

Abstract
A highly efficient novel traceless solid-phase synthesis of 3,4-dihydropyrazino[1,2-b]indazoles and their 6-oxides was developed by using commercially available building blocks, diamines, 2-nitrobenzenesulfonyl chlorides, and bromoketones/bromoacetates. Mild reaction conditions, diversely substituted building blocks and high purity of crude products enabled effective combinatorial syntheses of libraries.
Introduction
Our ongoing research interest focuses on the development of efficient chemical transformations yielding high purity and yield of diverse drug-like heterocyclic compounds. At the same time, we prioritize chemical routes that use building blocks with diverse side-chains that are commercially available or can be prepared in a straightforward manner (for our recently published solid-phase syntheses cf. references1-4).
We attempted to prepare polymer-supported α-acylamino ketones via N-alkylation of resin-bound nitrobenzenesulfonyl (Nos) activated/protected amines by bromoketones (a variant of the Fukuyama method.5). We observed a striking difference in the reaction outcome between 2-Nos and 4-Nos derivatives (Scheme 1). The 4-Nos group was cleanly removed by traditional thiol/base (2-mercaptoethanol/DBU) reagent solution. Unexpectedly, the same treatment of 2-Nos derivatives led to tandem carbon-carbon followed by nitrogen-nitrogen bonds formation and yielded indazole oxides of excellent purity.4 Here, we describe the extension of this tandem reaction for traceless solid-phase synthesis of pyrazino[1,2-b]indazoles.
Scheme 1.
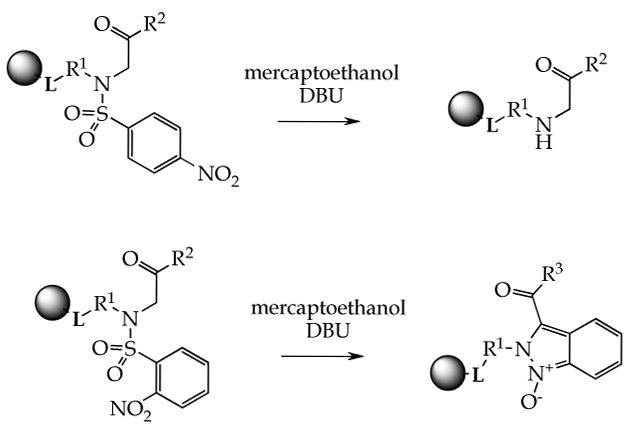
Different reactivities of 2- and 4-nitrobenzenesulfonylamides
To our knowledge, synthesis of 3,4-dihydropyrazino[1,2-b]indazole was not reported. Tapolcsányi et al.6 observed the formation of pyrazino[1,2-b]indazole by thermal cyclization of 2-(2-azidophenyl)pyrazine, prepared via Suzuki coupling of 2-chloropyrazine with 2-substituted phenylboronic acid (Scheme 2).
Scheme 2.
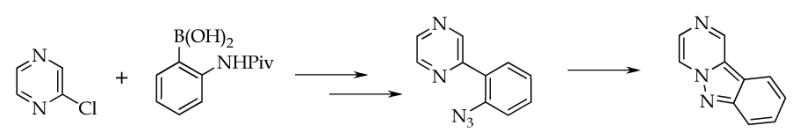
Synthesis of pyrazino[1,2-b]indazole6
Pyrido[1,2-b]indazole skeleton (formally a “carba analog” of pyrazino[1,2-b]indazole) was present in fused tetracyclic derivatives, such as indazolo[2,3-b]isoquinoline, prepared by Timári et al.7 from isoquinoline-3-O-triflate via Suzuki coupling. This planar heteroaromatic compound, an analog of ellipticine, was found to be an effective inhibitor of reverse transcriptase.7 An isomer, indazolo[2,3-a]quinoline, was synthesized by Csányi et al.8 from 2-(2-azidophenyl)quinoline, in an analogous manner to the route portrayed in Scheme 2. Compounds composed from four fused rings and comprising pyrido[1,2-b]indazole motif were found to be potent antibacterial,9 antiinflammatory,10 and antiplasmodial11 agents.
