

Abstract
Background
Cardiac resynchronization therapy (CRT) is widely applied in patients with heart failure and dyssynchronous contraction (DHF), but the electrophysiological consequences of CRT in heart failure remain largely unexplored.
Methods and Results
Adult dogs underwent left bundle-branch ablation and either right atrial pacing (190 to 200 bpm) for 6 weeks (DHF) or 3 weeks of right atrial pacing followed by 3 weeks of resynchronization by biventricular pacing at the same pacing rate (CRT). Isolated left ventricular anterior and lateral myocytes from nonfailing (control), DHF, and CRT dogs were studied with the whole-cell patch clamp. Quantitative polymerase chain reaction and Western blots were performed to measure steady state mRNA and protein levels. DHF significantly reduced the inward rectifier K+ current (IK1), delayed rectifier K+ current (IK), and transient outward K+ current (Ito) in both anterior and lateral cells. CRT partially restored the DHF-induced reduction of IK1 and IK but not Ito, consistent with trends in the changes in steady state K+ channel mRNA and protein levels. DHF reduced the peak inward Ca2+ current (ICa) density and slowed ICa decay in lateral compared with anterior cells, whereas CRT restored peak ICa amplitude but did not hasten decay in lateral cells. Calcium transient amplitudes were depressed and the decay was slowed in DHF, especially in lateral myocytes. CRT hastened the decay in both regions and increased the calcium transient amplitude in lateral but not anterior cells. No difference was found in CaV1.2 (α1C) mRNA or protein expression, but reduced CaVβ2 mRNA was found in DHF cells. DHF reduced phospholamban, ryanodine receptor, and sarcoplasmic reticulum Ca2+ ATPase and increased Na+-Ca2+ exchanger mRNA and protein. CRT did not restore the DHF-induced molecular remodeling, except for sarcoplasmic reticulum Ca2+ ATPase. Action potential durations were significantly prolonged in DHF, especially in lateral cells, and CRT abbreviated action potential duration in lateral but not anterior cells. Early afterdepolarizations were more frequent in DHF than in control cells and were reduced with CRT.
Conclusions
CRT partially restores DHF-induced ion channel remodeling and abnormal Ca2+ homeostasis and attenuates the regional heterogeneity of action potential duration. The electrophysiological changes induced by CRT may suppress ventricular arrhythmias, contribute to the survival benefit of this therapy, and improve the mechanical performance of the heart.
Keywords: ion channels, remodeling, heart failure, resynchronization, electrophysiology
Nearly 5 million Americans have heart failure (HF), and >250 000 die annually. Up to 50% of deaths in patients with HF are sudden and unexpected, mainly because of lethal ventricular arrhythmias such as ventricular tachycardia and ventricular fibrillation1; however, the mechanisms of ventricular tachycardia and ventricular fibrillation in patients with HF remain controversial.1,2
As HF progresses, the heart adapts to intrinsic and extrinsic stresses through a complex process of chamber remodeling and molecular modifications of myocyte structure and function. The structural remodeling and alteration of cell-to-cell coupling are associated with heterogeneous conduction delays in the ventricular myocardium that can lead to dyssynchronous ventricular contraction.3 Dyssynchronous contraction adversely influences the electrical phenotype of the failing heart and has been associated with regional changes in calcium (Ca2+) handling protein expression4 and exaggerated heterogeneities in conduction5 and repolarization,6-8 which enhances the susceptibility to ventricular arrhythmias in HF.9 Predictably, advanced HF with dyssynchronous cardiac contraction (DHF) is associated with a poor prognosis.10 Cardiac resynchronization therapy (CRT) with biventricular pacing improves symptoms,11 cardiac function, and mortality,12,13 presumably due to a reduction in stress-strain disparities and improvement in the efficiency of ventricular contraction. We have recently demonstrated that CRT reverses regional molecular remodeling and reduces apoptosis.14
Ion channel remodeling in HF underlies many of the changes in cellular electrophysiology and predisposes to atrial and ventricular arrhythmias in animal models and patients with HF.1,2,15-20 A consistent electrophysiological consequence of HF is a prolongation of ventricular repolarization, which is due in part to functional downregulation of outward potassium currents.2,15-17,19-21 However, little information is available about regional differences in ion channel function and expression or action potential (AP) profiles between early- and late-activated regions of dyssynchronous contracting left ventricles (LVs). Furthermore, the extent to which CRT reverses the DHF-induced electrophysiological remodeling remains unexplored.
Here, we tested the hypothesis that dyssynchronous mechanical contraction in HF contributes significantly to pathological remodeling at the cellular and molecular levels. We studied whether and to what extent remodeling could be reversed by biventricular pacing. We characterized the regional differences of AP, ionic currents, and [Ca2+]i transients (CaT) in myocytes isolated from the anterior and lateral LV myocardium of dogs with DHF and CRT and correlated the cellular electrophysiological changes with their subunit mRNA and protein expression in corresponding regions of the LV. These findings suggest distinct mechanisms of altered electrophysiological remodeling in DHF, as well as reverse remodeling by CRT. These data provide new mechanistic understanding of the therapeutic role of CRT and will help to identify new antiarrhythmic targets to prevent sudden death in patients with HF.
Methods
An expanded Methods section is available in the online-only Data Supplement.
Canine Tachypacing-Induced HF Model
All protocols followed US Department of Agriculture and National Institutes of Health guidelines and were approved by the Animal Care and Use Committee of the Johns Hopkins Medical Institutions. The canine models of DHF or CRT have been described previously.5,14,22 In brief, adult male mongrel dogs underwent left bundle-branch radiofrequency ablation and right atrial pacing (190 to 200 bpm) for 6 weeks (DHF dogs; n=7) or 3 weeks of right atrial pacing followed by 3 weeks of resynchronization by biventricular pacing at the same pacing rate (CRT dogs; n=7).
ECG, Echocardiography, and Hemodynamic Recordings
To monitor LV function during tachypacing, 2-dimensional echocardiography with tissue Doppler imaging was performed periodically together with recordings of ECGs. Briefly, standard 2-dimensional and color Doppler image data, triggered to the QRS complex, were saved in cine-loop format. LV volumes (end-systolic and end-diastolic) and LV ejection fraction were calculated from the conventional apical 2- and 4-chamber views. LV dyssynchrony was quantified with speckle-tracking radial strain analysis (Figure 1) as described previously.23 The LV dyssynchrony index was defined as the SD of time to peak systolic velocity of 12 segments. Standard 12-lead ECG and hemodynamic data (LV systolic and end-diastolic pressure, dP/dtmax) were recorded 6 weeks after the start of pacing.
Figure 1.
