

Abstract
Recently, biofilms have become a topic of interest in the study of the human pathogen group A Streptococcus (GAS). In this study, we sought to learn more about the make-up of these structures and gain insight into biofilm regulation. Enzymic studies indicated that biofilm formation by GAS strain MGAS5005 required an extracellular protein and DNA component(s). Previous results indicated that inactivation of the transcriptional regulator Srv in MGAS5005 resulted in a significant decrease in virulence. Here, inactivation of Srv also resulted in a significant decrease in biofilm formation under both static and flow conditions. Given that production of the extracellular cysteine protease SpeB is increased in the srv mutant, we tested the hypothesis that increased levels of active SpeB may be responsible for the reduction in biofilm formation. Western immunoblot analysis indicated that SpeB was absent from MGAS5005 biofilms. Complementation of MGAS5005Δsrv restored the biofilm phenotype and eliminated the overproduction of active SpeB. Inhibition of SpeB with E64 also restored the MGAS5005Δsrv biofilm to wild-type levels.
INTRODUCTION
Group A Streptococcus (GAS) is responsible for a wide range of human infections, including pharyngitis and tonsillitis, skin infections, sepsis, a toxic-shock syndrome, and necrotizing fasciitis (Cunningham, 2000; Musser & Krause, 1998; Reid et al., 2001). Until recently, there was little association of GAS and biofilms in the published literature. One of the first observations was reported by Akiyama et al. (2003) in a study examining GAS in skin infections. Confocal laser scanning microscopy of skin lesions revealed the presence of GAS microcolonies that appeared to be surrounded by a glycocalyx (Akiyama et al., 2003). A limited number of subsequent studies have demonstrated GAS biofilm formation in vitro and in vivo and microarray analyses have shown that gene transcription varies markedly in biofilms compared to planktonically grown GAS (Baldassarri et al., 2006; Cho & Caparon, 2005; Conley et al., 2003; Lembke et al., 2006; Manetti et al., 2007; Riani et al., 2007; Takemura et al., 2004). However, little is known about whether the ability to form biofilms is a characteristic of all GAS strains, what the biofilm matrix is composed of, or how this biofilm state is regulated.
In the present work, we demonstrate that a globally disseminated M1T1 serotype clone associated with invasive disease is capable of producing a structured, surface-attached community resembling a biofilm. In addition, we demonstrate that DNA and protein are critical to the formation of this structure. Furthermore, in an effort to gain insight into the regulation of the biofilm process, we examined the effects of loss of the virulence regulator Srv on GAS biofilm formation. Srv has been shown to be required for GAS virulence and to influence GAS gene expression (Reid et al., 2004, 2006). Here we show that Srv is required for biofilm formation and that increased activity of the cysteine protease SpeB in the absence of Srv contributes to the loss of the biofilm phenotype.
METHODS
Bacterial strains
MGAS5005 is a representative M1T1 serotype strain (speA+) which was isolated from a case of invasive GAS disease and has been the focus of numerous studies (reviewed by Musser & Krause, 1998; Reid et al., 2001). The MGAS5005 isogenic srv mutant strain (MGAS5005Δsrv) was generated by allelic replacement (Reid et al., 2004). MGAS5005Δsrv was complemented in trans [MGAS5005Δsrv(pIAβ8-srv)] as previously described (Doern et al., 2008).
Adherence assay
Overnight cultures of GAS grown at 37 °C (5 % CO2) in Todd–Hewitt Broth (TH) (Difco) supplemented with 0.2 % yeast extract (THY) (Fisher Scientific) were harvested and used to inoculate fresh THY. Cultures were then grown to an OD600 of 0.5. Tissue culture treated polystyrene six-well cell culture plates (Corning) were seeded with 3 ml of culture per well. Plates were incubated for 24 h at 37 °C, 5 % CO2. Medium was removed without disturbing the biofilm and wells were washed three times with distilled H2O (dH2O). Then 1 ml aliquots of 0.1 % crystal violet (CV) (Sigma-Aldrich) dissolved in dH2O were dispensed to each well. Surface-attached bacteria were allowed to stain for 30 min at room temperature and then washed three times with dH2O, after which 1 ml ethanol was added to each well to solubilize the CV. An A600 reading (measuring the colour intensity of the solubilized CV) was recorded for each sample. If staining was too dark for spectrophotometric analysis, the sample was diluted with additional ethanol and the dilution factor applied to the resulting reading (in these instances, the value is referred to as the A600 extrapolated, or A600e).
