

Abstract
Oxidative stress and endoplasmic reticulum (ER) stress have been implicated in cardiovascular diseases although the interplay between the two is not clear. This study was designed to examine the influence of oxidative stress through glutathione depletion on myocardial ER stress and contractile function in the absence or presence of the heavy metal scavenger antioxidant metallothionein (MT). FVB and MT overexpression transgenic mice received the GSH synthase inhibitor buthionine sulfoximine (BSO, 30 mM) in drinking water for 2 weeks. Oxidative stress, ER stress, apoptosis, cardiac function and ultrastructure were assessed using GSH/GSSG assay, reactive oxygen species (ROS), immunoblotting, caspase-3 activity, Langendorff perfused heart function (LVDP and ± dP/dt), and transmission electron microscopy. BSO led to a robust decrease in the GSH/GSSG ratio and increased ROS production, consolidating oxidative stress. Cardiac function and ultrastructure were compromised following BSO treatment, the effect of which was obliterated by MT. BSO promoted overt ER stress as evidenced by upregulated BiP, calregulin, phospho-IRE1α and phospho-eIF2α without affecting total IRE1α and eIF2α. BSO treatment led to apoptosis manifested as elevated expression of CHOP/GADD153, caspase-12 and Bax as well as caspase-3 activity, reduced Bcl-2 expression and JNK phosphorylation, all of which was ablated by MT. Moreover, both antioxidant N-acetylcysteine and the ER stress inhibitor tauroursodeoxycholic acid reversed the oxidative stress inducer menadione-elicited depression in cardiomyocyte contractile function. Taken together, these data suggested that ER stress occurs likely downstream of oxidative stress en route to cardiac dysfunction.
Keywords: Oxidative stress, ER stress, Glutathione, Hearts, Cardiomyocytes, Metallothionein
INTRODUCTION
Endoplasmic reticulum (ER) is an extensive intracellular membranous network involved in Ca2+ storage, Ca2+ signaling, glycosylation and trafficking of newly-synthesized membrane and secretory proteins. Perturbations of these processes by glucose and energy deprivation, viral infections, expression of mutant proteins, chemical triggers and cholesterol accumulation have been demonstrated to interfere with the proper functioning of ER, thus creating a condition defined as ER stress [1, 2]. ER stress leads to the activation of a complex signaling network called the unfolded protein response (UPR). The end result of ER stress and UPR, if sustained, is usually apoptotic cell death. ER stress has been shown to participate in the pathogenesis of a wide variety of diseases such as neurodegenerative disorders, diabetes, alcoholism, and ischemia reperfusion heart disease [3, 4]. Up to now, three classes of ER stress transducers have been identified including inositol-requiring protein-1 (IRE1), the protein kinase RNA (PKR)-like ER kinase (PERK)-translation initiation factor eIF-2α pathway and transcription factor-6 (ATF6) [3]. Similar to ER stress, oxidative stress, or accumulation of reactive oxygen species (ROS), is also closely associated with numerous diseases including heart diseases [1, 2, 5–7]. Oxidative stress is usually triggered by the disturbed prooxidant-antioxidant balance and promotes progression of heart dysfunction [8]. Recent evidence has shown that ER stress may trigger ROS production [9] and redox deviation [10] in the ER. Nonetheless, the precise interplay between oxidative stress and ER stress in the heart has been sparsely described.
Glutathione (GSH) is an important intracellular antioxidant to protect against oxidative damage [11, 12]. GSH is maintained predominantly in the reduced state through the glutathione redox cycle by glutathione reductase [12]. Depletion or loss of GSH contributes to oxidative injury [13]. Our earlier study revealed that cardiac overexpression of the heavy metal scavenger metallothionein effectively alleviated GSH depletion-induced oxidative cardiomyopathy [14] through reduction of ROS. However, whether ER stress plays a role in oxidative stress and antioxidant-elicited myocardial response remains largely unknown. Hence we took advantage of the GSH depletion murine model of oxidative stress using buthionine sulfoximine (BSO), a transition state inhibitor of γ-glutamyl cysteine synthetase to irreversibly inhibit GSH synthesis [15], to assess the role of ER stress in antioxidant-elicited cardioprotection against oxidative stress. GSH/GSSG ratio, ROS production, ER stress markers including BiP, calregulin, IRE1α, eIF2α and apoptosis markers such as caspase, CHOP (GADD153), JNK, Bax and Bcl2 were examined. Myocardial ultrastructure and contractile function were evaluated in wild-type FVB and metallothionein overexpression transgenic mice with or without BSO treatment. To further elucidate the sequential relationship between oxidative stress and ER stress, the oxidative stress inducer menadione-elicited changes in cardiomyocyte contractile function were examined in the absence or presence of the ER stress inhibitor tauroursodeoxycholic acid (TUDCA).
MATERIALS AND METHODS
Experimental animals
The animal procedures described in this study were approved by the University of Wyoming Institutional Animal Use and Care Committee. In brief, 5–6-month-old, female mice with a ten-fold cardiac-specific transgenic overexpression of the heavy metal scavenger metallothionein (MT) driven by the mouse α-MHC promoter [16] and wild-type Friend virus B (FVB) mice were used. All mice were maintained at 22°C with a 12/12-light/dark cycle and received lab chow and water ad libitum. A cohort of FVB and MT mice was given BSO (30 mM) in drinking water for 2 weeks [13].