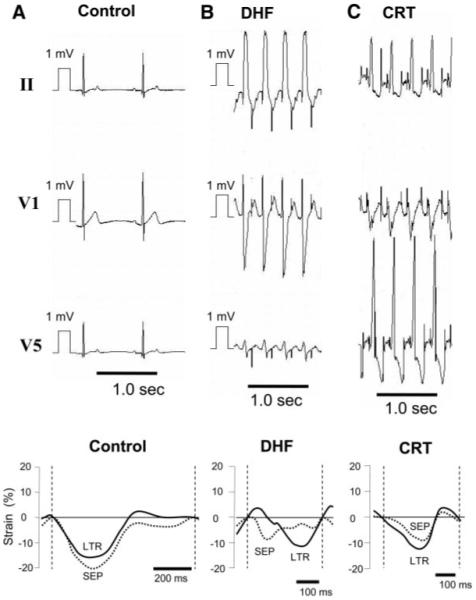
Representative ECGs and changes of LV septal (SEP) and lateral (LTR) strain from control (A), 6-week paced DHF (B), and CRT (C) dogs. ECGs from DHF and CRT dogs are shown during pacing, and control is shown during sinus rhythm. Biventricular pacing synchronized the strain patterns between LV septal and lateral walls and abbreviated the DHF-induced prolongation of QRS duration.
Patch Clamp and CaT
All electrophysiological and intracellular Ca2+ measurements were performed at physiological temperature (37°C). All methods for cell isolation, electrophysiological recording, and measurements are routine in our laboratory and have been described previously.4,8,16,17,20 Isolated ventricular myocytes from the anterior and lateral mid myocardium were current and voltage clamped by the standard whole-cell patch-clamp technique for measurement of APs, transient outward K+ current (Ito), inward rectifier K+ current (IK1), delayed rectifier K+ currents (IK), and Ca2+ current (ICa). Borosilicate glass electrodes with tip resistances of ≈3.0 MΩ when filled with the pipette solution were used.
CaTs were measured by indo-1 fluorescence excited at 365 nm with a xenon arc lamp, and emitted light at 405 and 495 nm was collected with a 2-channel photomultiplier tube assembly. Fluorescence signals were digitized and stored with electrophysiological recordings for offline analysis with custom software.24 The ratio of indo-1 fluorescence (R=F405 nm/F495 nm) was determined after subtraction of cellular autofluorescence. The rate of Ca2+ removal (τCa) was determined by fitting a single exponential to the Ca2+ time course.
Molecular Analysis
Canine Kir2.1, Kv4.3, KChIP2, Kv11.1 (ether a-go-go related gene [ERG]), Kv7.1 (KvLQT1), minK, Cav1.2 (α1C), Cavβ1, Cavβ2, sarcoplasmic reticulum Ca2+ATPase (SERCA2), phospholamban, ryanodine receptor, and Na+-Ca2+ exchanger steady state mRNA levels were measured by reverse-transcription polymerase chain reaction in tissue isolated from the LV anterior and lateral walls in control, DHF, and CRT dogs (online-only Data Supplement Table I). Kir2.1, Kv4.3, KChIP2, ERG, KvLQT1, Cav1.2 (α1C), SERCA2, phospholamban, ryanodine receptor, and Na+-Ca2+ exchanger proteins were measured by Western immunoblotting. Detailed methods are provided in the online-only Data Supplement.
Statistical Analysis
Differences among multiple groups were compared by ANOVA with Bonferroni test. Two-group analysis was performed by t test (paired or unpaired as appropriate). Differences in serial studies were assessed by repeated-measures ANOVA. Data are expressed as mean±SD or mean±SEM as indicated in each of the Figures. A value of P<0.05 was considered significant.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
ECG and Hemodynamic Changes
Figure 1 shows representative standard ECG and LV wall strain plots by tissue Doppler from a control (A), 6-week paced DHF (B), and CRT (C) dog. In the DHF dog, the QRS interval was wider, with a left bundle-branch block pattern compared with the control, and the tissue Doppler image showed dyssynchronous contraction between the septum and the lateral walls. CRT shortened the QRS duration and resynchronized the LV wall strain pattern so that it was similar to the control.
The Table summarizes the ECG, echocardiographic, and hemodynamic changes in control, 6-week paced DHF, and CRT dogs. In sinus rhythm, the R-R interval was shorter and the corrected QT and QRS durations were longer in both DHF and CRT dogs than in control dogs, but no significant difference was observed in these parameters between DHF and CRT dogs. However, during pacing, QRS width was significantly shorter in CRT than DHF dogs, which was consistent with the larger dyssynchrony index by tissue Doppler echocardiography in DHF compared with control or CRT dogs. Moreover, CRT significantly decreased the corrected QT interval compared with DHF. Conversely, LV diastolic and systolic volumes, as well as hemodynamic parameters such as LV end-diastolic and end-systolic pressure and dP/dtmax, were not statistically different between DHF and CRT. A trend was observed toward increased stroke volume and ejection fraction in CRT versus DHF dogs.
Table. ECG, Echocardiography, and Hemodynamic Parameters.
| Control | DHF | CRT | |
|---|---|---|---|
| ECG, ms | |||
| Sinus rhythm | |||
| RR | 608±115 | 438±34* | 428±34* |
| QT | 278±31 | 286±43 | 300±15* |
| QTc | 359±26 | 434±64* | 467±38* |
| QRS | 47±6 | 103±21* | 102±10* |
| Pacing | |||
| RR | . . . | 314±14 | 308±18 |
| QT | . . . | 280±23 | 239±39 |
| QTc | . . . | 489±35 | 429±58† |
| QRS | . . . | 112±16 | 64±6† |
| Echocardiography | |||
| DI, ms | 30±5 | 74±25* | 29±23† |
| LVEDV, mL | 52±6 | 87±18* | 93±20* |
| LVESV, mL | 17±4 | 64±17* | 63±15* |
| SV, % | 35±6 | 23±12* | 29±15 |
| LVEF, % | 67±7 | 25±12* | 30±11* |
| Hemodynamics | |||
| HR, bpm | 105±26 | 151±42* | 150±12* |
| LVEDP, mm Hg | 6±5 | 35±8* | 34±10* |
| LVESP, mm Hg | 142±27 | 103±19* | 101±10* |
| dP/dtmax mm Hg/s | 1900±467 | 831±246* | 846±133* |
QTc indicates corrected QT; DI, dyssynchrony index; LVEDV, LV end-diastolic volume; LVESV, LV end-systolic volume; SV, stroke volume; LVEF, LV ejection fraction; HR, heart rate; LVEDP, LV end-diastolic pressure; and LVESP, LV end-systolic pressure.
Values are mean±SD.
P<0.05 vs control
P<0.05 vs DHF.
Cell Capacitance
Cell capacitance was larger in DHF and CRT than in control dogs in cells isolated from both the anterior and lateral walls, but no regional difference was found between anterior and lateral cells. The capacitance of cells isolated from CRT dogs was not different from that of cells isolated from DHF dogs (online-only Data Supplement Table II).