Enzymic inhibition/disruption of biofilms
Methods were adapted from Wang et al. (2004). To test the ability to inhibit biofilm formation, 2 ml aliquots of exponentially growing GAS cultures (OD600 0.5) were mixed with 2 ml of one of the following reagents (final concn): 40 mM NaIO4 (metaperiodate), 200 μg DNase I ml−1 or 1 mg proteinase K ml−1. Enzyme-treated cultures were added to six-well tissue culture plates and incubated for 24 h at 37 °C, 5 % CO2. Wells were washed and stained with CV as described above. To test the ability to disrupt a pre-formed biofilm, each of the reagents described above was added to a 24-h-old biofilm and allowed to incubate for an additional 1 h. Wells were washed and stained as before.
Analysis of biofilm formation under continuous-flow conditions
Using a 25 gauge 1½ inch Precision Glide needle (Becton Dickinson), a 10 ml aliquot of exponentially growing GAS culture (OD600 0.5) was inoculated into a convertible flow-cell chamber (Stovall Life Sciences). Each inoculum was incubated for 3 h at 37 °C without flow to allow initial attachment within the chamber. Sterile THY medium was connected to the flow chamber through the pump head via Masterflex silicon tubing (peroxide treated) L/S14 (Cole-Parmer Instruments), and flow discharge was collected in a waste container downstream of the flow-cell chamber. Flow was initiated using a Masterflex Easy-Load II peristaltic pump (Cole-Parmer Instruments) with a flow rate of 0.7 ml min−1 and continued for 24 h at 37 °C.
Confocal laser scanning microscopy of flow-cell biofilms
To visually examine the health of the biofilm, samples were subjected to live/dead staining. Samples contained within flow-cell chambers were washed with PBS. A mixture of SYTO 9 and propidium iodide (LIVE/DEAD BacLight Bacterial Viability kit L7007; Molecular Probes) contained in 10 ml PBS was added to the chamber. The sample was allowed to incubate at room temperature for 1 h with the LIVE/DEAD stain and then washed with PBS. Samples were visualized using a Zeiss LSM510 confocal scanning laser microscope.
Scanning electron microscopy of flow-cell biofilms
Continuous flow-cell samples were fixed within the flow-cell chamber with 2.5 % glutaraldehyde in PBS. After fixation for a minimum of 1 h, samples were washed twice and subjected to dehydration, fixation, and critical-point drying for scanning electron microscopy. Flow-cell chamber slides were then trimmed, mounted, and coated with palladium. Biofilms were viewed on a Phillips SEM-515 scanning electron microscope.
RESULTS
Protein and DNA are significant components of the GAS biofilm
We first determined the level of biofilm formed by MGAS5005, a representative serotype M1T1 strain recently isolated from a case of invasive GAS disease. Population genetic analysis has indicated that M1T1 strains are among the most common causes of invasive GAS infections worldwide in most case studies (Musser & Krause, 1998; Reid et al., 2001). Dilution of the solubilized CV was necessary as the original CV staining was too dark for spectrophotometric analysis.
We next sought to learn more about the biofilm structure by examining the components present. We hypothesized that if proteins, DNA or polysaccharides are required, then biofilms should be inhibited or disrupted by proteinase K, DNase I, or metaperiodate, respectively. Addition of proteinase K or DNase I at the time six-well plates were seeded significantly inhibited biofilm formation (Fig. 1a). In addition, a 1 h incubation with either proteinase K or DNase I disrupted an existing 24 h old biofilm (Fig. 1b). Treatment with metaperiodate, a compound capable of oxidizing polysaccharides (Mack et al., 1996; Maira-Litran et al., 2002; Wang et al., 2004), led to a reduction in both the formation and the stability of the biofilm, but measurable biofilm was still present (Fig. 1a, b). As a control, we performed replicate plating to test the viability of the bacteria following treatment. C.f.u. numbers recovered following treatment with proteinase K or DNase I were equivalent to those following treatment with PBS (~109 c.f.u. ml−1). However, we recovered 106–107 c.f.u. ml−1 from metaperiodate-treated samples. This suggested that the decrease in biofilm observed may be due to the effects of metaperiodate on bacterial viability. Thus, the GAS biofilm requires a protein and a DNA component(s) for formation and stability. GAS polysaccharides may increase the mass of the biofilm when present, but periodate-sensitive polysaccharides do not appear to be required for GAS biofilm formation.
Fig. 1.

Enzymic inhibition or disruption of GAS biofilm formation. (a) To test the ability of specific enzymes to inhibit biofilm formation, proteinase K, DNase I or metaperiodate was added when the six-well plates were initially seeded with MGAS5005 in the exponential phase (OD600 0.5). After 24 h growth, strains incubated with proteinase K or DNase I produced significantly less, if any, biofilm. While metaperiodate curtailed biofilm formation, it did not abolish this ability. Each reported value is the mean±SD of at least six replicates and is adjusted by the dilution factor required to obtain a spectrophotometric reading (A600 extrapolated or A600e). (b) To test the ability of the same enzymes to disrupt an established GAS biofilm, proteinase K, DNase I or metaperiodate was added to cultures grown in six-well plates for 24 h. CV assays were completed after 1 h incubation with enzyme. Treatment with proteinase K or DNase I disrupted the biofilm and yielded a significantly reduced A600e. Treatment with metaperiodate reduced the amount of biofilm present, but was unable to completely disrupt the biofilm.
