

Abstract
The functional role of the interaction between c-Jun and simian virus 40 promoter factor 1 (Sp1) in epidermal growth factor (EGF)-induced expression of 12(S)-lipoxygenase gene in human epidermoid carcinoma A431 cells was studied. Coimmunoprecipitation experiments indicated that EGF stimulated interaction between c-Jun and Sp1 in a time-dependent manner. Overexpression of Ha-ras and c-Jun also enhanced the amount of c-Jun binding to Sp1. In addition, the c-Jun dominant negative mutant TAM-67 not only inhibited the coimmunoprecipitated c-Jun binding to Sp1 in a dose-dependent manner in cells overexpressing c-Jun but also reduced promoter activity of the 12(S)-lipoxygenase gene induced by c-Jun overexpression. Treatment of cells with EGF increased the interaction between the Sp1 oligonucleotide and nuclear c-Jun/Sp1 in a time-dependent manner. Furthermore, EGF activated the chimeric promoter consisting of 10 tandem GAL4-binding sites, which replaced the three Sp1-binding sites in the 12(S)lipoxygenase promoter only when coexpressed with GAL4-c-Jun () fusion proteins. These results indicate that the direct interaction between c-Jun and Sp1 induced by EGF cooperatively activated expression of the 12(S)-lipoxygenase gene, and that Sp1 may serve at least in part as a carrier bringing c-Jun to the promoter, thus transactivating the transcriptional activity of 12(S)-lipoxygenase gene.
Arachidonate 12-lipoxygenase (arachidonate/oxygen 12-oxidoreductase; EC 1.13.11.31) in the platelet was the first mammalian lipoxygenase discovered (1). It catalyzes the transformation of arachidonic acid into 12(S)-hydroperoxyeicosatetraenoic acid and is converted subsequently to 12(S)-hydroxyeicosatetraenoic acid [12(S)-HETE] by a glutathione-dependent peroxidase (2). Human platelet-type 12(S)-lipoxygenase was also found in human erythroleukemia cells (3, 4), epidermal cells (5), and epidermoid carcinoma A431 cells (6). 12(S)-HETE may play a significant role in the pathogenesis of some epidermal and epithelial inflammation, because a markedly elevated 12(S)-HETE level was found in psoriatic plague (7). In psoriatic lesions, overexpression of epidermal growth factor (EGF) receptors (8), transforming growth factor-α (TGFα) (9), and 12(S)-lipoxygenase (10) has been reported.
In studying the regulation of 12(S)-lipoxygenase, we reported that EGF, TGFα, and phorbol 12-myristate 13-acetate (PMA) induce expression of human 12(S)-lipoxygenase in A431 cells (11–13). To study the signal transduction of EGF-induced expression of 12(S)-lipoxygenase, we reported recently that EGF induced expression of c-Jun through activation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) signaling and the activity of transcription factor activator protein 1 (AP1) in cells, and that overexpression of c-Jun increased expression of 12(S)-lipoxygenase mRNA (14). We therefore conclude that EGF-induced expression of 12(S)-lipoxygenase in A431 cells was mediated through the Ras-Raf-MEK-ERK and Ras-Rac-JNK signaling pathways. Subsequent induction of c-Jun led by ERK and JNK activation was essential for this EGF response. However, neither the known AP1-binding sequence in the promoter region responsive to EGF nor the apparent binding between transcription factor AP1 and EGF-responsive promoter DNA was observed (15, 16). Although two simian virus 40 promoter factor 1 (Sp1)-binding sites residing at −158 to −150 bp and −123 to −114 bp were essential in the mediation of EGF induction of the 12(S)-lipoxygenase gene, the EGF response was caused neither by the increase in Sp1 biosynthesis nor by the dephosphorylation of Sp1, because no change of the binding between nuclear Sp1 proteins and promoter DNA was observed in control and EGF-treated cells (16). We therefore speculated that c-Jun might interact with Sp1 protein, which then leads to activation of the 12(S)-lipoxygenase gene promoter (14).
In this study, the functional interaction between c-Jun and Sp1 on EGF treatment in A431 cells was studied. The functional role of the interaction between c-Jun and Sp1 in promoter activation of the 12(S)-lipoxygenase gene was then elucidated. Evidence obtained from this study shows directly that the interaction between c-Jun and Sp1 mediated EGF-induced gene expression of human 12(S)-lipoxygenase, and that Sp1 might function as a carrier bringing c-Jun to the promoter region, thus activating expression of the 12(S)-lipoxygenase gene. The same mechanism was also observed in the activation of 12(S)-lipoxygenase gene expression induced by PMA in our preliminary study.
Materials and Methods
Materials.
Mouse EGF (natural, culture grade) was purchased from Collaborative Research. [γ-32P]ATP (5,000 Ci/mmol) was purchased from Amersham Pharmacia. o-Nitrophenyl-β-galactopyranoside and (octylphenoxy)polyethoxyethanol (IGEPAL CA-630) were from Sigma. The luciferase assay system was from Promega. Qiagen-tip 100 was from Qiagen (Hilden, Germany). β-Galactosidase plasmid driven by cytomegalovirus (pCMVβ) was from CLONTECH. Monoclonal antibodies against c-Jun were obtained from Transduction Laboratories (Lexington, KY). Polyclonal antibodies against c-Jun and Sp1, protein A-agarose and agarose conjugated to Sp1 or c-Jun antibodies were from Santa Cruz Biotechnology. Lipofectamine, DMEM, and Opti-MEM media were obtained from Life Technologies (Grand Island, NY). pFR-Luc, pFA2-c-Jun and pFC2-dbd plasmids were purchased from Stratagene. Vector TAM-67 was kindly provided by Michael J. Birrer of the National Institutes of Health. FBS was from HyClone. All other reagents used were of the highest purity obtainable.