Inward Rectifier K+ Current
Figure 2A shows whole-cell IK1 currents in lateral myocytes from control, DHF, and CRT dogs. The densities of IK1 from control and DHF myocytes were similar to data previously reported from our laboratory.16 IK1 density in DHF myocytes was reduced significantly compared with control, whereas CRT partially reversed the DHF-induced reduction of IK1 over a wide voltage range (Figure 2B). On the other hand, no regional difference was found in IK1 between anterior and lateral myocytes isolated from either DHF or CRT hearts (Figure 2C). The relevant component of IK1 for AP repolarization is the outward current “hump” observed at potentials positive to the K+ reversal potential. The peak outward component of IK1 at -70 and -60 mV was decreased modestly but significantly (P<0.05) in DHF myocytes (1.43±0.52 and 1.07±0.43 pA/pF, respectively) compared with control (1.78±0.54 and 1.26±0.51 pA/pF, respectively) and CRT (1.81±0.81 and 1.18±0.57 pA/pF, respectively). To investigate the molecular basis for changes in IK1, we measured Kir2.1 mRNA and protein levels; these were significantly downregulated in DHF compared with control (P<0.05), whereas those in CRT were decreased (although not statistically significant) compared with control (Figures 2D and 2E). No regional differences in Kir2.1 mRNA and protein expression were found among the 3 groups, which is consistent with the cellular electrophysiology.
Figure 2.

IK1 and Kir2.1 mRNA and protein levels in control, DHF, and CRT. A, Representative current traces in lateral myocytes isolated from control, DHF, and CRT canine ventricles elicited by the diagrammed voltage-clamp protocol (holding potential -40 mV, test pulse 500 ms in duration). B, Steady state current-voltage (I-V) relationship of IK1 and outward current portion of the I-V (right) in each group. The voltage-clamp protocol is shown in the inset. C, IK1 density at -140 and -70 mV in anterior (ANT) and lateral (LTR) myocytes of each group. D and E, Kir2.1 mRNA and protein expression in anterior and lateral myocytes of each group. A or ANT indicates anterior; L or LTR, lateral; and CSQ, calsequestrin. †P<0.05 vs control; #P<0.05 vs DHF. The values in parentheses are the number of cells or tissue samples studied in this and all remaining figures.
Transient Outward K+ Current
Figure 3A shows whole-cell Ito currents in lateral myocytes from control, DHF, and CRT dogs. The densities of Ito from control and DHF myocytes in this preparation are similar to those reported previously in our laboratory.16 Ito in DHF myocytes was reduced significantly compared with control (P<0.05), but CRT did not restore the DHF-induced reduction of Ito (Figure 3B). Interestingly, Ito current density was not different between anterior and lateral myocytes in any group (Figure 3C). To investigate the molecular basis for change in Ito, we measured the α-subunit (Kv4.3) and β-subunit (KChIP2) underlying this current (Figures 3D through 3G). Consistent with the Ito density, both Kv4.3 and KChIP2 mRNA and protein were significantly downregulated in both DHF and CRT, with no regional differences in each group.
Figure 3.

Ito, and its underlying subunit mRNA and protein expression in control, DHF, and CRT. A, Representative current traces in lateral myocytes from control, DHF, and CRT ventricles elicited by the voltage-clamp protocol shown in inset. B, Peak current-voltage relationship of Ito in each group. C, Ito density at +40 mV in anterior and lateral myocytes of each group. D through G, Kv4.3 and KChIP2 mRNA and protein expression in LV midmyocardial tissue isolated from control, DHF, and CRT dogs. A or ANT indicates anterior; L or LTR, lateral; and CSQ, calsequestrin. †P<0.05 vs control.
Delayed Rectifier K+ Currents
Figure 4A shows total IK in lateral myocytes from control, DHF, and CRT dogs. As shown in the superimposed currents elicited by a step pulse to +50 mV, the peak and tail IK currents were smaller in DHF than in control myocytes, and CRT partially restored the DHF-induced reduction of IK. Figure 4B displays the current-voltage relationship of the tail current density of IK in the 3 groups, showing that DHF reduced IK density by ≈50% compared with control, whereas CRT partially restored the DHF-induced reduction of IK (P<0.05). In addition, no regional difference was found in the reduction and restoration of IK by DHF and CRT (Figure 4C).
Figure 4.

IK and its underlying subunit mRNA and protein expression in control, DHF, and CRT hearts. A, Representative current traces from control, DHF, and CRT myocytes elicited by voltage-clamp protocol shown in the inset. B, Current-voltage relationship of the tail current of IK (IK tail) fitted to the Boltzmann equation: IK,tail=1/{1+exp[(V1/2-Vm)/k]}. Voltage-clamp protocol is shown in the inset. C, IK tail density at 40 mV between anterior and lateral myocytes in each group. D through H, KvLQT1, minK, and ERG mRNA and protein expression in mid myocardium of control, DHF, and CRT dogs. A or ANT indicates anterior; L or LTR, lateral; and CSQ, calsequestrin. †P<0.05 vs control; #P<0.05 vs DHF.
To investigate the molecular basis for changes in IK, we measured the expression of the underlying α-subunit (Kv7.1 [KvLQT1] for IKs and Kv11.1 [ERG] for IKr) and β-subunit (minK for IKs) mRNA and proteins. KvLQT1 mRNA and protein were reduced significantly in DHF compared with control myocytes; CRT did not alter KvLQT1 mRNA expression but increased the protein level such that it was not significantly different from control (Figure 4D and 4E). No significant differences were found in minK mRNA expression among the 3 groups (Figure 4F). ERG mRNA expression was reduced in DHF and completely restored by CRT (Figure 4G), but no significant difference was found in ERG protein expression in any of the groups (Figure 4H). In addition, consistent with the physiological data, no regional differences in gene or protein expression were found between anterior and lateral LV tissues from each group.
Inward Ca2+ Current (ICa) and CaT
Figure 5A shows ICa in LV midmyocardial myocytes isolated from control, DHF, and CRT hearts. In control, no difference was found in the peak ICa density or current decay between anterior and lateral myocytes; however, DHF reduced the peak ICa density and slowed the rate of current decay in lateral cells compared with anterior cells. Furthermore, in CRT, no regional difference was found in peak ICa density, but the rate of current decay was still slower in lateral than in anterior cells. The current-voltage relationships (Figure 5B) showed that peak ICa density was significantly less in lateral cells than in anterior cells in DHF at voltage steps from -15 to +20 mV (P<0.05). In contrast, CRT increased ICa density in lateral cells but not in anterior cells.
Figure 5.