Inactivation of the transcriptional regulator Srv results in a significant reduction in GAS biofilm formation
The observation that extracellular protein is important in the formation of the GAS biofilm led us to consider the possible role of the transcriptional regulator Srv. Our previous studies indicated that inactivation of srv resulted in a significant reduction in GAS virulence (Reid et al., 2004) and differential transcription of 51 genes encoding proven or putative extracellular proteins (Reid et al., 2006). The reduced expression of genes encoding extracellular proteins in the srv mutant strain led us to hypothesize that Srv may contribute to biofilm formation. Comparison of MGAS5005 and the srv isogenic mutant derivative MGAS5005Δsrv indicated that MGAS5005Δsrv produced significantly less biofilm (Fig. 2a). Complementation of srv in trans [MGAS5005Δsrv(pIAβ8-srv)] restored biofilm production to near wild-type levels (Fig. 2a).
Fig. 2.
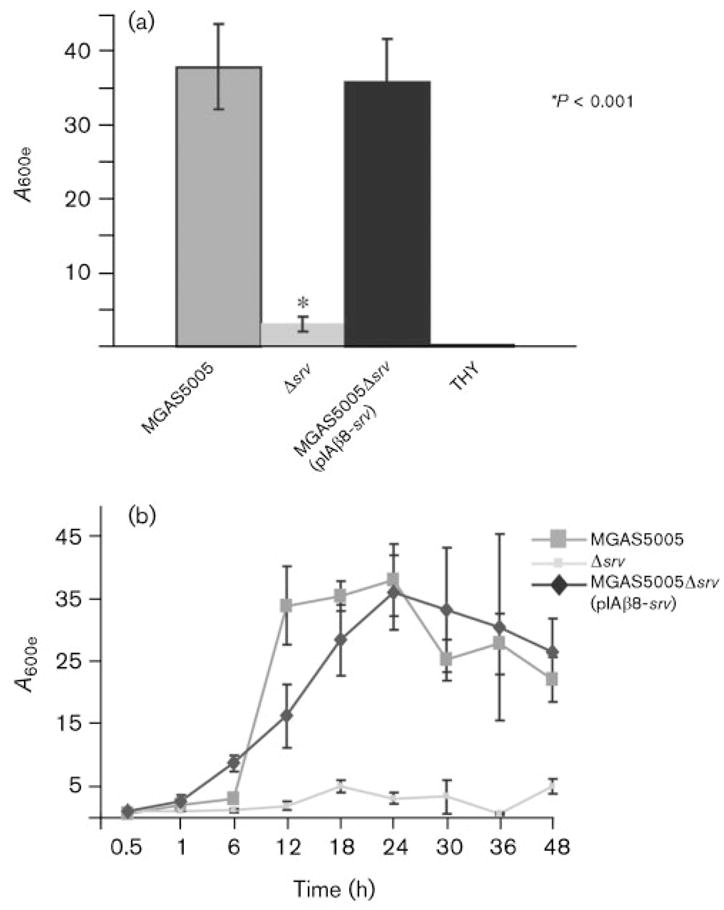
The transcriptional regulator Srv is required for maximal biofilm formation by MGAS5005. Each reported value for the CV assay is an average of at least six replicates and is adjusted by the dilution factor required to obtain a spectrophotometric reading (A600e). (a) A comparison of the wild-type (MGAS5005), the isogenic mutant lacking srv (Δsrv), and the complemented strain [MGAS5005Δsrv(pIAβ8-srv)]. The Δsrv mutant is significantly reduced in its ability to form biofilms. Complementation of srv in trans restores biofilm formation. The mean A600 (extrapolation unnecessary) for wells containing THY alone is provided as a negative control. (b) Kinetics of GAS biofilm formation. The A600e values of solubilized CV from surface-attached cells grown in six-well polystyrene plates were assayed over time.
To determine if the results observed were due to an impaired ability of MGAS5005Δsrv to form biofilms or an early disruption of the biofilm, we followed formation of the structure over time. MGAS5005 and MGAS5005Δsrv(pIAβ8-srv) produced biofilm at approximately the same rate and to the same level (Fig. 2b). Maximal biofilm formation was observed at 24 h, with levels tapering off thereafter. In contrast, CV staining of surface-attached MGAS5005Δsrv was significantly less at all time points measured (Fig. 2b).