Cell Culture.
Human epidermoid carcinoma A431 cells were grown at 37°C under 5% CO2 in 10-cm plastic dishes containing 10 ml of DMEM supplemented with 10% FBS, 100 μg/ml streptomycin, and 100 units/ml penicillin. In this series of experiments, cells were treated with 50 ng/ml EGF in culture medium supplemented with 10% FBS, unless stated otherwise.
Western Blotting.
An analytical 10% SDS-polyacrylamide slab gel electrophoresis was performed. The cell nuclear extracts were prepared from control and EGF-treated cells, and 30 μg of protein of each was analyzed as previously described (16). For immunoblotting, proteins in the SDS gels were transferred to a polyvinylidene difluoride membrane by an electroblot apparatus. Antibodies against human Sp1 and c-Jun were used as primary antibodies. Immunoblot analysis was carried out with mouse or rabbit IgG antibody coupled to horseradish peroxidase. An enhanced chemiluminescence kit (Amersham) was used for detection. The density of the immunoblots was determined by an image analysis system installed with the software bio-id (Vildber Lourmat, Marne-la-Valée, France).
Coimmunoprecipitation.
Two hundred micrograms of nuclear extracts was incubated with 10 μl of either anti-Sp1 or anti-c-Jun antibody-agarose conjugate in 300 μl of immunoprecipitation buffer [20 mM Hepes, pH 7.9/420 mM NaCl/1.5 mM MgCl2/0.2 mM EDTA/25% glycerol (vol/vol)/0.5 mM phenylmethylsulfonyl/1 mM orthovanadate/2 μg/ml pepstatin A/2 μg/ml leupeptin] under gentle shaking at 4°C overnight. Beads were pelleted at 7,500 × g for 2 min and washed three times with RIPA buffer [50 mM Tris⋅HCl, pH 7.5/1% IGEPAL CA-630 (vol/vol)/150 mM NaCl/0.5% sodium deoxycholate]. Protein was removed from the beads by boiling in sample buffer (120 mM Tris⋅HCl, pH 6.8/10% glycerol/3% SDS/20 mM DTT/0.4% bromophenol blue) for 5 min and was subjected to SDS/PAGE on a 10% gel. Western blot analysis was carried out as described above.
Transfection with Lipofectamine and Reporter Gene Assay.
Cells were transfected with plasmids by lipofection by using Lipofectamine according to the manufacturer's instruction with a slight modification. A431 cells were replated 36 h before transfection at a density of 3 × 105 cells in 2 ml of fresh culture medium in a 3.5-cm plastic dish. For use in transfection, 12.5 μl of Lipofectamine was incubated with 0.5 μg of pXLO luciferase plasmid, 0.2 μg of β-galactosidase plasmid, or indicated plasmids, as described in each experiment, in 1 ml of Opti-MEM medium for 30 min at room temperature. Cells were transfected by changing the medium with 1 ml of Opti-MEM medium containing the plasmids and Lipofectamine and then incubated at 37°C in a humidified atmosphere of 5% CO2 for 24 h. After the change of Opti-MEM medium to 2 ml of fresh culture medium, cells were incubated for an additional 36 h, unless stated otherwise. The luciferase and β-galactosidase activities in cell lysate were determined as described previously (16).
Assay for Binding of the c-Jun/Sp1 Complex to Sp1 Consensus Sites.
Sp1 oligonucleotides, 5′-ATTCGATCGGGGCGGGGCGAGC-3′ and Sp1 mutant 5′-CTTGCGAGGAGTTATTTTGCCGCAGAC-3′ (SPM), were end labeled with [γ-32P]ATP and T4 polynucleotide kinase. The binding reaction was performed in 60 μl of reaction mixture containing 0.8 μg of poly(dI-dC).poly(dI-dC), 20 mM Hepes, pH 7.9, 0.1 mM KCl, 2 mM MgCl2, 15 mM NaCl, 0.2 mM EDTA, 5 mM DTT, 10% (vol/vol) glycerol, 2% (wt/vol) polyvinyl alcohol, 60 μg of the cell nuclear extracts, and the radiolabeled probe (2.5 × 106 cpm). The mixture was incubated at room temperature for 30 min and then incubated with 35 μl of anti-c-Jun antibody-agarose conjugate or protein A-agarose in 300 μl of immunoprecipitation buffer [20 mM Hepes, pH 7.9/420 mM NaCl/1.5 mM MgCl2/0.2 mM EDTA/25% glycerol (vol/vol)/0.5 mM phenylmethylsulfonyl/1 mM orthovanadate/2 μg/ml pepstatin A/2 μg/ml leupeptin] under gentle shaking at 4°C overnight. Beads were pelleted at 7,500 × g for 2 min and washed three times with RIPA buffer. Radioactivity in pelleted beads was determined by a scintillation counter.
Plasmid Construction.
For the generation of vector pXLO-8G containing GAL4-binding sites connected to the 12(S)-lipoxygenase gene promoter fragment (−100 bp) in vector pXLO-8 (15), the fragment of GAL4-binding sites was amplified by PCR by using pFR-Luc plasmid as template and the following primers: GAL4a, 5′-CGCGGATCCCATGCCTGCAGGT-3′ and GAL4b, 5′-CGCGGATCCTACCCTCTAGAGT-3′. PCR products containing ×10 GAL4-binding sites were cleaved with BamHI and inserted into the BamHI site in pXLO-8 to generate pXLO-8G. Sequence of the vector was confirmed by DNA sequencing. The vector contains ×10 GAL4-binding sites but no Sp1 consensus sequence.