3,4-Dihydropyrazino[1,2-b]indazoles can be regarded as 2,3-substituted-2H-indazoles with an additional ring closed between substituents at positions 2 and 3. Synthetic compounds comprising indazole core have recently become an increasingly frequent subject of biological studies: indazole derivatives possessed significant potency in a wide range of biological targets. A recently published excellent review article by Cerecetto and colleagues12 portrayed the diversity of biological activities exhibited by indazoles.
Results and Discussion
To carry out the traceless solid-phase synthesis of pyrazino[1,2-b]indazole we took advantage of our recently discovered tandem reaction that transformed N-alkyl-2-nitro-N-(2-oxo-2-aryl-ethyl)-benzenesulfonamides to 2H-indazoles (Scheme 1).4 The target indazoles contained a carbonyl functionality attached to the side-chain of substituent at position 2 of the indazole ring. Its presence enabled to close a fused pyrazine ring via intramolecular nucleophile located on the R1 side-chain.
Briefly, 1,2-diamines were attached to Wang resin13 via carbamate linkage using carbonyldiimidazole (CDI) activation14,15 and provided resin 1 (Scheme 3). Symmetric as well as asymmetric 1,2-diamines were used. The reaction of CDI-treated Wang resin with 1,2-diaminopropane yielded predominantly (∼95%) one isomer, and the diamine was immobilized via the amino-group attached to the primary carbon. The structure was unequivocally determined at the stage of the target compound by 2D NMR experiments. Polymer-supported primary amines 1 were reacted with four different 2-nitrobenzenesulfonyl chlorides (Figure 1) to yield sulfonamides 2. To evaluate the sensitivity of the reaction outcome towards electronic properties, we used 2-nitrobenzenesulfonyl chlorides with electron-donating as well as electron-withdrawing groups and all substitutions were tolerated. Resin-bound sulfonamides 2 were alkylated with five different bromoketones/bromoacetates to yield intermediates 3. While alkylation of non-substituted sulfonamides and sulfonamides bearing electron-withdrawing group on benzene ring in the para position was completed after overnight reaction, alkylation of sulfonamides bearing electron-donating group required 2 days. DBU-mediated cyclization cleanly afforded resin-bound indazole oxides 4. At this stage the resins were split and one half of resin-bound indazole oxides were deoxygenated using methanesulfonyl chloride in the presence of triethylamine4,16 to provide polymer-supported 2H-indazoles 5.
Scheme 3.

Synthesis of 3,4-dihydropyrazino[1,2-b]indazolesa
aReagents and conditions: (i) CDI, substituted ethylenediamine, pyridine, DCM, 5 h; (ii) 2-nitrobenzenesulfonyl chloride, lutidine, DCM, overnight; (iii) bromoketone/bromoacetate, DIEA, DMF, overnight; (iv) DBU, DMF, 30 min; (v) methanesulfonyl chloride, TEA, DCM, overnight; (vi) 50% TFA in DCM, 1 h; (vii) AcOH, 2 h; (viii) TEA, MeOH, overnight
Figure 1.
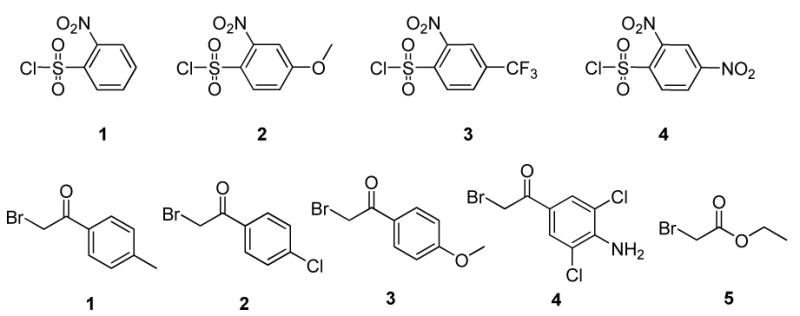
2-Nitrobenzenesulfonyl chlorides and bromoketones/bromoacetates used for the synthesis.
