treated) phosphorylation levels for 3-wks sTAC are shown for comparison. As previously reported, SIL depresses this activation [8].
Acknowledgments
Sources of Funding: This study was supported by National Institute of Health Grants: HL-07227; HL-089297, HL-077180; HL-084986; and HL-59408; The Abraham and Virginia Weiss Professorship, Belfer Research Laboratory (DAK), a grant from Daiichi-Sankyo Inc. (TN), and (DAK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rosamond W, Flegal K, Friday G, Furie K, Go A, Greenlund K, Haase N, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O’Donnell CJ, Roger V, Rumsfeld J, Sorlie P, Steinberger J, Thom T, Wasserthiel-Smoller S, Hong Y. Heart disease and stroke statistics--2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:e69–171. doi: 10.1161/CIRCULATIONAHA.106.179918. [DOI] [PubMed] [Google Scholar]
- 2.Mudd JO, Kass DA. Tackling heart failure in the twenty-first century. Nature. 2008;451:919–928. doi: 10.1038/nature06798. [DOI] [PubMed] [Google Scholar]
- 3.McKinsey TA, Kass DA. Small-molecule therapies for cardiac hypertrophy: moving beneath the cell surface. Nature Reviews Drug Discovery. 2007;6:1–18. doi: 10.1038/nrd2193. [DOI] [PubMed] [Google Scholar]
- 4.Feil R, Lohmann SM, de Jonge H, Walter U, Hofmann F. Cyclic GMP-dependent protein kinases and the cardiovascular system: insights from genetically modified mice. Circ Res. 2003;93:907–916. doi: 10.1161/01.RES.0000100390.68771.CC. [DOI] [PubMed] [Google Scholar]
- 5.Kass DA, Takimoto E, Nagayama T, Champion HC. Phosphodiesterase regulation of nitric oxide signaling. Cardiovasc Res. 2007;75:303–314. doi: 10.1016/j.cardiores.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 6.Ghofrani HA, Osterloh IH, Grimminger F. Sildenafil: from angina to erectile dysfunction to pulmonary hypertension and beyond. Nat Rev Drug Discov. 2006;5:689–702. doi: 10.1038/nrd2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kass DA, Champion HC, Beavo JA. Phosphodiesterase type 5: expanding roles in cardiovascular regulation. Circ Res. 2007;101:1084–1095. doi: 10.1161/CIRCRESAHA.107.162511. [DOI] [PubMed] [Google Scholar]
- 8.Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214–222. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- 9.Nagayama T, Zhang M, Hsu S, Takimoto E, Kass DA. Sustained soluble guanylate cyclase stimulation offsets nitric-oxide synthase inhibition to restore acute cardiac modulation by sildenafil. J Pharmacol Exp Ther. 2008;326:380–387. doi: 10.1124/jpet.108.137422. [DOI] [PubMed] [Google Scholar]
- 10.Takimoto E, Champion HC, Belardi D, Moslehi J, Mongillo M, Mergia E, Montrose DC, Isoda T, Aufiero K, Zaccolo M, Dostmann WR, Smith CJ, Kass DA. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ Res. 2005;96:100–109. doi: 10.1161/01.RES.0000152262.22968.72. [DOI] [PubMed] [Google Scholar]
- 11.Takimoto E, Belardi D, Tocchetti CG, Vahebi S, Cormaci G, Ketner EA, Moens AL, Champion HC, Kass DA. Compartmentalization of cardiac beta-adrenergic inotropy modulation by phosphodiesterase type 5. Circulation. 2007;115:2159–2167. doi: 10.1161/CIRCULATIONAHA.106.643536. [DOI] [PubMed] [Google Scholar]
- 12.Salloum FN, Abbate A, Das A, Houser JE, Mudrick CA, Qureshi I, Hoke NN, Roy SK, Brown WR, Prabhakar S, Kukreja RC. Sildenafil (Viagra) Attenuates IschemicCardiomyopathy and Improves Left VentricularFunction in Mice. Am J Physiol Heart Circ Physiol. 2008 doi: 10.1152/ajpheart.91438.2007. [DOI] [PubMed] [Google Scholar]