ICa and CaT in control, DHF, and CRT dogs. A, Representative ICa recorded from anterior and lateral myocytes. B, Peak current-voltage relationships for ICa,L in control, DHF, and CRT myocytes. Voltage-clamp protocol is shown in the inset. C, Voltage dependence of ICa activation in control, DHF, and CRT myocytes was fit with a Boltzmann equation of the form G/Gmax=1/[1+exp(V1/2-V)/k]. The voltage at which GCa was half-maximal (V1/2) did not differ among the 3 groups and regions. D, Plot of the time constant of the fast (major) component of current decay measured at 0 mV. E, Plot of the charge carried by ICa during the first 200 ms of the voltage step. F, Representative superimposed CaTs recorded in anterior and lateral myocytes from control, DHF, and CRT dogs stimulated at 0.5 Hz as measured with indo-1 fluorescence. G and H, Average peak fluorescence values and average decay τ of CaT measured at 0.5-Hz stimulation frequency in anterior and lateral myocytes from control, DHF, and CRT dogs. ANT indicates anterior; LTR, lateral. †P<0.05 vs control; #P<0.05 vs DHF by ANOVA; *P<0.05 by t test, anterior vs lateral.
Although the voltage dependence of activation of ICa (Figure 5C) was not altered significantly in either DHF or CRT compared with control in either anterior or lateral cells, the rate of current decay (Figure 5D) in DHF was significantly slower (41.7±9.5 versus 33.9±5.4 ms, P<0.05) in lateral cells and significantly faster (25.6±4.0 versus 32.1±5.3 ms, P<0.05) in anterior cells than in control cells, and this produced a robust difference in current decay between anterior and lateral cells in DHF. Moreover, CRT slowed the DHF-induced faster current decay in anterior cells (32.8±7.4 ms, P<0.05) but did not affect the rate of ICa decay in lateral cells (40.4±9.3 ms). Therefore, the total charge carried by ICa (Figure 5E) was not altered significantly by DHF in either anterior or lateral cells. On the other hand, CRT significantly increased the charge carried by ICa in lateral (from 0.14±0.04 to 0.19±0.06 pC/pF, P<0.05) but not anterior cells compared with control.
CaTs (Figure 5F) were not different in anterior and lateral cells from control hearts; however, DHF significantly reduced the CaT amplitude and slowed the rate of decay of the CaT most prominently in lateral myocytes. CRT hastened the DHF-induced slowing of the decay of the CaT without changing the amplitude in anterior cells, whereas CaT amplitude was increased and decay hastened significantly in lateral cells. The changes in amplitude and rate of decay of CaT in each group are shown in Figure 5G and 5H, respectively. Notably, CRT almost fully reversed the DHF-induced smaller amplitude and longer decay of CaT.
Molecular Basis for Abnormal Ca2+ Homeostasis
The molecular basis for changes in ICa and CaT was investigated by determining the steady state levels of Cav1.2 (Cavα1C), Cavβ1, Cavβ2, ryanodine receptor, phospholamban, SERCA2, and Na+-Ca2+ exchanger mRNA and protein (Figure 6). No significant differences in Cav1.2 mRNA and protein or Cavβ1 subunit mRNA expression were found among control, DHF, and CRT hearts (Figure 6A through 6C). On the other hand, Cavβ2 mRNA was decreased significantly in DHF but not CRT myocytes compared with control (Figure 6D). Steady state ryanodine receptor and phospholamban mRNA and protein expression were consistently lower in both DHF and CRT myocytes than in control (Figure 6E through 6H), and SERCA2 mRNA and protein levels were reduced significantly in DHF myocytes but were not decreased significantly in CRT (Figure 6I and 6J). On the other hand, Na+-Ca2+ exchanger mRNA expression (Figure 6K) was increased significantly in CRT (P<0.05) compared with control, and protein levels were increased in both DHF and CRT (Figure 6L). However, no regional difference was found in the steady state levels of Ca2+ channel or homeostasis-related genes and proteins in each group.
Figure 6.

Ca2+ handling-related mRNA and protein expression in control, DHF, and CRT myocytes. A and B, Cav1.2 (α1c) mRNA and protein. C and D, Cavβ1 and Cavβ2 mRNA. E and F, Ryanodine receptor (RyR2) mRNA and protein. G and H, Phospholamban (PLN) mRNA and protein. I and J, SERCA2 mRNA and protein. K and L, Na+-Ca2+ exchanger (NCX) mRNA and protein. A or ANT indicates anterior; L or LTR, lateral; and CSQ, calsequestrin. †P<0.05 vs control.
APs and Early Afterdepolarizations
Figure 7A shows superimposed APs elicited at pacing cycle lengths of 0.5, 1.0, 2.0, and 4.0 seconds in anterior and lateral myocytes from control, DHF, and CRT dogs. Compared with control, DHF prolonged the action potential duration (APD) at all pacing cycle lengths, more prominently in lateral than in anterior cells. In contrast, CRT partially reversed the DHF-induced prolongation of APD in lateral cells but not in anterior cells. The relationship between APD and pacing cycle length of each group (Figure 7B) revealed that APD in both DHF and CRT cells was similarly prolonged in anterior cells, and the slope of the APD-cycle length relationship was increased, whereas in lateral cells, a prominent prolongation of APD in DHF was found, particularly at long pacing cycle lengths, which resulted in a significant difference in APD between anterior and lateral cells (P<0.05). By selectively shortening the APD of lateral myocytes, CRT reduced the regional heterogeneity in APD compared with DHF. The resting membrane potential (Figure 7C) was not significantly different between control, DHF, and CRT dogs, whereas the phase 1 notch depth (Figure 7D) was significantly attenuated in DHF and CRT dogs, consistent with no restoration of Ito by CRT.
Figure 7.

APD in LV myocytes from control, DHF, and CRT hearts. A, Representative superimposed APs recorded at pacing cycle lengths (CL) of 0.5, 1.0, 2.0, and 4.0 seconds, B, Relationship between pacing CL and APD at 90% recovery (APD90) from anterior and lateral myocytes in each group. C and D, Bar plot of resting membrane potential (RMP; C) and phase 1 notch amplitude (D) at pacing CL of 2.0 seconds. †P<0.05 vs control; #P<0.05 vs DHF; *P<0.05 vs anterior. ANT or A indicates anterior; LTR or L, lateral.
Early afterdepolarizations (EADs) were observed more frequently in DHF than in control myocytes (P<0.001), and CRT significantly reduced the frequency of EADs compared with DHF (P<0.05; Figure 8A); however, no regional differences were found in development of EADs between anterior and lateral cells in each group. Figure 8B shows representative APs with [EAD(+)] or without [EAD(-)] EADs in myocytes from DHF hearts. The APD at 90% recovery (APD90) was modestly but significantly longer in EAD(+) myocytes than in EAD(-) myocytes; however, the APD at 20% recovery (APD20) was markedly shorter in EAD(+) myocytes than in EAD(-) myocytes. Therefore, we examined the relationship between the APD20, APD90, and APD20/APD90 ratio and EAD development in the failing myocytes (Figure 8C; online-only Data Supplement Table III). APD20 was shorter and APD90 longer in EAD(+) cells than in EAD(-) cells, which resulted in a dramatically smaller APD20/APD90 ratio in EAD(+) myocytes.
Figure 8.