Cleavage from the resins 4 using trifluoroacetic acid (TFA) in dichloromethane (DCM) provided acyclic intermediates 6. Most of the compounds spontaneously cyclized to 3,4-dihydropyrazino[1,2-b]indazole 6-oxides 7. Indazoles 6 that did not spontaneously cyclize were dissolved and cyclized in AcOH (Table 1). Cleavage from the resins 5 using TFA in DCM provided directly 3,4-dihydropyrazino[1,2-b]indazoles 8. Compound 5(1,1,5) prepared using ethyl bromoacetate as an alkylating agent, provided, after cleavage from resin, acyclic intermediates 9 which were cyclized by triethylamine (TEA) in methanol (MeOH) to yield 3,4-dihydropyrazino[1,2-b]indazol-1(2H)-one 10(1,1,5).
Table 1.
Synthesized compounds 7, 8, 10
| Product | R1 | R2 | R3 | Cyclization | Puritya [%] |
Isolation | Yieldb [%] |
Deox [days] |
MS ESI+ |
|---|---|---|---|---|---|---|---|---|---|
| 7(1,1,1) | H | H | 4-Me-Ph | TFA | 99 | B | 72 | - | 278 |
| 7(1,1,2) | H | H | 4-Cl-Ph | TFA | 99 | B | 43 | - | 298 |
| 7(1,1,3) | H | H | 4-OMe-Ph | TFA | 99 | B | 50 | - | 294 |
| 7(1,1,4) | H | H | 4-NH2-3,5-diCl-Ph | AcOH | 99 | A | 96 | - | 347 |
| 7(1,2,1) | H | 4-OMe | 4-Me-Ph | TFA | 85 | C | 16 | - | 308 |
| 7(1,2,2) | H | 4-OMe | 4-Cl-Ph | TFA | 56 | C | 16 | - | 328 |
| 7(1,2,3) | H | 4-OMe | 4-OMe-Ph | AcOH | 88 | C | 20 | - | 324 |
| 7(1,2,4) | H | 4-OMe | 4-NH2-3,5-diCl-Ph | AcOH | 67 | C | 20 | - | 377 |
| 7(1,3,1) | H | 4-CF3 | 4-Me-Ph | TFA | 99 | B | 78 | - | 346 |
| 7(1,3,2) | H | 4-CF3 | 4-Cl-Ph | TFA | 99 | B | 62 | - | 366 |
| 7(1,3,3) | H | 4-CF3 | 4-OMe-Ph | AcOH | 99 | A | 81 | - | 362 |
| 7(1,3,4) | H | 4-CF3 | 4-NH2-3,5-diCl-Ph | AcOH | 99 | A | 58 | - | 415 |
| 7(1,4,1) | H | 4-NO2 | 4-Me-Ph | TFA | 97 | B | 60 | - | 323 |
| 7(1,4,2) | H | 4-NO2 | 4-Cl-Ph | TFA | 99 | B | 59 | - | 343 |
| 7(1,4,3) | H | 4-NO2 | 4-OMe-Ph | AcOH | 99 | A | 75 | - | 339 |
| 7(1,4,4) | H | 4-NO2 | 4-NH2-3,5-diCl-Ph | AcOH | 99 | A | 67 | - | 392 |
| 7(2,1,1) | CH3 | H | 4-Me-Ph | TFA | 90 | C | 78 | - | 292 |
| 8(1,1,1) | H | H | 4-Me-Ph | TFA | 90 | B | 54 | 2 | 262 |
| 8(1,1,2) | H | H | 4-Cl-Ph | TFA | 99 | B | 60 | 1 | 282 |
| 8(1,1,3) | H | H | 4-OMe-Ph | TFA | 99 | B | 34 | 1 | 278 |
| 8(1,1,4) | H | H | 4-NH2-3,5-diCl-Ph | TFA | 93 | B | 99 | 1 | 331 |
| 8(1,2,1) | H | 4-OMe | 4-Me-Ph | TFA | 71 | C | 21 | 1 | 292 |
| 8(1,2,2) | H | 4-OMe | 4-Cl-Ph | TFA | 47 | C | 86 | 3 | 312 |
| 8(1,2,3) | H | 4-OMe | 4-OMe-Ph | TFA | 46 | C | 36 | 1 | 308 |