Determination of reduced and oxidized glutathione (GSH and glutathione disulfide [GSSG])
The heart glutathione contents were measured as described previously [17]. Tissue samples (□ 50 mg) were sonicated in picric acid and centrifuged at 13,500 × g for 20 min. The supernatant was then divided into two aliquots. One was directly used for total GSH assay and the other for GSSG. 100 μl of supernatant fractions with 2 μl vinyl pyridine were incubated at room temperature for 1 hr to scavenge GSH for the GSSG determination. The GSSG was then subtracted from the total glutathione to evaluate the GSH levels. GSH was determined by the DTNB-glutathione reductase recycling mechanism [18].
Cardiomyocyte isolation and in vitro drug treatment
Murine cardiomyocytes were isolated as described [14]. After ketamine/xylazine sedation, hearts were removed and perfused with Ca2+ -free Tyrode’s solution containing (in mM): NaCl 135, KCl 4.0, MgCl2 1.0, HEPES 10, NaH2PO4 0.33, glucose 10, butanedione monoxime 10, and the solution was gassed with 5% CO2/95% O2. Hearts were digested with Liberase Blendzyme 4 (Hoffmann-La Roche Inc., Indianapolis, IN, USA) for 20 min. Left ventricles were removed and minced before being filtered. Tissue pieces were gently agitated and pellet of cells was resuspended. Extracellular Ca2+ was added incrementally back to 1.20 mM over a period of 30 min. Isolated myocytes were used within 8 hrs of isolation. Normally, a yield of 50 – 60% viable rod-shaped cardiomyocytes with clear sarcomere striations was achieved. Only rod-shaped myocytes with clear edges were selected for mechanical study. To directly assess the role of ER stress on cardiomyocyte contractile function in response to oxidative stress, cardiomyocytes were treated with the oxidative stress inducer menadione (30 μM) [19] at 37°C for 2 hrs in the absence or presence of the antioxidant N-acetylcysteine (NAC, 500 μM) [20] or the ER stress inhibitor tauroursodeoxycholic acid (TUDCA, 500 μM) [21] prior to mechanical function assessment.
Detection of intracellular ROS
ROS were detected in isolated cardiomyocytes by analyzing the fluorescence intensity of the intracellular fluoroprobe 5-(6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA). In brief, cardiomyocytes were loaded with 10 μM non-fluorescent dye 2,7-dichlorodihydrofluorescein diacetate (H2DCFDA, Molecular Probes, Eugene, OR) at 37°C for 30 min. After rinsing with the Krebs-Henseleit buffer (KHB), the fluorescent intensity was measured using a fluorescent microplate reader at an excitation wavelength of 480 nm and an emission wavelength of 530 nm. Untreated cells showed no fluorescence and were used to determine the background fluorescence. The final fluorescent intensity was normalized to the protein content in each group [22].
Caspase-3 assay
The caspase-3 activity was determined according to our previously published method [23]. Briefly, 1 ml of PBS was added to heart tissues. Tissues were homogenized and centrifuged at 10,000 × g at 4°C for 10 min. The supernatant was discarded, and pellets were lysed in 100 μl of ice-cold lysis buffer [50 mM HEPES, pH 7.4, 0.1% CHAPS, 1 mM dithiothreitol (DTT), 0.1 mM EDTA, 0.1% NP40]. The assay for caspase-3 activity was carried out in a 96-well plate. Each well contained 30 μl of lysate, 70 μl of assay buffer (50 mM HEPES, pH 7.4, 0.1% CHAPS, 100 mM NaCl, 10 mM DTT, and 1 mM EDTA) and 20 μl of caspase-3 colorimetric substrate Ac-DEVD-pNA (Sigma Chemicals, St. Louis, MO). The 96-well plate was incubated at 37°C for 2 hrs, during which time caspase in the sample was allowed to cleave the chromophore p-NA from the substrate molecule. Absorbance readings were obtained at 405 nm with the caspase-3 activity being directly proportional to the colorimetric reaction. Protein content was determined using the Bradford method [24].
Langendorff perfused heart function
The Langendorff perfused heart function was assessed using the ADInstruments PowerLab® system. In brief, mice were anesthetized with ketamine/xylazine [25] and the hearts were perfused with KHB containing 7 mM glucose, 0.4 mM oleate, 1% BSA, and a low fasting concentration of insulin (10 μU/ml). The perfusion was initiated in the retrograde mode through the cannulated aorta [26]. Hearts were perfused at a constant aortic pressure of 4 ml/min at baseline for 60 min. A fluid-filled latex balloon connected to a solid-state pressure transducer was inserted into the left ventricle to measure the pressure. Left ventricular developed pressure (LVDP), the first derivative of LVDP, namely, the maximum rate of left ventricular pressure development (+ dP/dt) and the maximum rate of left ventricular pressure decline (− dP/dt) were recorded using a digital acquisition system at a balloon volume which resulted in a baseline LV end-diastolic pressure of 5 mmHg.
Transmission electron microscopy
Ultrastructural changes in murine ventricles were detected by transmission electron microscopy according to our previously published method [22]. In brief, left ventricular tissues from each group were cut into small pieces (< 3 mm3) and immediately fixed with 2.5% glutaraldehyde for at least 1 hour before postfixation with 1% uranyl acetate for 1 hour. Following PBS rinse, tissues were dehydrated with graded ethanol from 70 to 100% followed by 100% propylene oxide (PO), 1:1 mixture of PO and the embedding resin, and 100% embedding resin as transition. Tissues were then embedded in a fresh change of 100% embedding media in the oven at 60°C overnight. Ultra thin sections (50 nm) were cut on the ultramicrotome, stained with uranyl acetate, followed by lead citrate, and viewed on a Hitachi H-7000 transmission electron microscope equipped with a 4 k×4 k cooled charge coupled device (CCD) digital camera.