Results
Coimmunoprecipitation of c-Jun and Sp1 in EGF-Treated Cells.
Expression of Sp1 and c-Jun in nuclear extracts prepared from cells treated with EGF for 1–9 h was studied by using immunoblot analysis. No difference of Sp1 expression between control and EGF-treated cells was observed (Fig. 1A). Expression of c-Jun was observed in cells treated with EGF for 1 h and was sustained at least up to 9 h after EGF treatment (Fig. 1B). Interaction between Sp1 and c-Jun was then studied by coimmunoprecipitation by using Sp1 antibodies. No change of the immunoprecipitated Sp1 between control and EGF-treated cells was observed (Fig. 1C). The coimmunoprecipitated c-Jun increased in a time-dependent manner in EGF-treated cells. As shown in Fig. 1D, a significant binding of c-Jun to Sp1 was observed in cells treated with EGF for 1 h. The binding significantly increased in cells treated with EGF for up to 9 h. To verify further the interaction between Sp1 and c-Jun, coimmunoprecipitation was performed by using c-Jun antibodies. As shown in Fig. 1E, the immunoprecipitated c-Jun was observed significantly in cells treated with EGF for 1 h and increased significantly up to 9 h of EGF treatment. An increase in coimmunoprecipitated Sp1 was observed similar to that of the immunoprecipitated c-Jun (Fig. 1 E and F), supporting the presumption that on EGF treatment, c-Jun interaction with Sp1 is enhanced.
Figure 1.

Time-dependent effect of EGF on the interaction between Sp1 and c-Jun in vivo. Confluent cells were starved for 24 h in serum-free culture medium before EGF treatment and then treated with EGF in culture medium without serum for a different time period, as indicated. Nuclear extracts were prepared and subjected to Western blotting by using antibodies specific for Sp1 and c-Jun (A and B). Nuclear extracts from EGF-treated cells were immunoprecipitated (IP) with antibodies (Ab) against Sp1 or c-Jun bound to protein A-agarose. Immunoprecipitates were subjected to 10% SDS/PAGE followed by Western blotting with antibodies against Sp1 (C and F) and c-Jun (D and E), respectively.
Coimmunoprecipitation of c-Jun and Sp1 in Cells Overexpressing c-Jun or Ha-ras.
Dose-dependent expression of c-Jun in cells was observed after transient transfection with a vector containing c-Jun (Fig. 2A). Interaction between c-Jun and Sp1 in c-Jun-transfected cells was then studied by coimmunoprecipitation by using Sp1 antibodies. As shown in Fig. 2B, no change of immunoprecipitated Sp1 between control and c-Jun-overexpressed cells was observed. However, coimmunoprecipitated c-Jun increased in a dose-dependent manner in cells overexpressing c-Jun. We previously reported that activation of the human 12(S)-lipoxygenase gene promoter by Ha-ras overexpression was similar to that induced by EGF (17). Therefore, the effect of Ha-ras overexpression on the interaction between c-Jun and Sp1 was studied by coimmunoprecipitation by using Sp1 antibodies. Transient transfection with the expression vector of Ras induced expression of c-Jun in a dose-dependent manner in cells (data not shown). An increase in interaction between c-Jun and Sp1 in a dose-dependent manner was observed in cells overexpressing Ras, as shown in Fig. 2C.
Figure 2.
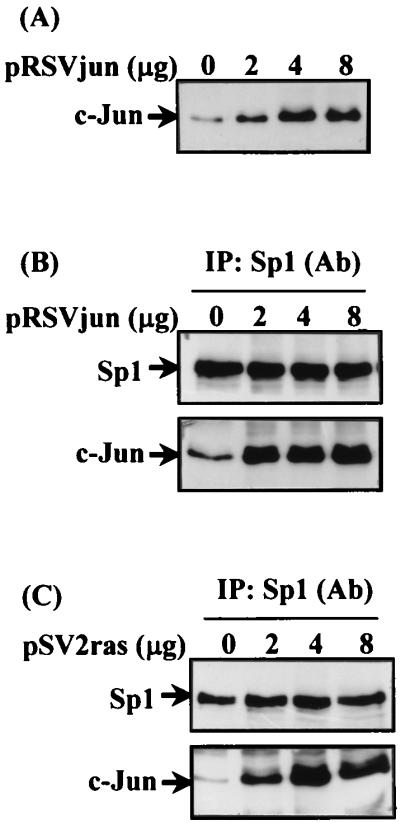
Binding of c-Jun and Sp1 in cells overexpressing c-Jun or Ha-ras. Cells were transfected with a different amount of pRSVjun or pSV2ras by the lipofection method. After the change of Opti-MEM medium to 3 ml of fresh culture medium in a 6-cm plastic dish, cells were incubated for an additional 36 h. Expression of c-Jun protein (A) and the coimmunoprecipitated c-Jun/Sp1 complex by using anti-Sp1 antibodies (B and C) was analyzed by Western blot with anti-c-Jun and anti-Sp1 antibodies.
Effect of c-Jun Dominant Negative Mutant on c-Jun/Sp1 Interaction and 12(S)-Lipoxygenase Promoter Activation in Cells Overexpressing c-Jun.