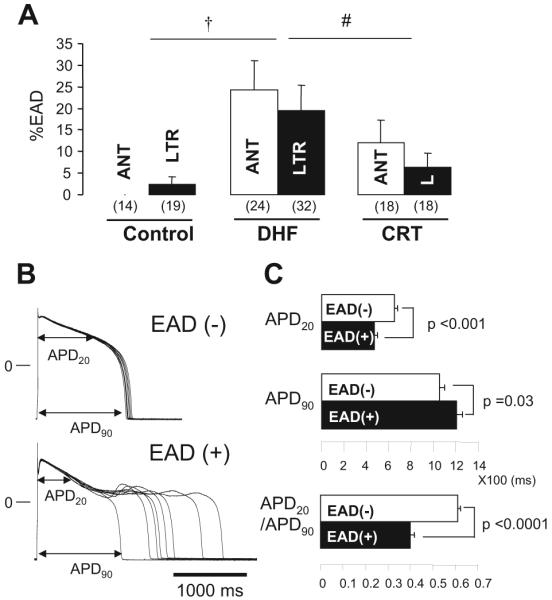
EADs in myocytes from control, DHF, and CRT hearts. A, Bar plot of frequency of EADs (%EADs indicates fraction of APs with EADs). B, Representative superimposed APs recorded in myocytes isolated from lateral wall of DHF hearts with [EAD(+)] or without [EAD(-)] EADs. C, Bar plots of APD20, APD90, and ratio of APD20 to APD90 (APD20/APD90) in failing myocytes. All data were obtained during pacing at 0.25 Hz. †P<0.05 vs control; #P<0.05 vs DHF. ANT or A indicates anterior; LTR or L, lateral.
Discussion
Our studies reveal the regional cellular electrophysiological consequences of synchronous and dyssynchronous ventricular contraction in the failing heart and include several novel insights into electrophysiological remodeling in HF. First, CRT partially reverses DHF-induced K+ channel remodeling (IK1 and IK)in both the anterior and lateral LV. An interesting divergence in the remodeling of K+ currents can be seen: CRT has no effect on DHF-induced downregulation of Ito or on the expression of Kv4.3 or KChIP2 mRNA or protein. Second, Ca2+ current remodeling and Ca2+ handling were significantly different in the anterior and lateral LV in DHF, and CRT significantly improved Ca2+ homeostasis, especially in the lateral wall. Third, the APD was significantly prolonged in DHF, especially in cells isolated from the lateral LV, and CRT abbreviated the APD in lateral cells and reduced the regional gradient of APD. Finally, EADs were more frequent in DHF, were significantly but not completely reduced to near control levels in CRT, and were associated with a modestly prolonged APD90 and a markedly reduced APD20/APD90 ratio. It is important to recognize that the model that we used corrects dyssynchronous contraction but does not affect tachycardia-induced LV dysfunction. Thus, the reversal of DHF-induced electrical and Ca2+ handling remodeling by CRT is due to the effect of electrical resynchronization by biventricular pacing.
K+ Channel Remodeling
Downregulation of K+ currents is the most consistent ionic current change in animal models1,16,20,21 and human HF.15 K+ current downregulation may promote ventricular tachycardia and ventricular fibrillation,21 either by direct prolongation of AP19 in the voltage range at which ICa,L reactivation occurs, which predisposes to the development of EADs,25 or by heterogeneous reduction of the repolarization reserve and the promotion of functional reentry. Although expressed cardiac K+ channels vary in different species, Ito downregulation is the most consistent ionic current change in failing mammalian hearts.15,16,20,21 In the present study, Ito and its related genes, Kv4.3 and KChIP2, were downregulated homogenously by DHF and were not affected by CRT. The data suggest that tachycardia, HF, or altered ventricular activation is more important in downregulation of Ito than mechanical synchrony. Reduced IK1 density in HF16,18,20 may contribute to prolongation of APD and enhanced susceptibility to spontaneous membrane depolarization.26 Although small but significant changes in IK1 density were observed in the outward current component, and the largest changes were observed at very negative voltages (Figure 2B and 2C), the resting membrane potential of myocytes did not differ in any of the groups (Figure 7C), because the major voltage range of altered IK1 was beyond physiological potentials. On the other hand, in tachypacing-induced HF models, IKs is downregulated, but IKr is less consistent.18,21 In the present study, both IK1 and IK densities were reduced in a regionally homogenous fashion by DHF in spite of the higher wall stress and myocyte stretch in the late-activated lateral wall than in the early-activated anterior wall27; this was partially but significantly restored by CRT without a change in global LV function, which suggests that K+ channel remodeling was not directly associated with the mechanical stress caused by dyssynchronous ventricular contraction.
The dichotomy in the regulation of Ito compared with IK1 and IK in CRT is remarkable. These currents share regulatory mechanisms that are altered in the failing heart and are differentially remodeled by CRT.14 The detailed regulation by the autonomic nervous system, renin-angiotensin-aldosterone signaling, and reactive oxygen species are distinct for each of the K+ currents studied and may explain the differences in response to CRT. In addition, biventricular tachypacing in the present study improved the synchrony of mechanical contraction, but global LV function as assessed by LV ejection fraction or end-diastolic pressure was still significantly depressed. It is likely that the molecular mechanisms for the altered functional expression of each current are mixed. The steady state levels of Kv4.3 and KChIP2 mRNA are consistently downregulated by tachypacing in the presence and absence of mechanical synchrony; thus, the balance between transcription and RNA degradation is altered. Furthermore, altered ventricular activation by CRT may suppress Ito expression independent of the presence of HF.28,29 Concordant changes in protein levels suggest the possibility of a pretranslational mechanism. However, the reduction in Kv4.3 and KChIP2 proteins in particular is not as pronounced as the magnitude of the current reduction, which suggests an additional posttranslational mechanism of functional downregulation of Ito. The changes in IK1 and Kir2.1 mRNA and protein in DHF and CRT appear to be more consistent with but not proof of a pretranslational mechanism of downregulation. Changes in KvLQT1 and ERG mRNA and protein are more variable and appear to be partially altered by CRT. There appear to be distinct differences in the mechanism of down-regulation of individual K+ currents in DHF and variable degrees and mechanisms of restoration of expression in CRT.
Altered ICa and Ca2+ Handling
Changes in ICa functional expression in HF are variable.30 Depending on the model and stage of HF, some studies showed a decrease in ICa density, whereas others reported no change.2,16,31 In the present study, the peak ICa density and current decay in DHF were regionally different between myocytes isolated from the lower-stress anterior and higher-stress lateral walls. Furthermore, CRT regionally restored the DHF-induced changes in ICa, increasing the peak ICa density in lateral cells and slowing the decay in anterior cells; CRT thus served to mitigate the DHF-induced regional heterogeneities in ICa density and gating.