Western blot analysis
The total protein was prepared as described [22]. In brief, tissue samples from mouse ventricles were removed and homogenized in a lysis buffer containing 20 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, 0.1% SDS, and protease inhibitor cocktail. Samples were then sonicated for 15 sec and centrifuged at 13,000x g for 20 min at 4°C. The protein concentration of the supernatant was determined using Protein Assay Reagent (Bio-Rad Laboratories, Richmond, CA, USA). Protein samples were then mixed 1:2 with Laemmli sample buffer with 5% 2-mercaptoethanol and heated at 95°C for 5 min. Equal amounts (50 μg protein/lane) of the protein mixture, or the SeeBlue Plus2 PreStained markers (Invitrogen, Carlsbad, CA, USA) were separated on 10% or 15% SDS-polyacrylamide gels in a minigel apparatus (Mini-PROTEAN II, Bio-Rad); then were transferred electrophoretically to Nitrocellulose membranes (0.2 μm pore size, Bio-Rad Laboratories, Inc, Hercules, CA, USA). Membranes were incubated for 1 hr at room temperature in a blocking solution containing 5% milk in Tris-buffered saline (TBS). After TBS washed, membranes were incubated overnight at 4°C with primary antibody including rabbit anti-BiP (1:1,000), goat anti-calregulin (calreticulin) (1:1,000), rabbit anti-IRE1α (1:500), rabbit anti-phospho-IRE1α (1:1,000), rabbit anti-eIF2α (1:1,000), rabbit anti-phospho-eIF2α (1:500), rabbit anti-Caspase-12 (1:500), rabbit anti-CHOP (GADD153) (1:1,000), rabbit-anti JNK (1:1,000), mouse anti-phospho-JNK (1:1000), mouse anti-Bcl2 (1:1,000), rabbit anti-Bax (1:500) and rabbit anti-GAPDH (as loading control, 1:1,000) antibodies. After three washes with TBS-T to remove excessive primary antibody binding, blots were incubated with horseradish peroxidase (HRP)–conjugated secondary antibody (1:5,000) for 1 hr at room temperature. The antigens were detected by the luminescence method. Quantification of band density was determined using Quantity One software (Bio-Rad, version 4.4.0) and reported in optical density per square millimeter.
Cell shortening/relengthening
Mechanical properties of cardiomyocytes were assessed using a SoftEdge MyoCam system (IonOptix, Milton, MA, USA). In brief, cells were placed in a Warner chamber mounted on the stage of an inverted microscope (Olympus IX-70) and superfused (~1 ml/min at 25°C) with a buffer containing (in mM) 131 NaCl, 4 KCl, 1 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES at pH 7.4. The cells were field stimulated with suprathreshold voltage at a frequency of 0.5 Hz using a pair of platinum wires placed on opposite sides of the chamber connected to a FHC stimulator (Brunswick, NE, USA). The myocyte being studied was displayed on the computer monitor using an IonOptix MyoCam camera. An IonOptix SoftEdge software was used to capture changes in cell length during shortening and relengthening. Cell shortening and relengthening were assessed using the following indices: resting cell length, peak shortening (PS), time-to-PS (TPS), time-to-90% relengthening (TR90), and maximal velocity of shortening/relengthening (± dL/dt) [14].
Statistical analysis
Data were presented as mean ± SEM. Statistical significance (p < 0.05) for each variable was estimated by a one-way analysis of variance (ANOVA) followed by a Tukey’s multiple comparison test as the post hoc analysis.
RESULTS
General characteristics of mice, ROS levels and caspase-3 activity following BSO treatment
Neither BSO nor metallothionein overexpression significantly affected body and organ weights. As expected, BSO dramatically reduced GSH levels and the GSH/GSSG ratio but not the GSSG levels in hearts, the effect of which was unaffected by metallothionein (Table 1). In addition, GSH depletion significantly increased ROS production in isolated cardiomyocytes as measured by the intracellular fluoroprobe CM-H2DCFDA, the effect of which was counteracted by metallothionein. Metallothionein itself did not elicit any significant effect on ROS generation (Fig. 1A). To examine if apoptosis plays any role in metallothionein- and BSO-induced cardiac response, caspase-3 activity was assessed in myocardium from FVB and metallothionein mice with or without BSO treatment. Results shown in Fig. 1B depict significantly elevated caspase-3 activity in hearts from BSO-treated FVB mice. Consistent with its effect on ROS production, metallothionein significantly attenuated the BSO-elicited elevation in caspase-3 activity.
Table 1.
General feature of FVB and MT mice with or without BSO treatment for 2 weeks.
| Parameter | FVB (7) | FVB+BSO (9) | MT (6) | MT+BSO (9) |
|---|---|---|---|---|
| Body Weight (g) | 24.9 ± 0.7 | 23.9 ± 0.5 | 26.0 ± 0.6 | 24.5 ± 0.7 |
| Heart Weight (mg) | 140 ± 8 | 132 ± 6 | 153 ± 14 | 136 ± 9 |
| Heart/Body Weight (mg/g) | 5.64 ± 0.32 | 5.52 ± 0.20 | 5.86 ± 0.43 | 5.51 ± 0.25 |
| Liver Weight (g) | 1.15 ± 0.02 | 1.13 ± 0.03 | 1.22 ± 0.05 | 1.21 ± 0.05 |
| Kidney Weight (mg) | 323 ± 13 | 320 ± 9 | 360 ± 17 | 340 ± 16 |
| Heart GSH (nmol/mg tissue) | 63.0 ± 2.62 | 34.3 ± 0.92* | 66.4 ± 1.16 | 36.5 ± 0.58*$ |
| Heart GSSG (nmol/mg tissue) | 16.9 ± 0.15 | 17.2 ± 0.44 | 17.0± 0.12 | 16.8 ± 0.23 |
| Heart GSH/GSSG | 3.73 ± 0.13 | 1.99 ± 0.04* | 3.91 ± 0.04 | 2.18 ± 0.03*$ |
Mean ± SEM,
p < 0.05 vs. FVB group,
p < 0.05 vs. MT transgenic group, number of mice per group is given in parentheses.