The expression vector of the c-Jun dominant negative mutant TAM-67 (18) was used to study whether interaction between c-Jun and Sp1 mediates activation of the 12(S)-lipoxygenase gene promoter. Dominant negative c-Jun expressed by vector TAM-67 is a 29-kDa protein, which is an N-terminal truncated protein of wild-type c-Jun. Its molecular mass is 10 kDa smaller than that of wild type. c-Jun and its dominant negative mutant TAM-67 are therefore distinguishable by Western analysis. As shown in Fig. 3A, the polyclonal antibodies of c-Jun recognized the expression of c-Jun and its dominant negative mutant TAM-67. Expression of the dominant negative mutant TAM-67 was in a dose-dependent manner in cells transfected with 2, 4, and 8 μg of dominant negative vector (Fig. 3A). Coimmunoprecipitation of Sp1 and c-Jun was performed by using Sp1 antibodies. As shown in Fig. 3B, cotransfection of the c-Jun dominant negative vector with the expression vector of c-Jun in cells inhibited the coimmunoprecipitated c-Jun with Sp1 in a dose-dependent manner, and concurrently the dominant negative mutant protein coimmunoprecipitated with Sp1 increased in a dose-dependent manner. Changes in c-Jun and in dominant mutant c-Jun coimmunoprecipitated with Sp1 correlated in a mirror-image fashion. These results indicate that the dominant negative mutant c-Jun induced inhibition of c-Jun/Sp1 interaction in cells overexpressing c-Jun by competing with c-Jun to interact with Sp1. The promoter activity of the 12(S)-lipoxygenase gene induced by c-Jun overexpression was also inhibited by the c-Jun dominant negative mutant in a dose-dependent manner, as shown in Fig. 4. The profile of the inhibitory effect on promoter activation (Fig. 4) was similar to that of the inhibitory effect on the interaction between c-Jun and Sp1 (Fig. 3B).
Figure 3.

Effect of c-Jun dominant negative mutant on c-Jun/Sp1 interaction in pRSVjun-treated cells. Cells were transfected with 2 μg of pRSVjun and a different amount of c-Jun dominant negative vector TAM-67 by the lipofection method. After the change of Opti-MEM medium to 3 ml of fresh culture medium in a 6-cm plastic dish, cells were incubated for an additional 36 h. Expression of c-Jun and TAM-67 proteins (A) and the coimmunoprecipitated c-Jun/Sp1 and TAM-67/Sp1 complex by using anti-Sp1 antibodies (B) was analyzed by Western blot with anti-c-Jun and anti-Sp1 antibodies. pcDNA3.1 was used as a vector to adjust for the same amount of plasmids in each experiment.
Figure 4.

Effect of c-Jun dominant negative mutant on the promoter activity of 12-lipoxygenase in pRSVjun-treated cells. Cells were transfected with 0.5 μg of luciferase plasmid pXLO-7–1 bearing 12(S)-lipoxygenase gene promoter (−224 bp), 0.2 μg of β-galactosidase, 0.5 μg of pRSVjun, and a different amount of TAM-67 by the lipofection method. After the change of Opti-MEM medium to 2 ml of fresh culture medium in a 3.5-cm plastic dish, cells were incubated for an additional 36 h. The luciferase and β-galactosidase activities were then determined. Values are means ± SEM of three determinations. pcDNA3.1 was used as a vector to adjust for the same amount of plasmids in each experiment.
Effect of EGF on the Interaction Between Sp1 Oligonucleotide and c-Jun/Sp1.
To study directly the binding of nuclear c-Jun/Sp1 with Sp1-binding sites in the 12(S)-lipoxygenase gene promoter, an immunoprecipitation method was developed in this study. An Sp1 oligonucleotide radiolabeled with 32P-ATP was incubated with cell nuclear extract. A complex of Sp1 oligonucleotide and c-Jun/Sp1 was immunoprecipitated with agarose bearing c-Jun antibodies, and the immunoprecipitated radiolabeled Sp1 oligonucleotide was measured. As shown in Fig. 5, treatment of cells with EGF increased interaction between the Sp1 oligonucleotide and nuclear c-Jun/Sp1 in a time-dependent manner. A significant effect was observed in cells treated with EGF for 1 h, and a maximum increase was found in cells treated with EGF for 3 h and was sustained up to 9 h after EGF treatment.
Figure 5.

Effect of EGF on the interaction between Sp1 oligonucleotides and c-Jun/Sp1. Nuclear extracts from EGF-treated cells were prepared and subjected to the assay for binding of the c-Jun/Sp1 complex to Sp1 consensus sites. The 32P-radiolabeled Sp1 oligonucleotide (●) and Sp1 mutant SPM (□) were used as a probe for binding, respectively. For background control, protein A-agarose was used to substitute anti-c-Jun antibody–agarose conjugate in the assay (■). Values are means ± SEM of three determinations.
Activation of a Chimeric Reporter Bearing GAL4-Binding Sites by GAL4-c-Jun.