Inhibition of SpeB in MGAS5005Δsrv restores biofilm formation
Our data suggested that Srv-mediated gene transcription was required for proper biofilm formation. This may be due to reduced transcription of genes encoding extra-cellular proteins required for attachment and/or aggregation, or to the effects of some other product that is directly or indirectly affected by Srv. Previously, we discovered that inactivation of srv led to increased production of the GAS cysteine protease SpeB (Reid et al., 2006). We can visualize the increased activity of SpeB in a casein agar assay, in which strains are stab inoculated into casein agar and allowed to grow at 37 °C under microaerophilic conditions. The resulting zone of translucence surrounding the stab site is indicative of SpeB caseinolytic activity. This assay demonstrated that complementation with srv in trans restored SpeB activity to wild-type levels (Fig. 3).
Fig. 3.
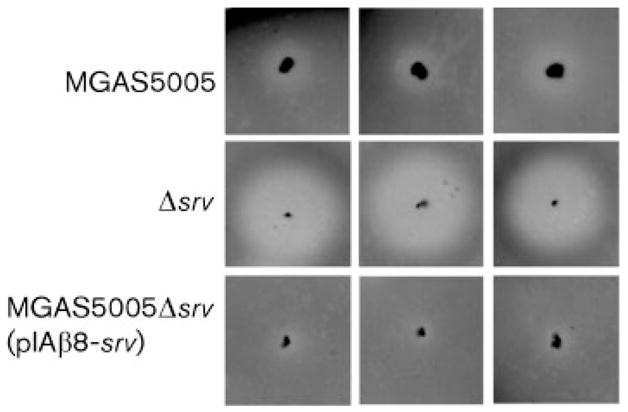
Casein agar assay for SpeB proteolytic activity. MGAS5005, Δsrv and MGAS5005Δsrv(pIAβ8-srv) were stab inoculated into agar plates containing skim milk and incubated for 18 h microaerophilically. Protease activity is manifest as a zone of translucence surrounding the stab sites. There is a marked increase in the amount of SpeB proteolysis exhibited by Δsrv compared to MGAS5005 or MGAS5005Δsrv(pIAβ8-srv).
To test the hypothesis that an increase in SpeB may be responsible for the MGAS5005Δsrv biofilm defect, we examined biofilm formation in the presence of an inhibitor of SpeB activity, E64. E64 is a potent and highly selective cysteine protease inhibitor that irreversibly binds to an active thiol group to form a thioether linkage. Previous groups have used concentrations of ~30 μM to 10 mM E64 (Kansal et al., 2003; Vincents et al., 2004). However, in these studies the inhibitor was added to assays using GAS supernatants or purified proteins. Since we wished to add E64 to living cells and allow the culture to incubate for 24 h, we elected to use a final concentration of 333 μM. Addition of E64 at the time the plates were seeded restored the ability of MGAS5005Δsrv to form biofilms (Fig. 4a). To determine that the observed results were due to the inhibition of SpeB and not another protease, we examined the biofilm formation of a speB isogenic mutant (Lukomski et al., 1998) with and without the addition of E64 (Fig. 4a). No difference was observed in the ability of the ΔspeB strain to form biofilms. Examination of the attached bacteria prior to solubilization with ethanol revealed that the CV stain was not as uniform in E64-treated MGAS5005Δsrv as in the treated wild-type, but there was a clear increase in attached bacteria compared to untreated MGAS5005Δsrv (Fig. 4b).
Fig. 4.
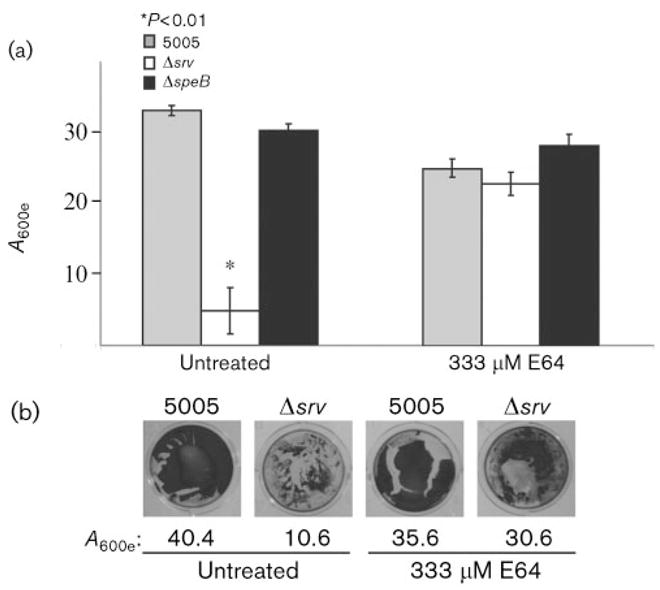
Inhibition of SpeB activity restores GAS biofilm formation. (a) Graphical representation of CV assays of biofilm-forming ability in the presence and absence of the cysteine protease inhibitor E64. While addition of E64 may reduce the biofilm capacity of the wild-type slightly, biofilm formation of the Δsrv strain is restored to comparable levels. E64 did not alter biofilm formation by the ΔspeB isogenic mutant strain, suggesting that the alteration in biofilm phenotype is due to SpeB activity. The data are means±SD of at least six replicates. (b) Single-well images of CV assays prior to solubilization in ethanol. After image capture, the CV from surface-attached cells was solubilized and the A600e values are shown. Note the increase in stained material in the Δsrv well in the presence of E64.