Fig. 1.

Panel A: ROS levels in isolated cardiomyocytes from FVB and metallothionein (MT) transgenic mice treated with or without BSO; Panel B: Myocardial apoptosis assessed using Caspase-3 activity in myocardial tissues from FVB and MT mice treated with or without BSO. Mean ± SEM, n = 4–9 mice per group, * p < 0.05 vs. FVB group, # p < 0.05 vs. FVB-BSO group.
Effect of MT on BSO-elicited response on Langendorff perfused heart function
Cardiac performance was evaluated using the Langendorff perfused heart system. Mechanical indices including left ventricular developed pressure (LVDP), maximal rate of pressure development (+ dP/dt) and decline (− dP/dt) were similar between FVB and MT mice in the absence of BSO treatment. BSO treatment significantly depressed LVDP and ± dP/dt in FVB mice although such effect was significantly attenuated or obliterated with the overexpression of metallothionein (Fig. 2). These data suggest that metallothionein may protect against oxidative stress-induced cardiac contractile dysfunction.
Fig. 2.

Effect of BSO treatment on myocardial contractility using the Langendorff perfused heart system (Panel A: LVDP; Panel B: + dP/dt; and Panel C: − dP/dt) in FVB and metallothionein (MT) transgenic mice treated with or without BSO. Mean ± SEM, n = 3 independent mice per group, * p < 0.05 vs. FVB group, # p < 0.05 vs. FVB-BSO group, $ p < 0.05 vs. MT group.
Electron microscopic characteristics
Ultrastructural changes in myocardial tissues from FVB and MT mice following BSO treatment were examined by transmission electron microscope (TEM). As shown in Fig. 3, there were no overt ultrastructural differences (regular structure, mitochondria and myofilaments with uninterrupted sarcomeres) between FVB and MT groups in the absence of BSO treatment (Fig. 3 A–B, E–F). BSO treatment triggered dramatic cytoarchitectonic changes including mitochondrial disorganization of cristae and matrix clear-out, irregular myofilaments and disruption of sarcomeres in left ventricles of FVB mice (Fig. 3 C–D). BSO also triggered generation of bulbiform bodies indicating lipid droplets. Interestingly, the BSO-induced ultrastructural changes were significantly alleviated by metallothionein. Micrographs from the MT-BSO mice displayed similar myocardial morphology compared with the non-BSO treated groups (Fig. 3 G–H).
Fig. 3.
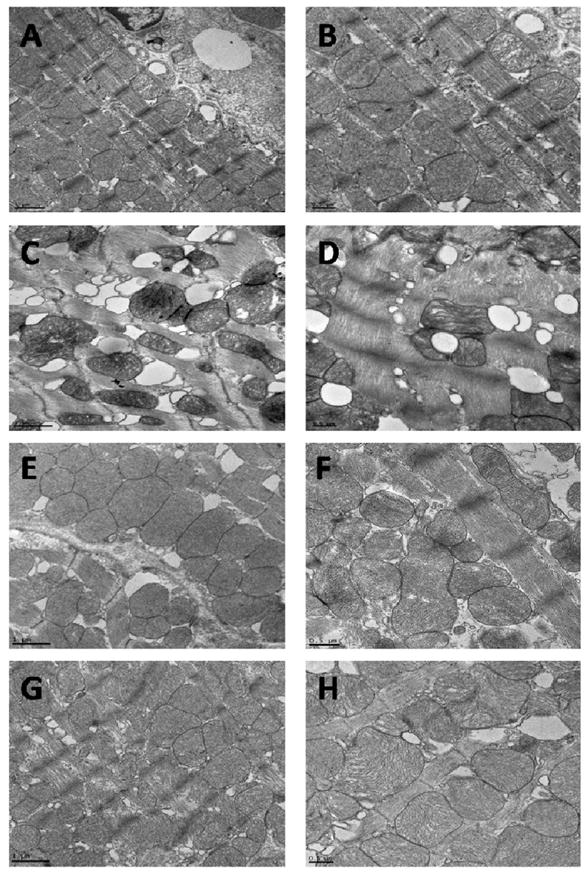
Transmission electron microscopic micrographs of left ventricular tissues from FVB and metallothionein (MT) transgenic mice treated with or without BSO. Panel A–B: FVB; Panel C–D: FVB-BSO; Panel E–F: MT; and Panel G–H: MT-BSO. Tissues in A (× 5000), B (× 8000), E (× 5000) and F (× 8000) show regular structure, mitochondria and myofilaments with uninterrupted sarcomeres; The BSO-treated myocardial tissues (C) (× 5000) and (D) (× 8000) displayed altered mitochondria, irregular myofilaments and the bulbiform bodies. These ultrastructural changes were less obvious in MT group (G) (× 5000) and (H) (× 8000).