Nuclear c-Jun/Sp1 binding to the Sp1 consensus sequence in the promoter seems to play a crucial role in transcriptional activation of 12(S)-lipoxygenase in EGF-treated cells, thus we studied the functional role of Sp1 in this process. Sp1 may function as a carrier bringing c-Jun to the gene promoter. To study this possibility, a chimeric promoter (pXLO-8G) consisting of 10 tandem GAL4-binding sites, which replace the three Sp1-binding sites in the promoter of 12(S)-lipoxygenase, was constructed as shown in Fig. 6A. It was then used to test promoter activation of pXLO-8G by a fusion protein consisting of GAL4 and the N terminus of c-Jun, which was overexpressed by vector pFA2-c-Jun through the mediation of binding between the GAL4 protein and its binding sites in the chimeric promoter of pXLO-8G. A vector pFC2-dbd that overexpresses only GAL4 protein was used as a negative control in this assay system. Results from this assay system are indicated in Fig. 6B. Overexpression of the fusion protein GAL4-c-Jun induced a 1-fold increase in the promoter activity of pXLO-8G compared with control cells. EGF treatment of cells activated the effect of fusion protein GAL4-c-Jun on the promoter activity of pXLO-8G in a time-dependent manner. Treatment of cells with 50 ng/ml EGF for 6, 12, and 18 h resulted in 25, 116, and 283% increases in the stimulatory effect of GAL4-c-Jun on the promoter activity. To study the essential role of binding between GAL4 and GAL4-binding sites of promoter in transactivation of GAL4-c-Jun, pFC2-dbd overexpressing only GAL4 protein was cotransfected with the expression vector of GAL4-c-Jun for the competition of binding to GAL4-binding sites. As shown in Fig. 6B, the promoter activity of vector pXLO-8G induced by GAL4-c-Jun overexpression in EGF-treated cells was inhibited by cotransfection with pFC2-dbd overexpressing GAL4 protein. These results indicate clearly that, in EGF-treated A431 cells, c-Jun recruited to the gene promoter through mediation between GAL4 of fusion protein and its binding sites in the chimeric promoter was also able to transactivate the promoter activity of vector pXLO-8G and to support the notion that Sp1 may serve at least in part as a carrier bringing c-Jun to the promoter to transactivate the transcriptional activity of the 12(S)-lipoxygenase gene.
Figure 6.

Transactivation of the 12(S)-lipoxygenase promoter consisting of ×10 GAL4-binding sites by GAL4-c-Jun in EGF-treated cells. Constructs of luciferase reporters and expression vectors used in these experiments are indicated (A). The transactivation effect of GAL4-c-Jun on a chimeric promoter bearing GAL4-binding sites was analyzed (B). Cells were transfected with 0.5 μg of pXLO-8G, 0.2 μg of β-galactosidase, 10 ng of pFA2-c-Jun, or 50 ng of pFC2dbd by the lipofection method, as indicated. After the change of Opti-MEM medium to 2 ml of fresh culture medium in a 3.5-cm plastic dish, cells were incubated for an additional 24 h and then treated with 50 ng/ml EGF. After 30 min of EGF treatment, the medium was removed, and the cells were further cultured in fresh culture medium for 6–18 h. Luciferase and β-galactosidase activities were then determined, respectively. Values are means ± SEM of three determinations.
Discussion
In this study, two pieces of evidence were provided to indicate that c-Jun induced by EGF activated gene expression of 12(S)-lipoxygenase by interacting with Sp1 in EGF-treated A431 cells. First, an interaction between c-Jun and Sp1 induced by EGF was observed. The amount of c-Jun interacting with Sp1 induced by EGF was proportional to the induction of c-Jun expression (Fig. 1), indicating that induction of c-Jun expression by EGF was a prerequisite for the following interaction between c-Jun and Sp1 in EGF-treated cells. This theory was supported further by studies with the overexpression vectors of c-Jun and Ha-ras. Overexpression of c-Jun and Ha-ras, respectively, in cells also enhanced the amount of c-Jun interacting with Sp1 (Fig. 2). Second, the dominant negative mutant of c-Jun blocked both the interaction between c-Jun and Sp1 and the activation of the 12(S)-lipoxygenase gene promoter in a similar fashion in cells overexpressing c-Jun (Figs. 3 and 4). These results indicated that direct interaction between c-Jun and Sp1 cooperatively activated expression of the 12(S)-lipoxygenase gene. We found previously that Sp1 binding to the two Sp1 consensus sites residing at −158 to −150 bp and −123 to −114 bp of the promoter is essential not only for EGF induction but also for basal expression of the 12(S)-lipoxygenase gene (16). Nevertheless, no change in binding between the nuclear Sp1 protein and the promoter DNA between control and EGF-treated cells was observed (16). In this report, we confirmed further that EGF treatment has no effect on the expression of Sp1 in A431 cells (Fig. 1A). Taking these results together, we conclude that Sp1 was a basic transcription factor required for expression of the 12(S)-lipoxygenase gene. However, it may not be a target protein directly modified by EGF-induced signaling in A431 cells.