HF causes significant changes in Ca2+-handling proteins.2,32 In failing human hearts, the steady state level of the Cav1.2(α1C) mRNA was reported to be decreased33 or unchanged.17 Moreover, Vanderheyden et al34 recently suggested the possibility of reverse molecular remodeling of SERCA2 in CRT responders. In the present study, Cav1.2(α1C) mRNA and protein were not different between control, DHF, and CRT dogs, consistent with our previous data in human myocardium.17
Cavβ-subunit expression has been positively correlated with peak Cav1.2 current density.35 The present data showed that Cavβ1 mRNA levels were unchanged in DHF and CRT, whereas steady state Cavβ2 mRNA levels were reduced in DHF and partially restored by CRT. In DHF, ryanodine receptor, phospholamban, and SERCA2a mRNA and protein levels were downregulated, whereas Na+-Ca2+ exchanger was upregulated, consistent with previous studies.4,31,36,37 No regional differences in mRNA and protein expression were found in any of these mediators of Ca2+ handling in DHF and CRT, which suggests that the regional differences of Ca2+ handling in DHF and its restoration by CRT are posttranslational.
The mechanisms underlying the differences in regional remodeling of K+ currents and Ca2+ handling in DHF remain obscure. Plotnikov et al7 reported that cardiac dyssynchrony by LV pacing (120 to 150 bpm for 3 weeks) produced a slower decay of ICa inactivation, consistent with the present results. Moreover, this phenomenon was suppressed by β-adrenergic blockade. These findings suggest that DHF-induced changes of ICa inactivation kinetics might be mediated by regionally heterogeneous β-adrenergic receptor stimulation. Furthermore, the Ca2+-handling proteins are functionally regulated by phosphorylation, prominently by the key intracellular enzymes protein kinase A and Ca2+-cal-modulin-dependent protein kinase II (CaMKII),38-40 as well as a variety of phosphatases that may be regionally regulated.14
Prolongation of APD and Development of EADs
The APD is consistently prolonged in human and animal models of HF.2,15-17,19-21 A recent study using a canine model of dyssynchrony suggests that after 4 weeks, the APD is prolonged in the late-activated compared with the early-activated regions.6 In the present work, CRT produced a partial but statistically significant shortening of DHF-induced prolongation of APD selectively in lateral cells. Although the peak ICa density in DHF was decreased in lateral cells compared with anterior cells, the decrease was modest and not significant compared with control lateral myocytes (Figure 5B). On the other hand, ICa decay was slowed significantly, which might contribute to regional AP prolongation in the setting of homogenous K+ current down-regulation, although Ca2+ flux through L-type channels during a square pulse protocol does not fully reflect that during an AP. These data provide some insight into the mechanism of regional AP remodeling in DHF and CRT.
Moreover, on a molecular level, we have shown that tumor necrosis factor-α and CaMKII were increased in DHF, prominently in the lateral wall, and these differences were absent in CRT.14 Tumor necrosis factor-α decreases Ito and prolongs APD in rat ventricular myocytes.41 CaMKII influences Ca2+ current and sarcoplasmic reticulum function39,40 and increases persistent Na+ current,42,43 which results in prolongation of APD.44 It is possible, and indeed likely, that other regional alterations in Ca2+ handling (Figure 5F) or an increased persistent Na+ current contribute to regional differences in the APD and AP profile in DHF and to the regionally specific effects of biventricular pacing on this phenotype. Furthermore, EADs were observed more frequently in DHF than in control myocytes, and CRT reduced the occurrence of EADs in both anterior and lateral cells.
Prolongation of APD90 was associated with an increased frequency of EADs, but notably, a smaller APD20/APD90 ratio was even more strongly correlated with the appearance of EADs (Figure 8C). Thus, APD prolongation alone may not be sufficient to generate EADs in failing myocytes.44 The APD20/APD90 ratio is an empiric metric but suggests that long APs with a reduced plateau voltage are the most likely to exhibit EADs. This type of AP profile that is generated by the reduction in K+ current density and reduced ICa density with slowed kinetics is highly susceptible to EADs that result from reactivation of ICa.
Study Limitations
The model of DHF and CRT used in the present study is a limitation. Six weeks of tachypacing (200 bpm) reproducibly induces dilated cardiomyopathy with LV enlargement, increased LV end-diastolic pressure, and decreased dP/dtmax; however, the study was designed to examine the effects of CRT with ongoing HF, and thus, tachypacing was maintained. This differs from CRT in patients, which is performed at lower heart rates. Therefore, the processes of both remodeling and the reversal of remodeling in this circumstance are likely to be different from those in human DHF.
The second limitation is that the present study focused on the cellular and molecular bases of electrophysiological remodeling in DHF and restoration by CRT. It did not evaluate the susceptibility to and frequent development of arrhythmia in vivo or in a whole-heart model.
Clinical Implications
CRT has emerged as an effective pacing/mechanical therapy for patients with HF and a prolonged QRS duration. CRT has been associated with improved cardiac function, symptomatology, and exercise capacity and, when combined with defibrillator therapy, reduced mortality.12,13 The role of CRT in preventing arrhythmias45,46 or reversing adverse electrical remodeling remains controversial. The present study provides novel information on the altered expression and function of ionic currents and CaTs in DHF and after CRT. Understanding the fundamental mechanisms of the altered ion channel function and Ca2+ homeostasis, as well as the electrophysiological remodeling in DHF, and the capacity for restoration by CRT will not only help define the therapeutic role of CRT but will help to identify antiarrhythmic targets that can be exploited by other therapeutic strategies designed to prevent sudden death.
Supplementary Material
Acknowledgments
The authors thank Yanli Tian, Deborah DiSilvestre, Victoria Halperin, and Richard S. Tunin for excellent technical support.
Sources of Funding
The work was supported by National Institutes of Health grants P01 HL 077180 (to Drs Kass and Tomaselli) and HL 072488 (to Dr Tomaselli) and a grant from Medtronic Japan (to Dr Aiba). Dr Tomaselli is the Michel Mirowski, MD, Professor in Cardiology.
Footnotes
Disclosures
Dr Tomaselli has received a research grant from Boston Scientific. The remaining authors report no conflicts.
CLINICAL PERSPECTIVE
Cardiac resynchronization therapy (CRT) with biventricular pacing improves symptoms, cardiac function, and exercise capacity, and when combined with defibrillator therapy, it reduces mortality in patients with heart failure who have dyssynchronous contraction (DHF). CRT reduces stress-strain disparities and thus improves the efficiency of contraction of the ventricle. However, the role of CRT in preventing arrhythmias or reversing adverse electrical remodeling remains controversial. The present study provides novel information on the altered regional expression and function of ionic currents and calcium (Ca2+) transients in DHF and partial restoration by CRT in a canine pacing-induced DHF model. CRT partially restores the DHF-induced reduction of selected K+ currents and significantly improves Ca2+ homeostasis, especially in the lateral wall of the left ventricle. The overall effect of CRT is abbreviation of the DHF-induced prolongation of action potential duration in cells isolated from the lateral left ventricle, thus reducing the regional action potential duration gradient and the frequency of potentially arrhythmogenic early afterdepolarizations compared with DHF. Thus, CRT partially reverses both the cellular triggers and substrate for arrhythmias in DHF.