- 13.Das A, Xi L, Kukreja RC. Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J Biol Chem. 2005;280:12944–12955. doi: 10.1074/jbc.M404706200. [DOI] [PubMed] [Google Scholar]
- 14.Zhang M, Koitabashi N, Nagayama T, Rambaran R, Feng N, Takimoto E, Koenke T, O’Rourke B, Champion HC, Crow MT, Kass DA. Expression, activity, and pro-hypertrophic effects of PDE5A in cardiac myocytes. Cell Signal. 2008 doi: 10.1016/j.cellsig.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borlaug BA, Melenovsky V, Marhin T, Fitzgerald P, Kass DA. Sildenafil inhibits beta-adrenergic-stimulated cardiac contractility in humans. Circulation. 2005;112:2642–2649. doi: 10.1161/CIRCULATIONAHA.105.540500. [DOI] [PubMed] [Google Scholar]
- 16.Nagayama T, Hsu S, Zhang M, Koitabashi N, Bedja D, Gabrielson K, Takimoto E, Kass DA. Sildenafil stops progressive chamber, cellular, and molecular remodeling and improves calcium handling and function in hearts with pre-existing advanced hypertrophy due to pressure-overload. J Am Coll Cardiol. 2009 doi: 10.1016/j.jacc.2008.08.069. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sayed N, Kim DD, Fioramonti X, Iwahashi T, Duran WN, Beuve A. Nitroglycerin-induced S-nitrosylation and desensitization of soluble guanylyl cyclase contribute to nitrate tolerance. Circ Res. 2008;103:606–614. doi: 10.1161/CIRCRESAHA.108.175133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim D, Rybalkin SD, Pi X, Wang Y, Zhang C, Munzel T, Beavo JA, Berk BC, Yan C. Upregulation of phosphodiesterase 1A1 expression is associated with the development of nitrate tolerance. Circulation. 2001;104:2338–2343. doi: 10.1161/hc4401.098432. [DOI] [PubMed] [Google Scholar]
- 19.Liu CQ, Leung FP, Lee VW, Lau CW, Yao X, Lu L, Huang Y. Prevention of nitroglycerin tolerance in vitro by T0156, a selective phosphodiesterase type 5 inhibitor. Eur J Pharmacol. 2008;590:250–254. doi: 10.1016/j.ejphar.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 20.MacPherson JD, Gillespie TD, Dunkerley HA, Maurice DH, Bennett BM. Inhibition of phosphodiesterase 5 selectively reverses nitrate tolerance in the venous circulation. J Pharmacol Exp Ther. 2006;317:188–195. doi: 10.1124/jpet.105.094763. [DOI] [PubMed] [Google Scholar]
- 21.Fischmeister R, Castro LR, Abi-Gerges A, Rochais F, Jurevicius J, Leroy J, Vandecasteele G. Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res. 2006;99:816–828. doi: 10.1161/01.RES.0000246118.98832.04. [DOI] [PubMed] [Google Scholar]
- 22.Castro LR, Verde I, Cooper DM, Fischmeister R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation. 2006;113:2221–2228. doi: 10.1161/CIRCULATIONAHA.105.599241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holtwick R, van Eickels M, Skryabin BV, Baba HA, Bubikat A, Begrow F, Schneider MD, Garbers DL, Kuhn M. Pressure-independent cardiac hypertrophy in mice with cardiomyocyte-restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase-A. J Clin Invest. 2003;111:1399–1407. doi: 10.1172/JCI17061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zahabi A, Picard S, Fortin N, Reudelhuber TL, Deschepper CF. Expression of constitutively active guanylate cyclase in cardiomyocytes inhibits the hypertrophic effects of isoproterenol and aortic constriction on mouse hearts. J Biol Chem. 2003;278:47694–47699. doi: 10.1074/jbc.M309661200. [DOI] [PubMed] [Google Scholar]