Sp1 belongs to a zinc finger family of transcription factors that recognize the GC-rich DNA sequence (19), and it interacts with several transcription factors that bind to their respective response elements in the regulation of responsive genes. For example, YY1 (20), the p65 subunit of NFκB (21), GATA1 (22), and HLTF (23) have been shown to interact functionally with the zinc domain of Sp1. In the present studies, we found that interaction between c-Jun and Sp1 played a pivotal role in EGF-induced expression of the 12(S)-lipoxygenase gene. Because no AP1-binding site is present in the gene promoter of 12(S)-lipoxygenase (15), the activation mechanism of the 12(S)-lipoxygenase gene is therefore different from the genes regulated by a transcription factor that binds to its own responsive element, followed by interaction with Sp1 in the gene promoter. Our present results clearly indicated the nuclear c-Jun biosynthetically induced by EGF in A431 cells interacted directly with Sp1 in the activation of 12(S)-lipoxygenase expression by EGF. Another important finding from this study is the demonstration of an increase in binding of nuclear c-Jun/Spl to the Sp1-binding oligonucleotide in cells on EGF treatment (Fig. 5). This evidence supports strongly our theory that recruitment of c-Jun through direct interaction with Sp1 to the gene promoter may play a pivotal role in the transactivation of 12(S)-lipoxygenase on EGF treatment. Kardassis et al. reported recently that overexpression of c-Jun transactivates the human p21WAF1 gene, which carries TATA box in the promoter, in HepG2 cells via a short proximal region with multiple binding sites of Sp1 but without the AP1-binding site on it (24). Their results suggest that a mechanism of activation of Sp1 by c-Jun is based on the physical and functional interaction between these two transcription factors. In the present report, we presented more concrete evidence to indicate that, in EGF-treated cells, there is direct interaction between c-Jun and Sp1 as shown by coimmunoprecipitation and an increase in binding of nuclear c-Jun/Sp1 to Sp1-binding oligonucleotide with the sequence present in the 12(S)-lipoxygenase gene promoter. Our results demonstrated that in the transcriptional regulation of a gene that has no TATA box in the promoter, an interaction between c-Jun and Sp1 was required functionally to activate gene expression. In addition to EGF, treatment of A431 cells with PMA also stimulated expression of human 12(S)-lipoxygenase (13). We reported previously that activation of the human 12(S)-lipoxygenase gene promoter by PMA was similar to that induced by EGF (25). Recently, we also found that PMA treatment induced expression of c-Jun mainly through activation of the Raf-MEK-ERK signaling pathway and partially through that of the Ras-Raf-MEK-ERK signaling pathway and then induced interaction between c-Jun and Sp1 in the same fashion as that induced by EGF, indicating that protein kinase C activation could also induce interaction between c-Jun and Sp1 (our unpublished data).
c-Jun contains a C-terminal basic region leucine zipper DNA-binding domain and an N-terminal transactivation domain, and its transactivation activity can be regulated through phosphorylation of clusters of serine/threonine residues in N terminus (26, 27). Two major serine phosphoacceptor sites have been identified within the N-terminal domain at Ser-63 and Ser-73 that are activated through ERK and JNK signaling (28, 29). The C-terminal domain at Thr-231 and Ser-249 has been found to be phosphorylated by activation of casein kinase II, which is a constitutive protein kinase (30). It is noteworthy that phorbol ester and overexpression of Ras cause maximum activation of c-Jun by promoting both phosphorylation of the N-terminal sites and dephosphorylation of the C-terminal sites, resulting in a rapid and significant increase in the activity of this key transcription factor (31–33). Another interesting piece of evidence obtained from this study is the demonstration that recruitment of c-Jun to the gene promoter through the binding between GAL4 of fusion protein GAL4-c-Jun and GAL4-binding sites in the chimeric promoter with no Sp1-binding sites on it was also able to stimulate reporter activity, and that reporter activity was enhanced by EGF treatment in a time-dependent fashion (Fig. 6B). This EGF response was similar to that of the EGF effect on promoter activation of 12(S)-lipoxygenase (34). These results support the notion that recruitment of c-Jun to the promoter was a crucial step in EGF-induced gene activation of 12(S)-lipoxygenase, and that Sp1 might function as a carrier in this process. Enhancement of reporter activity in this chimeric promoter system by EGF treatment, as shown in Fig. 6B, might be because of the increase in transactivation activity of c-Jun in fusion protein GAL4-c-Jun through N terminus phosphorylation. Because the C terminus was reported to be the binding domain of c-Jun for interaction with Sp1 by using an in vitro binding assay (24), it will therefore be interesting to investigate the functional role of dephosphorylation of c-Jun at Thr-231 and Ser-249 of the C-terminal domain in the interaction between c-Jun and Sp1 in gene activation of 12(S)-lipoxygenase induced by EGF and PMA in the intact cell.
AP1 is formed by either the homodimer of c-Jun or the heterodimer of c-Jun and c-Fos. We reported previously that, in the treatment of A431 cells with EGF, the expression of both c-Jun and c-Fos was induced, but the induction of c-Jun was more significant than that of c-Fos protein (14). The maximum induction of c-Jun was observed in cells treated with EGF and then sustained for at least up to 9 h after EGF treatment (Fig. 1B). The kinetic changes in the formation of the nuclear c-Jun/Sp1 complex (Fig. 1 C and D), and in the binding of the nuclear c-Jun/Sp1 complex to the Sp1-binding oligonucleotide (Fig. 5) were similar to the induction of c-Jun biosynthesis (Fig. 1B). The maximum induction in these three parameters was observed in cells treated with EGF for 1 h and then sustained up to at least 9 h after EGF treatment. This evidence strongly supports the functional role of interaction between c-Jun and Sp1 in EGF-induced expression of the 12(S)-lipoxygenase gene promoter. The functional role of c-Fos in EGF-induced expression of 12(S)-lipoxygenase was not as evident as that of c-Jun. Although a slight induction of promoter activity of 12(S)-lipoxygenase was observed in cells overexpressing c-Fos, it was only one-eighth of the response of c-Jun overexpression (14, 35). In contrast to the long-term expression of c-Jun, the maximum expression of c-Fos was observed in cells treated with EGF for 1 h, but the induction then declined and almost disappeared in cells treated with EGF for 6 h (14). A plateau interaction between nuclear c-Jun and Sp1 was still present in cells treated with EGF for 6 and 9 h (Fig. 1). Therefore, the induction of c-Jun might contribute more than that of c-Fos in EGF-induced expression of 12(S)-lipoxygenase and thus c-Fos may play little functional role in the interaction between nuclear c-Jun and Sp1 in the activation of the 12(S)-lipoxygenase gene promoter in A431 cells treated with EGF.