References
- 1.Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res. 2004;95:754–763. doi: 10.1161/01.RES.0000145047.14691.db. [DOI] [PubMed] [Google Scholar]
- 2.Nattel S, Maguy A, Le Bouter S, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007;87:425–456. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- 3.Grines CL, Bashore TM, Boudoulas H, Olson S, Shafer P, Wooley CF. Functional abnormalities in isolated left bundle branch block: the effect of interventricular asynchrony. Circulation. 1989;79:845–853. doi: 10.1161/01.cir.79.4.845. [DOI] [PubMed] [Google Scholar]
- 4.Spragg DD, Leclercq C, Loghmani M, Faris OP, Tunin RS, DiSilvestre D, McVeigh ER, Tomaselli GF, Kass DA. Regional alterations in protein expression in the dyssynchronous failing heart. Circulation. 2003;108:929–932. doi: 10.1161/01.CIR.0000088782.99568.CA. [DOI] [PubMed] [Google Scholar]
- 5.Spragg DD, Akar FG, Helm RH, Tunin RS, Tomaselli GF, Kass DA. Abnormal conduction and repolarization in late-activated myocardium of dyssynchronously contracting hearts. Cardiovasc Res. 2005;67:77–86. doi: 10.1016/j.cardiores.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 6.Jeyaraj D, Wilson LD, Zhong J, Flask C, Saffitz JE, Deschenes I, Yu X, Rosenbaum DS. Mechanoelectrical feedback as novel mechanism of cardiac electrical remodeling. Circulation. 2007;115:3145–3155. doi: 10.1161/CIRCULATIONAHA.107.688317. [DOI] [PubMed] [Google Scholar]
- 7.Plotnikov AN, Yu H, Geller JC, Gainullin RZ, Chandra P, Patberg KW, Friezema S, Danilo P, Jr, Cohen IS, Feinmark SJ, Rosen MR. Role of L-type calcium channels in pacing-induced short-term and long-term cardiac memory in canine heart. Circulation. 2003;107:2844–2849. doi: 10.1161/01.CIR.0000068376.88600.41. [DOI] [PubMed] [Google Scholar]
- 8.Pak PH, Nuss HB, Tunin RS, Kaab S, Tomaselli GF, Marban E, Kass DA. Repolarization abnormalities, arrhythmia and sudden death in canine tachycardia-induced cardiomyopathy. J Am Coll Cardiol. 1997;30:576–584. doi: 10.1016/s0735-1097(97)00193-9. [DOI] [PubMed] [Google Scholar]
- 9.Akar FG, Spragg DD, Tunin RS, Kass DA, Tomaselli GF. Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ Res. 2004;95:717–725. doi: 10.1161/01.RES.0000144125.61927.1c. [DOI] [PubMed] [Google Scholar]
- 10.Bader H, Garrigue S, Lafitte S, Reuter S, Jais P, Haissaguerre M, Bonnet J, Clementy J, Roudaut R. Intra-left ventricular electromechanical asynchrony: a new independent predictor of severe cardiac events in heart failure patients. J Am Coll Cardiol. 2004;43:248–256. doi: 10.1016/j.jacc.2003.08.038. [DOI] [PubMed] [Google Scholar]
- 11.Abraham WT, Fisher WG, Smith AL, Delurgio DB, Leon AR, Loh E, Kocovic DZ, Packer M, Clavell AL, Hayes DL, Ellestad M, Trupp RJ, Underwood J, Pickering F, Truex C, McAtee P, Messenger J. Cardiac resynchronization in chronic heart failure. N Engl J Med. 2002;346:1845–1853. doi: 10.1056/NEJMoa013168. [DOI] [PubMed] [Google Scholar]
- 12.Cleland JG, Daubert JC, Erdmann E, Freemantle N, Gras D, Kappenberger L, Tavazzi L. The effect of cardiac resynchronization on morbidity and mortality in heart failure. N Engl J Med. 2005;352:1539–1549. doi: 10.1056/NEJMoa050496. [DOI] [PubMed] [Google Scholar]
- 13.Bristow MR, Saxon LA, Boehmer J, Krueger S, Kass DA, De Marco T, Carson P, DiCarlo L, DeMets D, White BG, DeVries DW, Feldman AM. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med. 2004;350:2140–2150. doi: 10.1056/NEJMoa032423. [DOI] [PubMed] [Google Scholar]
- 14.Chakir K, Daya SK, Tunin RS, Helm RH, Byrne MJ, Dimaano VL, Lardo AC, Abraham TP, Tomaselli GF, Kass DA. Reversal of global apoptosis and regional stress kinase activation by cardiac resynchronization. Circulation. 2008;117:1369–1377. doi: 10.1161/CIRCULATIONAHA.107.706291. [DOI] [PubMed] [Google Scholar]
- 15.Beuckelmann DJ, Nabauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73:379–385. doi: 10.1161/01.res.73.2.379. [DOI] [PubMed] [Google Scholar]
- 16.Kaab S, Nuss HB, Chiamvimonvat N, O’Rourke B, Pak PH, Kass DA, Marban E, Tomaselli GF. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ Res. 1996;78:262–273. doi: 10.1161/01.res.78.2.262. [DOI] [PubMed] [Google Scholar]
- 17.Kaab S, Dixon J, Duc J, Ashen D, Nabauer M, Beuckelmann DJ, Steinbeck G, McKinnon D, Tomaselli GF. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation. 1998;98:1383–1393. doi: 10.1161/01.cir.98.14.1383. [DOI] [PubMed] [Google Scholar]
- 18.Li GR, Lau CP, Ducharme A, Tardif JC, Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol. 2002;283:H1031–H1041. doi: 10.1152/ajpheart.00105.2002. [DOI] [PubMed] [Google Scholar]
- 19.Akar FG, Rosenbaum DS. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res. 2003;93:638–645. doi: 10.1161/01.RES.0000092248.59479.AE. [DOI] [PubMed] [Google Scholar]
- 20.Rose J, Armoundas AA, Tian Y, DiSilvestre D, Burysek M, Halperin V, O’Rourke B, Kass DA, Marban E, Tomaselli GF. Molecular correlates of altered expression of potassium currents in failing rabbit myocardium. Am J Physiol Heart Circ Physiol. 2005;288:H2077–H2087. doi: 10.1152/ajpheart.00526.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsuji Y, Zicha S, Qi XY, Kodama I, Nattel S. Potassium channel subunit remodeling in rabbits exposed to long-term bradycardia or tachycardia: discrete arrhythmogenic consequences related to differential delayed-rectifier changes. Circulation. 2006;113:345–355. doi: 10.1161/CIRCULATIONAHA.105.552968. [DOI] [PubMed] [Google Scholar]
- 22.Leclercq C, Faris O, Tunin R, Johnson J, Kato R, Evans F, Spinelli J, Halperin H, McVeigh E, Kass DA. Systolic improvement and mechanical resynchronization does not require electrical synchrony in the dilated failing heart with left bundle-branch block. Circulation. 2002;106:1760–1763. doi: 10.1161/01.cir.0000035037.11968.5c. [DOI] [PubMed] [Google Scholar]