Extracellular SpeB is absent from 24 h GAS biofilms
Previously, we demonstrated that SpeB was detected by Western immunoblotting in culture supernatants of MGAS5005 following 8 h of planktonic growth (Reid et al., 2006). By contrast, SpeB was detectable in MGAS5005Δsrv supernatants after just 2 h of growth (Reid et al., 2006). For the experiments presented above, cultures were allowed to grow planktonically for 3 h prior to seeding of the six-well plates. Biofilms were then allowed to develop for 24 h. If SpeB inhibits or disperses the GAS biofilm, then it must be present at very low levels if at all in the biofilm. To test this hypothesis, GAS biofilms and their supernatants were collected, treated as a single sample (biofilm and supernatant together), and probed by Western immunoblot analysis. Abundant SpeB was detected in the sample from the biofilm-deficient MGAS5005Δsrv (Fig. 5). However, SpeB was undetectable in MGAS5005 samples, and only trace amounts of SpeB-zymogen (inactive) were detected in MGAS5005Δsrv(pIAβ8-srv) samples (Fig. 5). As a negative control, samples from MGAS5005ΔspeB were probed (Fig. 5) (Lukomski et al., 1998).
Fig. 5.

Detection of SpeB in GAS biofilms. Western immunoblot analysis failed to detect the presence of SpeB in supernatants collected from MGAS5005 and MGAS5005ΔspeB after growth for 24 h in a biofilm state. Elevated levels of SpeB were detected in supernatants collected from MGAS5005Δsrv. Low levels of the inactive form of SpeB (zymogen) were detected in supernatants collected from the srv-complemented strain MGAS5005Δsrv(pIAβ8-srv).
Analysis of GAS biofilm formation using continuous flow
Research has indicated that the growth and structures of bacterial biofilms are affected by hydrodynamic shear that may be generated by the flow of fluids over epithelial surfaces in the host (Lappin-Scott & Bass, 2001; Purevdorj et al., 2002; Stoodley et al., 2001). While our data indicate that GAS is capable of forming dense, surface-attached communities under static conditions, we sought to determine the ability of GAS to form biofilms under continuous flow. The convertible flow-cell chamber was inoculated with 10 ml of the wild-type, the srv mutant or the complemented strain, harvested at OD600 0.5, and the bacteria were allowed to attach for 3 h at 37 °C. Flow of fresh THY medium was then initiated at a rate of ~0.7 ml min−1, and the chamber was incubated for an additional 24 h. After 24 h of growth, the MGAS5005 and the MGAS5005Δsrv(pIAβ8-srv) flow chambers were completely filled with a viscous material (Fig. 6a). Plating of a sample of this material revealed GAS and no contaminants (data not shown). As observed under static conditions, MGAS5005Δsrv was largely unable to form a biofilm (Fig. 6a). Small pockets of attachment were observed for MGAS5005Δsrv, but these pockets were washed away when the chamber was flushed with PBS in preparation for microscopic analyses. Taken together, these data and our analysis of static biofilms over time (Fig. 2b) suggested that MGAS5005Δsrv was deficient in attachment.
Fig. 6.

Continuous-flow system for study of GAS biofilm formation. (a) Inactivation of srv eliminates the ability of GAS to form biofilms under continuous-flow conditions. Complementation of srv in trans [MGAS5005Δsrv(pIAβ8-srv)] restores the biofilm phenotype. (b) Confocal microscopy of a 24 h continuous-flow biofilm. The image is an X–Y micrograph of MGAS5005 biofilm stained with LIVE/DEAD reagent. Cells with intact membranes (living) are stained green while cells with disrupted membranes are stained red. Areas of overlap appear yellow. Fifty X–Y plane micrographs indicate that the biofilm is approximately 80 μm thick. Vertical stacked Z images (margins) show living cells dispersed throughout the macroscopic structure. (c, d) Scanning electron microscopy of a 24 h continuous-flow biofilm; (c) shows the thickness of the biofilm formed within the chamber. At high magnification (d), chains of GAS can be clearly seen. Note the matrix material that appears to cover the bacterial chains. White arrows indicate the presence of fibrous strands.