Epidermal keratinocyte hyperproliferation is a major component of the psoriatic phenotype. TGFα, which is the natural ligand for EGF receptors present on keratinocytes, is overexpressed in psoriatic skin (9). The increased production of TGFα may be an important contribution to keratinocyte hyperplasia, because TGFα is a potent mitogen for keratinocytes (9). Application of phorbol ester to mouse skin also induces psoriasiform hyperplasia (36). Our results in this series of studies indicated that all EGF, TGFα, and PMA induced expression of 12(S)-lipoxygenase in human epidermoid cells and provided a mechanism to explain the formation of 12(S)-HETE in psoriatic lesion. In an in vivo study, unequivocal growth promotion in addition to the inflammation reaction was observed on 12(S)-HETE activation in guinea-pig skin (37). Taking these results together, the high concentration of 12(S)-HETE because of induction of 12(S)-lipoxygenase expression may contribute at least in part to abnormal epidermal hyperproliferation and inflammatory changes in the development of a psoriatic plaque.
Results from the present study, together with our previous reports, provided an example in an intact cell system to demonstrate how c-Jun, induced by extracellular stimulators such as growth factors, cytokines, or stress signals, by cooperatively interacting with Sp1, which binds to the Sp1-binding sites of the gene promoter, was able to participate directly in the regulation of some housekeeping genes lacking TATA box in the promoter. The signal transduction pathway of EGF-induced gene expression of 12(S)-lipoxygenase in A431 cells can be summarized as follows. The induction of Ras by EGF is mediated by EGF receptors binding to Grb2 and Sos (38). EGF efficiently activated Ras-Raf-MEK-ERK and Ras-Rac-JNK pathways, which then activate AP1. Biosynthesis of c-Jun was enhanced, subsequently interacted with Sp1, and transactivated promoter activation of the 12(S)-lipoxygenase gene, followed by an increase in mRNA transcription and enzyme translation of 12(S)-lipoxygenase. Our studies delineated the mechanisms by which EGF mediates transcriptional activation of 12(S)-lipoxygenase in epidermal cells. Induction of the interaction between c-Jun and Spl may be an important link between the overexpression of EGF receptors and TGFα and 12(S)-lipoxygenase expression in human psoriatic lesions.
Acknowledgments
We are greatly indebted to Drs. Shozo Yamamoto (Tokushima University, Tokushima, Japan), Hsia-Sheng Liu (National Cheng Kung University, Tainan, Taiwan), Hsin-fang Yang-Yen (Academia Sinica, Taipei, Taiwan), and Michael J. Birrier (National Institutes of Health) for providing plasmids pXLO, pSV2ras, pRSVjun, and TAM-67, respectively. Thanks are also due to Drs. H. S. Liu, B. C. Yang, C. C. Lu, T. H. Leu and W. M. Kan for their valuable discussions, and to Dr. T. P. Su for a critical review of the manuscript. The secretarial assistance of Y. L. Chang is also appreciated. This work was supported in part by grants NSC 87–2314-B-006–105, NSC 87–2314-B-006–025 and NSC 88–2314-B-006–001 from the National Science Council of the Republic of China.
Abbreviations
- 12(S)-HETE
12(S)-hydroxyeicosatetraenoic acid
- EGF
epidermal growth factor
- PMA
phorbol 12-myristate 13-acetate
- ERK
extracellular signal-regulated kinase
- JNK
c-Jun N-terminal kinase
- AP1
activator protein 1
- Sp1
simian virus 40 promoter factor 1
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.180321497.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.180321497
References
- 1.Hamberg M, Samuelsson B. Proc Natl Acad Sci USA. 1974;71:3400–3404. doi: 10.1073/pnas.71.9.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang W C, Nakao J, Orimo H, Murota S. Biochem J. 1982;202:771–776. doi: 10.1042/bj2020771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Funk C D, Furic L, FitzGerald G A. Proc Natl Acad Sci USA. 1990;87:5638–5642. doi: 10.1073/pnas.87.15.5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Izumi T, Hoshiko S, Radmark O, Samuelssson B. Proc Natl Acad Sci USA. 1990;87:7477–7481. doi: 10.1073/pnas.87.19.7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takahashi Y, Reddy G R, Ueda N, Yamamoto S, Arase S. J Biol Chem. 1993;268:16443–16448. [PubMed] [Google Scholar]
- 6.Chang W C, Liu Y W, Ning C C, Suzuki H, Yoshimoto T, Yamamoto S. J Biol Chem. 1993;268:18734–18739. [PubMed] [Google Scholar]
- 7.Hammarstrom S, Hamberg M, Samuelsson B, Duell E A, Stawiski M, Voorhees J J. Proc Natl Acad Sci USA. 1975;72:5130–5134. doi: 10.1073/pnas.72.12.5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nanney L B, Stoscheck C M, Magid M, King L E. J Invest Dermatol. 1986;86:260–265. doi: 10.1111/1523-1747.ep12285389. [DOI] [PubMed] [Google Scholar]