Western blot analysis of ER stress markers
To explore the potential mechanism responsible for metallothionein-offered protection against BSO-elicited myocardial damage, expression of the ER stress markers BiP, calregulin (calreticulin), IRE1α, and eIF2α was evaluated. The immunoblotting results revealed no significant change in the total IRE1α and eIF2α expression in FVB and MT groups with or without BSO treatment. However, BSO significantly up-regulated the expression of BiP and calregulin, as well as phosphorylation of IRE1α and eIF2α in FVB mice, the effects of which were significantly alleviated by metallothionein. Metallothionein itself did not affect the expression of these ER stress markers and chaperones (Fig. 4).
Fig. 4.
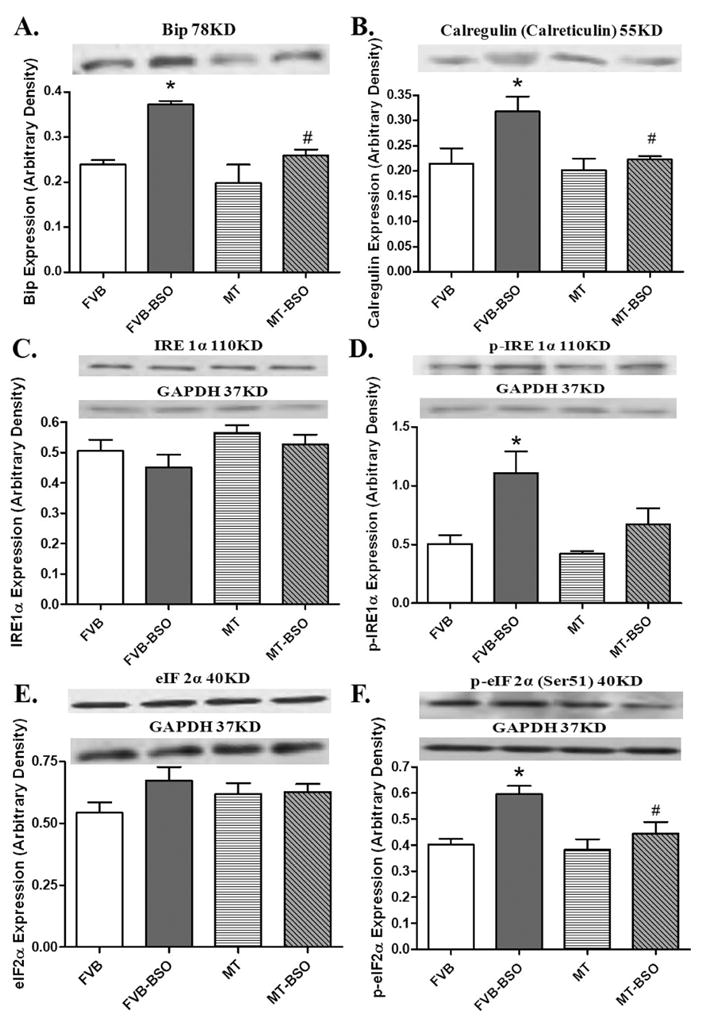
Effect of BSO on the expression of BiP (panel A), calregulin (calreticulin, panel B), IRE 1α (panel C), phosphorylated IRE 1α (panel D), eIF 2α (panel E) and phosphorylated eIF 2α (Panel F) in FVB and metallothionein (MT) transgenic mice. Inset: representative gels of BiP, calregulin (calreticulin), IRE1α, p-IRE1α, eIF2α and p-eIF2α proteins using specific antibodies. GAPDH was used as the internal loading control. Mean ± SEM, n = 4–8 mice per group, * p < 0.05 vs. FVB group, # p < 0.05 vs. FVB-BSO group.
Western blot analysis of the ER apoptotic makers
To further explore the effect of BSO and metallothionein on ER stress-mediated apoptotic pathway, we evaluated the ER-related apoptotic proteins CHOP/GADD153, caspase-12, c-Jun NH2-terminal kinase (JNK), phosphorylated JNK, Bax and Bcl-2. Our data shown in Fig. 5 displayed that BSO significantly upregulated CHOP/GADD153, caspase-12 and Bax while it downregulated phospho-JNK and Bcl-2. Total JNK expression was unaffected. Although metallothionein itself did not affect the expression of these ER apoptotic proteins, it obliterated the BSO-induced changes in CHOP/GADD153, caspase-12, Bax, phospho-JNK and Bcl-2.
Fig. 5.

Western blot analysis of the CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP/GADD153, panel A); caspase-12 (panel B); c-Jun NH2-terminal kinase (JNK, panel C); phosphorylated JNK (panel D); Bax (panel E) and Bcl-2 (panel F) from FVB and metallothionein (MT) transgenic mice treated with or without BSO. Insets: representative gel blots of these proteins using respective antibodies. Mean ± SEM, n = 4–8 mice per group, * p < 0.05 vs. FVB group; # p < 0.05 vs. FVB-BSO group.
Effect of ER stress inhibition on oxidative stress-induced changes in cell shortening
To further examine the role of ER stress in oxidative stress-induced cardiac contractile defects, freshly isolated cardiomyocytes from wild-type FVB mice were treated with the oxidative stress inducer menadione (30 μM) [19] for 2 hrs in the absence or presence of the ER stress inhibitor TUDCA (500 μM) [21] or the antioxidant NAC (500 μM, used as a positive control). Somewhat similar to our previous finding [27], the oxidative stress inducer menadione significantly decreased PS and ± dL/dt without affecting resting cell length, TPS and TR90 in murine cardiomyocytes. Interestingly, both TUDCA and NAC ablated menadione-induced mechanical defects without eliciting any effects on cardiomyocyte mechanics themselves (Fig. 6). These data provided direct evidence for a likely role of ER stress in the oxidative stress-induced cardiac contractile dysfunction.