To determine the viability of cells within the biofilm, the MGAS5005 within the flow-cell was stained with a commercial reagent (LIVE/DEAD, Molecular Probes) and visualized by confocal microscopy (Fig. 6b). By this method, living cells with intact membranes are stained green with SYTO 9 while dead cells are stained red with propidium iodide. Colocalization of both signals gives a yellow colour. After 24 h, the majority of cells in the biofilm were alive. Fifty consecutive micrographs of an X–Y plane indicated that the biofilm was approximately 80 μm thick. Vertical stacked Z images (margins of Fig. 6b) showed living cells dispersed throughout the macroscopic structure.
Electron microscopy revealed a densely packed population of MGAS5005 that appeared to be encased in an extracellular matrix (Fig. 6c, d). An angled image of the biofilm removed from the corner of the flow chamber revealed the three-dimensional nature of the structure (Fig. 6c). Higher magnification allowed the visualization of chains of bacteria which appeared to be coated in matrix (Fig. 6d). Fibrous strands could be seen running within the matrix (Fig. 6d, white arrows). Some of these appeared to be sheared, probably during the fixation process.
DISCUSSION
In a recent study, Baldassarri et al. (2006) examined a total of 289 GAS isolates from different clinical sources and demonstrated that 90 % of those strains were capable of forming biofilms. In this investigation, we sought to learn more about the make-up of these structures and gain insight into biofilm regulation. Enzymic treatment of GAS biofilms demonstrated a requirement for DNA and protein in the structure, but suggested only a passive role for carbohydrate. This is in contrast to numerous systems, for example Pseudomonas aeruginosa, for which several genetically encoded polysaccharides have been shown to be required (Donlan, 2001; Donlan & Costerton, 2002; Hall-Stoodley et al., 2004; Ryder et al., 2007). Cho & Caparon (2005) found that the GAS hyaluronic acid capsule (HasA) was not required for biofilm formation in a static system (CV assay), but that following initial attachment, the hasA mutant was unable to aggregate under flow conditions. Given the volume of enzyme that would be required, we were unable to test inhibition or disruption under flow conditions. Thus, hyaluronic acid does not appear necessary for initial biofilm attachment, but it may be needed for aggregation and biofilm maturation. Alternatively, differences between the serotype M1 strain used here and the Cho M5 strain may account for the observed differences. Further assessment in vivo will address these possibilities.
Increasing evidence suggests not only that DNA is a major component of bacterial biofilms, but that genetically encoded systems controlling programmed cell death provide a mechanism for the release of bacterial DNA (reviewed by Bayles, 2007). Therefore, it is not surprising that we demonstrated a role for DNA in GAS biofilms. However, the process by which GAS releases DNA is not yet known. GAS does not appear to encode cid/lrg homologues which control programmed cell death in other bacterial species, although the presence of additional or diverged genes encoding holins cannot be ruled out.
DNA, or more appropriately the lack of extracellular DNA, has been linked to severe GAS infection. Evidence has been presented that MGAS5005 has a mutation within the GAS control of virulence operon (covRS/csrRS) which limits or abolishes speB expression. This is hypothesized to allow the production of a GAS extracellular DNase (Sda1), a protein that would normally be degraded by SpeB (Buchanan et al., 2006; Sumby et al., 2005; Walker et al., 2007). According to this model, Sda1 facilitates the escape from neutrophil extracellular traps (NETs) by degrading the associated neutrophil DNA (Brinkmann et al., 2004; Walker et al., 2007). The infecting strain is then free to disseminate and potentially cause severe invasive disease (Sumby et al., 2006; Walker et al., 2007). Our data indicate that DNA is required for MGAS5005 biofilm formation in vitro. This suggests that production of SdaI is not solely dependent on the absence of SpeB, but that it requires a signal encountered in vivo. This idea is in concert with the data of Sumby et al. (2006), who found naturally occurring examples of the covS mutation following passage in a mouse model.