- 25.Fiedler B, Lohmann SM, Smolenski A, Linnemuller S, Pieske B, Schroder F, Molkentin JD, Drexler H, Wollert KC. Inhibition of calcineurin-NFAT hypertrophy signaling by cGMP-dependent protein kinase type I in cardiac myocytes. Proc Natl Acad Sci U S A. 2002;99:11363–11368. doi: 10.1073/pnas.162100799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takimoto E, Koitabashi N, Hsu S, Ketner EA, Zhang M, Nagayama T, Bedja D, Gabrielson KL, Blanton R, Siderovski DP, Mendelsohn ME, Kass DA. RGS2 mediates cardiac compensation to pressure-overload and anti-hypertrophic effects of PDE5 inhibition. J Clin Invest. 2009;53(2) doi: 10.1172/JCI35620. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang KM, Wang GR, Lu P, Karas RH, Aronovitz M, Heximer SP, Kaltenbronn KM, Blumer KJ, Siderovski DP, Zhu Y, Mendelsohn ME. Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat Med. 2003;9:1506–1512. doi: 10.1038/nm958. [DOI] [PubMed] [Google Scholar]
- 28.Hao J, Michalek C, Zhang W, Zhu M, Xu X, Mende U. Regulation of cardiomyocyte signaling by RGS proteins: differential selectivity towards G proteins and susceptibility to regulation. J Mol Cell Cardiol. 2006;41:51–61. doi: 10.1016/j.yjmcc.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 29.Takimoto E, Koitabashi N, Hsu S, Ketner EA, Nagayama T, Bedja D, Gabrielson K, Blanton R, Siderovski DP, Mendelsohn ME, Kass DA. RGS2 mediates cardiac compensation to pressure-overload and anti-hypertrophic effects of PDE5 inhibition. J Clin Invest. 2008 doi: 10.1172/JCI35620. (revision under review) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mulhearn TJ, El-Haddad H, Mahmud M, Gobska M, Haile A, Bridges M, Bedja D, Gabrielson K, Hemmnes AR, Kass DA, Champion HC. PDE5A inhibition suppresses maladaptive but not physiological cardiac hypertrophy. Circulation. 2007;114(Suppl II):II–5. [Google Scholar]
- 31.Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM, Richardson JA, Hill JA, Olson EN. Activated glycogen synthase-3 beta suppresses cardiac hypertrophy in vivo. Proc Natl Acad Sci U S A. 2002;99:907–912. doi: 10.1073/pnas.231619298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta 20 mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Das A, Xi L, Kukreja RC. Protein kinase G dependent cardioprotective mechanism of phosphodiesterase-5 inhibition involves phosphorylation of ERK and GSK3beta. J Biol Chem. 2008 doi: 10.1074/jbc.M801547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J. Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res. 2003;92:609–616. doi: 10.1161/01.RES.0000065442.64694.9F. [DOI] [PubMed] [Google Scholar]
- 35.Hirotani S, Zhai P, Tomita H, Galeotti J, Marquez JP, Gao S, Hong C, Yatani A, Avila J, Sadoshima J. Inhibition of glycogen synthase kinase 3beta during heart failure is protective. Circ Res. 2007;101:1164–1174. doi: 10.1161/CIRCRESAHA.107.160614. [DOI] [PubMed] [Google Scholar]
- 36.Lam CS, Roger VL, Rodeheffer RJ, Bursi F, Borlaug BA, Ommen SR, Kass DA, Redfield MM. Cardiac structure and ventricular-vascular function in persons with heart failure and preserved ejection fraction from Olmsted County, Minnesota. Circulation. 2007;115:1982–1990. doi: 10.1161/CIRCULATIONAHA.106.659763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melenovsky V, Borlaug BA, Rosen B, Hay I, Ferruci L, Morell CH, Lakatta EG, Najjar SS, Kass DA. Cardiovascular features of heart failure with preserved ejection fraction versus nonfailing hypertensive left ventricular hypertrophy in the urban Baltimore community: the role of atrial remodeling/dysfunction. J Am Coll Cardiol. 2007;49:198–207. doi: 10.1016/j.jacc.2006.08.050. [DOI] [PubMed] [Google Scholar]