Fig. 6.

Contractile properties of cardiomyocytes from FVB mice incubated for 2 hrs with the oxidative stress inducer menadione (30 μM) in the absence or presence of the antioxidant N-acetylcysteine (NAC, 500 μM) or the ER stress inhibitor tauroursodeoxycholic acid (TUDCA, 500 μM). Panel A: resting cell length; Panel B: Peak shortening (normalized to cell length); Panel C: Maximal velocity of shortening (+ dL/dt); Panel D: Maximal velocity of relengthening (− dL/dt); Panel E: Time-to-PS (TPS); and Panel F: Time-to-90% relengthening (TR90). Mean ± SEM, n = 43 – 49 cells from 3 mice per group, * p < 0.05 vs. control group, # p < 0.05 vs. menadione group.
DISCUSSION
The major findings of our study revealed that glutathione depletion leads to oxidative stress, apoptosis, ER stress, myocardial contractile and ultrastructural (mitochondrial swelling and myofilament aberrations) defect. The fact that the thiol-rich heavy metal scavenger metallothionein abrogates BSO-induced ultrastructural, functional and mitochondrial changes as well as ER stress favors that ER stress may occur downstream of oxidative stress in myocardial dysfunction. This notion received further consolidation from our in vitro cardiomyocyte mechanical study where inhibition of ER stress with TUDCA ablated oxidative stress-triggered cardiomyocyte mechanical dysfunction. These results have collectively prompted for a likely role of ER stress in oxidative stress-associated cardiac mechanical dysfunction and more importantly, the therapeutic value of ER stress as a potential drug target in myocardial dysfunction.
Our data suggested that glutathione depletion-associated oxidative stress is associated with the development of ER stress. A causal link has been implicated between protein folding and ROS generation [9, 28]. Neurologic studies have depicted that ER stress or UPR is likely downstream of 6-hydroxydopamine-induced oxidative stress en route to cell death [29, 30]. However, limited information is available with regards to the precise interplay between oxidative stress and ER stress in the hearts. Our present study revealed that glutathione depletion-induced oxidative stress may precede ER stress given that the antioxidant metallothionein reversed BSO-elicited ER stress in murine hearts. Several studies have implicated a unique role of metallothionein in the antioxidant defense system, where glutathione is an essential component [31, 32]. Our data of reduced GSH/GSSG ratio and increased ROS reflected confirmed oxidative stress elicited by BSO, an effective inducer for oxidative stress [13, 14]. The antioxidant metallothionein attenuated BSO-induced oxidative stress but not the depleted glutathione levels (GSH/GSSG ratio), indicating a role for metallothionein independent of the bioavailability of glutathione. We further observed that glutathione depletion prompted ER stress, which is supported by upregulated ER stress markers including BiP, calregulin, CHOP/GADD153, phospho-IRE1α and phospho-eIF2α without affecting IRE1α and eIF2α. Metallothionein alleviated BSO-induced ROS accumulation, cardiac dysfunction and ER stress, favoring a downstream role for ER stress in the oxidative stress-induced myocardial defects. Observation from our in vitro study further supports the notion that ER stress occurs downstream of oxidative stress in the regulation of cardiomyocyte contractile function. Furthermore, our result revealed that glutathione depletion resulted in apoptosis as evidenced by the upregulated caspase-3 activity, CHOP/GADD153, caspase-12, Bax and downregulated Bcl-2 expression, supporting the notion of oxidative stress/ER stress-elicited cell death in the hearts [33].
Our results provide evidence that oxidative stress induced by glutathione depletion may predispose the onset and progression of ER stress. BSO treatment directly promotes ER stress including BiP, calregulin, phosphorylation of IRE 1α and eIF 2α, the effect of which was attenuated by metallothionein. IRE1, PERK and ATF6 are the three ER sensor proteins which regulate the UPR through their respective signaling pathways, before binding to the immunoglobulin-binding protein (BiP) in unstressed cells [34]. BiP is a protein chaperone also known as the glucose regulated protein of 78 kDa (GRP78) [35]. In response to ER stress, unfolded proteins accumulate in the ER lumen and promote BiP release from UPR sensors. This may lead to the activation of IRE1 and PERK to phosphorylate eIF2α [28, 36], consistent with our present data. In addition, BSO treatment enhanced expression of calregulin, a ROS-sensitive ER chaperone governing ER-Golgi trafficking [37, 38]. Interestingly, all those changes in ER stress were effectively eliminated by metallothionein. Our data did not favor any direct response of the heavy metal scavenger metallothionein on ER stress markers. Furthermore, BSO treatment upregulated CHOP/GADD153, caspase-3, caspase-12, Bax while it downregulates Bcl-2. Consistent with its effect on cardiac function, ROS and ER stress, these apoptotic markers were reversed by metallothionein, indicating that a major role of oxidative stress in apoptosis. Last but not the least, glutathione depletion depressed activation of the stress signaling molecule JNK in FVB but not metallothionein mice. These data suggest that JNK pathway may not play a major role in glutathione depletion-elicited oxidative stress. The reduced JNK activation may reflect a compensatory response of JNK signal in response to BSO-induced glutathione depletion.