It is of note that our data also indicate that inactivation of srv in MGAS5005 compensates for the covS mutation and restores SpeB production in this strain. In addition, our data indicate that there is a necessary protein component for initial adherence and/or aggregation leading to biofilm maturation. We hypothesized that the high levels of SpeB may be responsible for the biofilm-deficient phenotype of the srv mutant. Addition of the cysteine protease inhibitor E64 restored the ability of MGAS5005Δsrv to form biofilms, suggesting that SpeB degrades GAS proteins needed for establishment of the biofilm (E64-treated MGAS5005ΔspeB produced wild-type levels of biofilm). This leads to the hypothesis that the timed production of SpeB might contribute to GAS biofilm dispersion. Under this model, GAS begins in a biofilm state. In reaction to an unknown stimulus, an alteration in Srv-mediated control occurs and results in the direct or indirect increase of SpeB production and/or secretion. SpeB degrades GAS and host proteins integral to the biofilm. This alone may disperse the biofilm or, as put forth by others, the increased activity of and resulting damage from SpeB is perceived as a signal (in addition to host signals) which drives selection for mutations in covS (Sumby et al., 2006; Walker et al., 2007). Inactive CovS leads to the repression of speB, induction of sdaI, and the degradation of host and bacterial DNA (which may be more accessible due to the prior cleavage of proteins by SpeB). GAS bacteria are now free to disperse and potentially adopt an invasive phenotype (Sumby et al., 2006; Walker et al., 2007). While this model is preliminary, we believe it is testable. Clearly, further study of GAS biofilm formation will provide new insights into the pathogenesis of GAS.
Acknowledgments
This project was supported by Public Health Service Grant R01AI063453 from the National Institutes of Health to S. D. R. We thank D. J. Wozniak for critical reading of the manuscript as well as Robert C. Holder for technical assistance.
Abbreviations
- CV
crystal violet
- GAS
group A Streptococcus
References
- Akiyama H, Morizane S, Yamasaki O, Oono T, Iwatsuki K. Assessment of Streptococcus pyogenes microcolony formation in infected skin by confocal laser scanning microscopy. J Dermatol Sci. 2003;32:193–199. doi: 10.1016/s0923-1811(03)00096-3. [DOI] [PubMed] [Google Scholar]
- Baldassarri L, Creti R, Recchia S, Imperi M, Facinelli B, Giovanetti E, Pataracchia M, Alfarone G, Orefici G. Therapeutic failures of antibiotics used to treat macrolide-susceptible Streptococcus pyogenes infections may be due to biofilm formation. J Clin Microbiol. 2006;44:2721–2727. doi: 10.1128/JCM.00512-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayles KW. The biological role of death and lysis in biofilm development. Nat Rev Microbiol. 2007;5:721–726. doi: 10.1038/nrmicro1743. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, Feramisco J, Nizet V. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol. 2006;16:396–400. doi: 10.1016/j.cub.2005.12.039. [DOI] [PubMed] [Google Scholar]
- Cho KH, Caparon MG. Patterns of virulence gene expression differ between biofilm and tissue communities of Streptococcus pyogenes. Mol Microbiol. 2005;57:1545–1556. doi: 10.1111/j.1365-2958.2005.04786.x. [DOI] [PubMed] [Google Scholar]
- Conley J, Olson ME, Cook LS, Ceri H, Phan V, Davies HD. Biofilm formation by group A streptococci: is there a relationship with treatment failure? J Clin Microbiol. 2003;41:4043–4048. doi: 10.1128/JCM.41.9.4043-4048.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham MW. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev. 2000;13:470–511. doi: 10.1128/cmr.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doern CD, Holder RC, Reid SD. Point mutations within the streptococcal regulator of virulence (Srv) alter protein–DNA interactions and Srv function. Microbiology. 2008;154:1998–2007. doi: 10.1099/mic.0.2007/013466-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlan RM. Biofilm formation: a clinically relevant microbiological process. Clin Infect Dis. 2001;33:1387–1392. doi: 10.1086/322972. [DOI] [PubMed] [Google Scholar]
- Donlan RM, Costerton JW. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev. 2002;15:167–193. doi: 10.1128/CMR.15.2.167-193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- Kansal RG, Nizet V, Jeng A, Chuang WJ, Kotb M. Selective modulation of superantigen-induced responses by streptococcal cysteine protease. J Infect Dis. 2003;187:398–407. doi: 10.1086/368022. [DOI] [PubMed] [Google Scholar]
- Lappin-Scott HM, Bass C. Biofilm formation: attachment, growth, and detachment of microbes from surfaces. Am J Infect Control. 2001;29:250–251. doi: 10.1067/mic.2001.115674. [DOI] [PubMed] [Google Scholar]