- 9.Elder J T, Fisher G J, Lindguist P B, Bennett G L, Pittelkow M R, Coffey R J, Ellingsworth L, Derynck R, Voorhees J J. Science. 1989;243:811–814. doi: 10.1126/science.2916128. [DOI] [PubMed] [Google Scholar]
- 10.Hussain H, Sornick L P, Shannon V R, Wilson J D, Funk C D, Pentland A P, Holtzman M J. Am J Physiol. 1994;266:C243–C253. doi: 10.1152/ajpcell.1994.266.1.C243. [DOI] [PubMed] [Google Scholar]
- 11.Chang W C, Ning C C, Lin M T, Huang J D. J Biol Chem. 1992;267:3657–3666. [PubMed] [Google Scholar]
- 12.Chen L C, Chen B K, Liu Y W, Chang W C. FEBS Lett. 1999;455:105–110. doi: 10.1016/s0014-5793(99)00865-0. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y W, Asaoka Y, Suzuki H, Yoshimoto T, Yamamoto S, Chang W C. J Pharmacol Exp Ther. 1994;271:567–573. [PubMed] [Google Scholar]
- 14.Chen B K, Kung H C, Tsai T Y, Chang W C. Mol Pharmacol. 2000;57:153–161. [PubMed] [Google Scholar]
- 15.Yoshimoto T, Arakawa T, Hada T, Yamamoto S, Takahashi E. J Biol Chem. 1992;267:24805–24809. [PubMed] [Google Scholar]
- 16.Liu Y W, Arakawa T, Yamamoto S, Chang W C. Biochem J. 1997;324:133–140. doi: 10.1042/bj3240133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen B K, Liu Y W, Yamamoto S, Chang W C. Biochim Biophys Acta. 1997;1344:270–277. doi: 10.1016/s0005-2760(96)00151-8. [DOI] [PubMed] [Google Scholar]
- 18.Dong Z, Crawford H C, Lavrovsky V, Taub D, Watts R, Matrisian L M, Colburn N H. Mol Carcinog. 1997;19:204–212. doi: 10.1002/(sici)1098-2744(199707)19:3<204::aid-mc8>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 19.Berg J M. Proc Natl Acad Sci USA. 1992;89:11109–11110. doi: 10.1073/pnas.89.23.11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee J S, Galvin K M, Shi Y. Proc Natl Acad Sci USA. 1993;90:6145–6149. doi: 10.1073/pnas.90.13.6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perkins N D, Agranoff A B, Pascal E, Nabel G J. Mol Cell Biol. 1994;14:6570–6583. doi: 10.1128/mcb.14.10.6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merika M, Orkin S H. Mol Cell Biol. 1995;15:2437–2447. doi: 10.1128/mcb.15.5.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding H, Benotmane A M, Suske G, Collen D, Belayew A. J Biol Chem. 1999;274:19573–19580. doi: 10.1074/jbc.274.28.19573. [DOI] [PubMed] [Google Scholar]
- 24.Kardassis D, Papakosta P, Pardali K, Moustakas A. J Biol Chem. 1999;274:29572–29581. doi: 10.1074/jbc.274.41.29572. [DOI] [PubMed] [Google Scholar]
- 25.Liaw Y W, Liu Y W, Chen B K, Chang W C. Biochim Biophys Acta. 1997;1389:23–33. doi: 10.1016/s0005-2760(97)00090-8. [DOI] [PubMed] [Google Scholar]
- 26.Abate C, Curran T. Semin Cancer Biol. 1990;1:19–26. [PubMed] [Google Scholar]
- 27.Karin M, Liu Zg, Zandi E. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 28.Pulverer B J, Kyriakis J M, Avruch J, Nikolakaki E, Woodgett J R. Nature (London) 1991;353:670–674. doi: 10.1038/353670a0. [DOI] [PubMed] [Google Scholar]
- 29.May G H, Allen K E, Clark W, Funk M, Gillespie D A. J Biol Chem. 1998;273:33429–33435. doi: 10.1074/jbc.273.50.33429. [DOI] [PubMed] [Google Scholar]
- 30.Lin A, Frost J, Deng T, Smeal T, al-Alawi N, Kikkawa U, Hunter T, Brenner D, Karin M. Cell. 1992;70:777–789. doi: 10.1016/0092-8674(92)90311-y. [DOI] [PubMed] [Google Scholar]
- 31.Boyle W J, Smeal T, Defize L H, Angel P, Woodgett J R, Karin M, Hunter T. Cell. 1991;64:573–584. doi: 10.1016/0092-8674(91)90241-p. [DOI] [PubMed] [Google Scholar]
- 32.Binetruy B, Smeal T, Karin M. Nature (London) 1991;351:122–127. doi: 10.1038/351122a0. [DOI] [PubMed] [Google Scholar]
- 33.Smeal T, Binetruy B, Mercola D A, Birrer M, Karin M. Nature (London) 1991;354:494–496. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- 34.Liu Y W, Chen B K, Chen C J, Arakawa T, Yoshimoto T, Yamamoto S, Chang W C. Biochim Biophys Acta. 1997;1344:38–46. doi: 10.1016/s0005-2760(96)00128-2. [DOI] [PubMed] [Google Scholar]
- 35.Chen B K, Chang W C. Biochem Biophys Res Commun. 1999;261:848–852. doi: 10.1006/bbrc.1999.1114. [DOI] [PubMed] [Google Scholar]
- 36.Horn F, Marks F, Fisher G J, Marcelo C L, Voorhees J J. J Invest Dermatol. 1987;88:220–222. doi: 10.1111/1523-1747.ep12525380. [DOI] [PubMed] [Google Scholar]
- 37.Chan C C, Duhame L, Ford-Hutchinson A W. J Invest Dermatol. 1985;85:333–334. doi: 10.1111/1523-1747.ep12276933. [DOI] [PubMed] [Google Scholar]
- 38.Chardin P, Camonis J J, Gale N W, Aelst L, van Schlessinger J, Wigler M H, Bar-Sagi D. Science. 1993;260:1338–1343. doi: 10.1126/science.8493579. [DOI] [PubMed] [Google Scholar]