In summary, data from our present study have shed some light on the sequential relationship between oxidative stress and ER stress in the maintenance and regulation of cardiac function. Nonetheless, the jury is still out with regards to the precise interplay between the two. Further study is warranted to examine the effect of oxidative stress inhibition on ER stress-induced cardiac contractile function, given that ER stress is known to initiate ROS production and redox deviation [9, 10, 33]. In addition, it is unclear exactly how oxidative stress triggers ER stress. Although it may be speculated that oxidative stress may initiate ER stress process through protein oxidative modification or inhibition of Ca2+-ATPase [33], further scrutiny is warranted to elucidate the precise mechanism behind the oxidative stress-elicited UPR and ER stress.
Acknowledgments
The authors wish to acknowledge Dr. Zhaojie Zhang from our Microscopic Facility for his technical guidance and Dr. Ji Li for his generosity for the ADInstruments PowerLab® system. This work was supported in part by grants from NIH Rocky Mountain Regional INBRE P20 RR16474, National Natural Science Foundation of China (30728023) and National Basic Research Program of China (973 Program 2007CB512106).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Haynes CM, Titus EA, Cooper AA. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol Cell. 2004 Sep 10;15(5):767–76. doi: 10.1016/j.molcel.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 2.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000 Apr 28;101(3):249–58. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 3.Glembotski CC. Endoplasmic reticulum stress in the heart. Circ Res. 2007 Nov 9;101(10):975–84. doi: 10.1161/CIRCRESAHA.107.161273. [DOI] [PubMed] [Google Scholar]
- 4.Li SY, Ren J. Cardiac overexpression of alcohol dehydrogenase exacerbates chronic ethanol ingestion-induced myocardial dysfunction and hypertrophy: role of insulin signaling and ER stress. J Mol Cell Cardiol. 2008 Jun;44(6):992–1001. doi: 10.1016/j.yjmcc.2008.02.276. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006 Apr 13;440(7086):944–8. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 6.Kaneto H, Katakami N, Kawamori D, Miyatsuka T, Sakamoto K, Matsuoka TA, et al. Involvement of oxidative stress in the pathogenesis of diabetes. Antioxid Redox Signal. 2007 Mar;9(3):355–66. doi: 10.1089/ars.2006.1465. [DOI] [PubMed] [Google Scholar]
- 7.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005 Oct;115(10):2656–64. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. 2000 Jun;18(6):655–73. doi: 10.1097/00004872-200018060-00002. [DOI] [PubMed] [Google Scholar]
- 9.Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, et al. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proceedings of the National Academy of Sciences of the United States of America. 2008 Nov 25;105(47):18525–30. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merksamer PI, Trusina A, Papa FR. Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell. 2008 Nov 28;135(5):933–47. doi: 10.1016/j.cell.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sagrista ML, Garcia AE, Africa De Madariaga M, Mora M. Antioxidant and pro-oxidant effect of the thiolic compounds N-acetyl-L-cysteine and glutathione against free radical-induced lipid peroxidation. Free Radic Res. 2002 Mar;36(3):329–40. doi: 10.1080/10715760290019354. [DOI] [PubMed] [Google Scholar]
- 12.Chakravarthi S, Jessop CE, Bulleid NJ. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006 Mar;7(3):271–5. doi: 10.1038/sj.embor.7400645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaziri ND, Wang XQ, Oveisi F, Rad B. Induction of oxidative stress by glutathione depletion causes severe hypertension in normal rats. Hypertension. 2000 Jul;36(1):142–6. doi: 10.1161/01.hyp.36.1.142. [DOI] [PubMed] [Google Scholar]
- 14.Ren J, Privratsky JR, Yang X, Dong F, Carlson EC. Metallothionein alleviates glutathione depletion-induced oxidative cardiomyopathy in murine hearts. Crit Care Med. 2008 Jul;36(7):2106–16. doi: 10.1097/CCM.0b013e31817bf925. [DOI] [PubMed] [Google Scholar]
- 15.Jain A, Martensson J, Mehta T, Krauss AN, Auld PA, Meister A. Ascorbic acid prevents oxidative stress in glutathione-deficient mice: effects on lung type 2 cell lamellar bodies, lung surfactant, and skeletal muscle. Proceedings of the National Academy of Sciences of the United States of America. 1992 Jun 1;89(11):5093–7. doi: 10.1073/pnas.89.11.5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang YJ, Chen Y, Yu A, Voss-McCowan M, Epstein PN. Overexpression of metallothionein in the heart of transgenic mice suppresses doxorubicin cardiotoxicity. J Clin Invest. 1997 Sep 15;100(6):1501–6. doi: 10.1172/JCI119672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aberle IN, Ren J. Experimental Assessment of the Role of Acetaldehyde in Alcoholic Cardiomyopathy. Biol Proced Online. 2003;5:1–12. doi: 10.1251/bpo41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969 Mar;27(3):502–22. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Azhar G, Nagano K, Wei JY. Differential vulnerability to oxidative stress in rat cardiac myocytes versus fibroblasts. Journal of the American College of Cardiology. 2001 Dec;38(7):2055–62. doi: 10.1016/s0735-1097(01)01665-5. [DOI] [PubMed] [Google Scholar]