- Lembke C, Podbielski A, Hidalgo-Grass C, Jonas L, Hanski E, Kreikemeyer B. Characterization of biofilm formation by clinically relevant serotypes of group A streptococci. Appl Environ Microbiol. 2006;72:2864–2875. doi: 10.1128/AEM.72.4.2864-2875.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukomski S, Burns EH, Jr, Wyde PR, Podbielski A, Rurangirwa J, Moore-Poveda DK, Musser JM. Genetic inactivation of an extracellular cysteine protease (SpeB) expressed by Streptococcus pyogenes decreases resistance to phagocytosis and dissemination to organs. Infect Immun. 1998;66:771–776. doi: 10.1128/iai.66.2.771-776.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack D, Fischer W, Krokotsch A, Leopold K, Hartmann R, Egge H, Laufs R. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear β-1,6-linked glucosaminoglycan: purification and structural analysis. J Bacteriol. 1996;178:175–183. doi: 10.1128/jb.178.1.175-183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maira-Litran T, Kropec A, Abeygunawardana C, Joyce J, Mark G, III, Goldmann DA, Pier GB. Immunochemical properties of the staphylococcal poly-N-acetylglucosamine surface polysaccharide. Infect Immun. 2002;70:4433–4440. doi: 10.1128/IAI.70.8.4433-4440.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manetti AG, Zingaretti C, Falugi F, Capo S, Bombaci M, Bagnoli F, Gambellini G, Bensi G, Mora M, et al. Streptococcus pyogenes pili promote pharyngeal cell adhesion and biofilm formation. Mol Microbiol. 2007;64:968–983. doi: 10.1111/j.1365-2958.2007.05704.x. [DOI] [PubMed] [Google Scholar]
- Musser JM, Krause RM. The revival of group A streptococcal diseases, with a commentary on staphylococcal toxic shock syndrome. In: Krause RM, editor. Emerging Infections. Academic Press; 1998. pp. 185–218. [Google Scholar]
- Purevdorj B, Costerton JW, Stoodley P. Influence of hydrodynamics and cell signaling on the structure and behavior of Pseudomonas aeruginosa biofilms. Appl Environ Microbiol. 2002;68:4457–4464. doi: 10.1128/AEM.68.9.4457-4464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid SD, Hoe NP, Smoot LM, Musser JM. Group A Streptococcus: allelic variation, population genetics, and host–pathogen interactions. J Clin Invest. 2001;107:393–399. doi: 10.1172/JCI11972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid SD, Montgomery AG, Musser JM. Identification of srv, a PrfA-like regulator of group A Streptococcus that influences virulence. Infect Immun. 2004;72:1799–1803. doi: 10.1128/IAI.72.3.1799-1803.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid SD, Chaussee MS, Doern CD, Chaussee MA, Montgomery AG, Sturdevant DE, Musser JM. Inactivation of the group A Streptococcus regulator srv results in chromosome wide reduction of transcript levels, and changes in extracellular levels of Sic and SpeB. FEMS Immunol Med Microbiol. 2006;48:283–292. doi: 10.1111/j.1574-695X.2006.00150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riani C, Standar K, Srimuang S, Lembke C, Kreikemeyer B, Podbielski A. Transcriptome analyses extend understanding of Streptococcus pyogenes regulatory mechanisms and behavior toward immunomodulatory substances. Int J Med Microbiol. 2007;297:513–523. doi: 10.1016/j.ijmm.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Ryder C, Byrd M, Wozniak DJ. Role of polysaccharides in Pseudomonas aeruginosa biofilm development. Curr Opin Microbiol. 2007;10:644–648. doi: 10.1016/j.mib.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoodley P, Jacobsen A, Dunsmore BC, Purevdorj B, Wilson S, Lappin-Scott HM, Costerton JW. The influence of fluid shear and AICI3 on the material properties of Pseudomonas aeruginosa PAO1 and Desulfovibrio sp. EX265 biofilms. Water Sci Technol. 2001;43:113–120. [PubMed] [Google Scholar]
- Sumby P, Barbian KD, Gardner DJ, Whitney AR, Welty DM, Long RD, Bailey JR, Parnell MJ, Hoe NP, et al. Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc Natl Acad Sci U S A. 2005;102:1679–1684. doi: 10.1073/pnas.0406641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog. 2006;2:e5. doi: 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura N, Noiri Y, Ehara A, Kawahara T, Noguchi N, Ebisu S. Single species biofilm-forming ability of root canal isolates on gutta-percha points. Eur J Oral Sci. 2004;112:523–529. doi: 10.1111/j.1600-0722.2004.00165.x. [DOI] [PubMed] [Google Scholar]
- Vincents B, von Pawel-Rammingen U, Björck L, Abrahamson M. Enzymatic characterization of the streptococcal endopeptidase, IdeS, reveals that it is a cysteine protease with strict specificity for IgG cleavage due to exosite binding. Biochemistry. 2004;43:15540–15549. doi: 10.1021/bi048284d. [DOI] [PubMed] [Google Scholar]
- Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, McArthur JD, Dinkla K, Aziz RK, et al. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med. 2007;13:981–985. doi: 10.1038/nm1612. [DOI] [PubMed] [Google Scholar]
- Wang X, Preston JF, III, Romeo T. The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J Bacteriol. 2004;186:2724–2734. doi: 10.1128/JB.186.9.2724-2734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]