- 23.Abraham TP, Dimaano VL, Liang HY. Role of tissue Doppler and strain echocardiography in current clinical practice. Circulation. 2007;116:2597–2609. doi: 10.1161/CIRCULATIONAHA.106.647172. [DOI] [PubMed] [Google Scholar]
- 24.O’Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, Marban E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ Res. 1999;84:562–570. doi: 10.1161/01.res.84.5.562. [DOI] [PubMed] [Google Scholar]
- 25.Aiba T, Shimizu W, Inagaki M, Noda T, Miyoshi S, Ding WG, Zankov DP, Toyoda F, Matsuura H, Horie M, Sunagawa K. Cellular and ionic mechanism for drug-induced long QT syndrome and effectiveness of verapamil. J Am Coll Cardiol. 2005;45:300–307. doi: 10.1016/j.jacc.2004.09.069. [DOI] [PubMed] [Google Scholar]
- 26.Nuss HB, Kaab S, Kass DA, Tomaselli GF, Marban E. Cellular basis of ventricular arrhythmias and abnormal automaticity in heart failure. Am J Physiol. 1999;277:H80–H91. doi: 10.1152/ajpheart.1999.277.1.H80. [DOI] [PubMed] [Google Scholar]
- 27.Helm RH, Byrne M, Helm PA, Daya SK, Osman NF, Tunin R, Halperin HR, Berger RD, Kass DA, Lardo AC. Three-dimensional mapping of optimal left ventricular pacing site for cardiac resynchronization. Circulation. 2007;115:953–961. doi: 10.1161/CIRCULATIONAHA.106.643718. [DOI] [PubMed] [Google Scholar]
- 28.Yu H, McKinnon D, Dixon JE, Gao J, Wymore R, Cohen IS, Danilo P, Jr, Shvilkin A, Anyukhovsky EP, Sosunov EA, Hara M, Rosen MR. Transient outward current, Ito1, is altered in cardiac memory. Circulation. 1999;99:1898–905. doi: 10.1161/01.cir.99.14.1898. [DOI] [PubMed] [Google Scholar]
- 29.Patberg KW, Plotnikov AN, Quamina A, Gainullin RZ, Rybin A, Danilo P, Jr, Sun LS, Rosen MR. Cardiac memory is associated with decreased levels of the transcriptional factor CREB modulated by angiotensin II and calcium. Circ Res. 2003;93:472–478. doi: 10.1161/01.RES.0000088785.24381.2F. [DOI] [PubMed] [Google Scholar]
- 30.Pitt GS, Dun W, Boyden PA. Remodeled cardiac calcium channels. J Mol Cell Cardiol. 2006;41:373–388. doi: 10.1016/j.yjmcc.2006.06.071. [DOI] [PubMed] [Google Scholar]
- 31.Armoundas AA, Rose J, Aggarwal R, Stuyvers BD, O’Rourke B, Kass DA, Marban E, Shorofsky SR, Tomaselli GF, William Balke C. Cellular and molecular determinants of altered Ca2+ handling in the failing rabbit heart: primary defects in SR Ca2+ uptake and release mechanisms. Am J Physiol Heart Circ Physiol. 2007;292:H1607–H1618. doi: 10.1152/ajpheart.00525.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi T, Allen PD, Lacro RV, Marks AR, Dennis AR, Schoen FJ, Grossman W, Marsh JD, Izumo S. Expression of dihydropyridine receptor (Ca2+ channel) and calsequestrin genes in the myocardium of patients with end-stage heart failure. J Clin Invest. 1992;90:927–935. doi: 10.1172/JCI115969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vanderheyden M, Mullens W, Delrue L, Goethals M, de Bruyne B, Wijns W, Geelen P, Verstreken S, Wellens F, Bartunek J. Myocardial gene expression in heart failure patients treated with cardiac resynchronization therapy: responders versus nonresponders. J Am Coll Cardiol. 2008;51:129–136. doi: 10.1016/j.jacc.2007.07.087. [DOI] [PubMed] [Google Scholar]
- 35.Miriyala J, Nguyen T, Yue DT, Colecraft HM. Role of CaVβ subunits, and lack of functional reserve, in protein kinase A modulation of cardiac CaV1.2 channels. Circ Res. 2008;102:e54–e64. doi: 10.1161/CIRCRESAHA.108.171736. [DOI] [PubMed] [Google Scholar]
- 36.Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- 37.Hobai IA, O’Rourke B. Enhanced Ca2+-activated Na+-Ca2+ exchange activity in canine pacing-induced heart failure. Circ Res. 2000;87:690–698. doi: 10.1161/01.res.87.8.690. [DOI] [PubMed] [Google Scholar]
- 38.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 39.Kohlhaas M, Zhang T, Seidler T, Zibrova D, Dybkova N, Steen A, Wagner S, Chen L, Brown JH, Bers DM, Maier LS. Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ Res. 2006;98:235–244. doi: 10.1161/01.RES.0000200739.90811.9f. [DOI] [PubMed] [Google Scholar]
- 40.Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKIIδC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–911. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 41.Fernandez-Velasco M, Ruiz-Hurtado G, Hurtado O, Moro MA, Delgado C. TNF-alpha downregulates transient outward potassium current in rat ventricular myocytes through iNOS overexpression and oxidant species generation. Am J Physiol Heart Circ Physiol. 2007;293:H238–H245. doi: 10.1152/ajpheart.01122.2006. [DOI] [PubMed] [Google Scholar]
- 42.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maltsev VA, Reznikov V, Undrovinas NA, Sabbah HN, Undrovinas A. Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am J Physiol Heart Circ Physiol. 2008;294:H1597–H1608. doi: 10.1152/ajpheart.00484.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, Roden DM, Passier R, Olson EN, Colbran RJ, Anderson ME. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–1293. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- 45.Medina-Ravell VA, Lankipalli RS, Yan GX, Antzelevitch C, Medina-Malpica NA, Medina-Malpica OA, Droogan C, Kowey PR. Effect of epicardial or biventricular pacing to prolong QT interval and increase transmural dispersion of repolarization: does resynchronization therapy pose a risk for patients predisposed to long QT or torsade de pointes? Circulation. 2003;107:740–746. doi: 10.1161/01.cir.0000048126.07819.37. [DOI] [PubMed] [Google Scholar]
- 46.Fish JM, Di Diego JM, Nesterenko V, Antzelevitch C. Epicardial activation of left ventricular wall prolongs QT interval and transmural dispersion of repolarization: implications for biventricular pacing. Circulation. 2004;109:2136–2142. doi: 10.1161/01.CIR.0000127423.75608.A4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.