- 20.Robin E, Guzy RD, Loor G, Iwase H, Waypa GB, Marks JD, et al. Oxidant stress during simulated ischemia primes cardiomyocytes for cell death during reperfusion. The Journal of biological chemistry. 2007 Jun 29;282(26):19133–43. doi: 10.1074/jbc.M701917200. [DOI] [PubMed] [Google Scholar]
- 21.Denk GU, Hohenester S, Wimmer R, Bohland C, Rust C, Beuers U. Role of mitogen-activated protein kinases in tauroursodeoxycholic acid-induced bile formation in cholestatic rat liver. Hepatol Res. 2008 Jul;38(7):717–26. doi: 10.1111/j.1872-034X.2008.00321.x. [DOI] [PubMed] [Google Scholar]
- 22.Privratsky JR, Wold LE, Sowers JR, Quinn MT, Ren J. AT1 blockade prevents glucose-induced cardiac dysfunction in ventricular myocytes: role of the AT1 receptor and NADPH oxidase. Hypertension. 2003 Aug;42(2):206–12. doi: 10.1161/01.HYP.0000082814.62655.85. [DOI] [PubMed] [Google Scholar]
- 23.Li SY, Gomelsky M, Duan J, Zhang Z, Gomelsky L, Zhang X, et al. Overexpression of aldehyde dehydrogenase-2 (ALDH2) transgene prevents acetaldehyde-induced cell injury in human umbilical vein endothelial cells: role of ERK and p38 mitogen-activated protein kinase. The Journal of biological chemistry. 2004 Mar 19;279(12):11244–52. doi: 10.1074/jbc.M308011200. [DOI] [PubMed] [Google Scholar]
- 24.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976 May 7;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 25.Ren J, Wold LE. Measurement of Cardiac Mechanical Function in Isolated Ventricular Myocytes from Rats and Mice by Computerized Video-Based Imaging. Biol Proced Online. 2001 Dec 11;3:43–53. doi: 10.1251/bpo22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verma S, Yuen VG, Badiwala M, Anderson TJ, McNeill JH. Working heart function in diabetes is not improved by spironolactone treatment. Can J Physiol Pharmacol. 2003 May;81(5):493–6. doi: 10.1139/y03-041. [DOI] [PubMed] [Google Scholar]
- 27.Esberg LB, Ren J. The oxygen radical generator pyrogallol impairs cardiomyocyte contractile function via a superoxide and p38 MAP kinase-dependent pathway: protection by anisodamine and tetramethylpyrazine. Cardiovascular toxicology. 2004;4(4):375–84. doi: 10.1385/ct:4:4:375. [DOI] [PubMed] [Google Scholar]
- 28.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007 Dec;9(12):2277–93. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 29.Holtz WA, Turetzky JM, Jong YJ, O’Malley KL. Oxidative stress-triggered unfolded protein response is upstream of intrinsic cell death evoked by parkinsonian mimetics. J Neurochem. 2006 Oct;99(1):54–69. doi: 10.1111/j.1471-4159.2006.04025.x. [DOI] [PubMed] [Google Scholar]
- 30.Yamamuro A, Yoshioka Y, Ogita K, Maeda S. Involvement of endoplasmic reticulum stress on the cell death induced by 6-hydroxydopamine in human neuroblastoma SH-SY5Y cells. Neurochem Res. 2006 May;31(5):657–64. doi: 10.1007/s11064-006-9062-6. [DOI] [PubMed] [Google Scholar]
- 31.Haidara K, Moffatt P, Denizeau F. Metallothionein induction attenuates the effects of glutathione depletors in rat hepatocytes. Toxicol Sci. 1999 Jun;49(2):297–305. doi: 10.1093/toxsci/49.2.297. [DOI] [PubMed] [Google Scholar]
- 32.Zhou G, Li X, Hein DW, Xiang X, Marshall JP, Prabhu SD, et al. Metallothionein suppresses angiotensin II-induced nicotinamide adenine dinucleotide phosphate oxidase activation, nitrosative stress, apoptosis, and pathological remodeling in the diabetic heart. Journal of the American College of Cardiology. 2008 Aug 19;52(8):655–66. doi: 10.1016/j.jacc.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 33.Yokouchi M, Hiramatsu N, Hayakawa K, Okamura M, Du S, Kasai A, et al. Involvement of selective reactive oxygen species upstream of proapoptotic branches of unfolded protein response. The Journal of biological chemistry. 2008 Feb 15;283(7):4252–60. doi: 10.1074/jbc.M705951200. [DOI] [PubMed] [Google Scholar]
- 34.Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007 Dec;18(6):716–31. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morris JA, Dorner AJ, Edwards CA, Hendershot LM, Kaufman RJ. Immunoglobulin binding protein (BiP) function is required to protect cells from endoplasmic reticulum stress but is not required for the secretion of selective proteins. J Biol Chem. 1997 Feb 14;272(7):4327–34. doi: 10.1074/jbc.272.7.4327. [DOI] [PubMed] [Google Scholar]
- 36.Buck TM, Wright CM, Brodsky JL. The activities and function of molecular chaperones in the endoplasmic reticulum. Semin Cell Dev Biol. 2007 Dec;18(6):751–61. doi: 10.1016/j.semcdb.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waser M, Mesaeli N, Spencer C, Michalak M. Regulation of calreticulin gene expression by calcium. J Cell Biol. 1997 Aug 11;138(3):547–57. doi: 10.1083/jcb.138.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ihara Y, Kageyama K, Kondo T. Overexpression of calreticulin sensitizes SERCA2a to oxidative stress. Biochem Biophys Res Commun. 2005 Apr 22;329(4):1343–9. doi: 10.1016/j.bbrc.2005.02.112. [DOI] [PubMed] [Google Scholar]