| 8(1,2,4) | H | 4-OMe | 4-NH2-3,5-diCl-Ph | TFA | 52 | C | 19 | 1 | 361 |
| 8(1,3,1) | H | 4-CF3 | 4-Me-Ph | TFA | 99 | A | 12 | 1 | 330 |
| 8(1,3,2) | H | 4-CF3 | 4-Cl-Ph | TFA | 96 | B | 61 | 3 | 350 |
| 8(1,3,3) | H | 4-CF3 | 4-OMe-Ph | TFA | 99 | B | 64 | 2 | 346 |
| 8(1,3,4) | H | 4-CF3 | 4-NH2-3,5-diCl-Ph | TFA | 99 | B | 98 | 3 | 399 |
| 8(1,4,1) | H | 4-NO2 | 4-Me-Ph | TFA | 93 | B | 56 | 4 | 307 |
| 8(1,4,2) | H | 4-NO2 | 4-Cl-Ph | TFA | 90 | B | 7 | 5 | 327 |
| 8(1,4,3) | H | 4-NO2 | 4-OMe-Ph | TFA | 98 | B | 56 | 2 | 323 |
| 8(1,4,4) | H | 4-NO2 | 4-NH2-3,5-diCl-Ph | TFA | 90 | B | 12 | 2 | 376 |
| 10(1,1,5) | H | H | NA | TEA/MeOH | 66 | C | 33 | 1 | 188 |
Purity of the crude product before purification;
Yield after purification, NA, not applicable
All library compounds were submitted for evaluation of biological activities to High Throughput Screening in the Molecular Libraries Probe Production Centers Network. The results are available in PubChem (http://pubchem.ncbi.nlm.nih.gov/).
Substituted ethylenediamines used in the first combinatorial step enabled to close six membered dihydropyrazine ring. Attempts to incorporate seven-membered diazepine ring were successful when 1,3-diaminopropane and ethyl bromoacetate were used as building blocks. The 2,3,4,5-tetrahydro-1H-[1,4]diazepino[1,2-b]indazol-1-one 11 was obtained after cyclization of the corresponding indazole precursor in TEA containing MeOH at 60 °C, although in a low yield (Scheme 4). Compounds prepared using bromoketones as alkylating agents were not stable; we observed an equilibrium between acyclic and cyclic (Schiff base) forms.
Scheme 4.
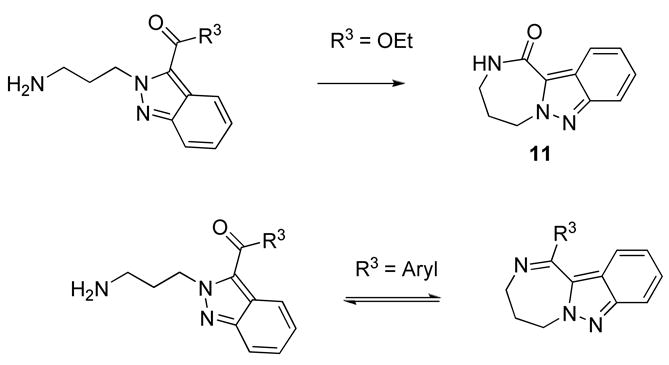
Synthesis of 2,3,4,5-tetrahydro-1H-[1,4]diazepino[1,2-b]indazol-1-one 11
Crude cyclic products were obtained by evaporation of solvent (TFA, AcOH, or MeOH). Isolation and purification of target compounds were carried out by three different methods depending on the purity of the crude product: precipitation using diethyl ether (method A), passing through a C18 cartridge (method B) or HPLC purification (method C). Results for individual compounds are summarized in Table 1. All synthesized compounds bear no residual functional group from the attachment to solid support; thus, this synthetic scheme represents a traceless synthesis of 3,4-dihydropyrazino[1,2-b]indazoles. Figure 2 portrays excellent purities of crude preparations by LC traces of a subset of library compounds.
Figure 2.
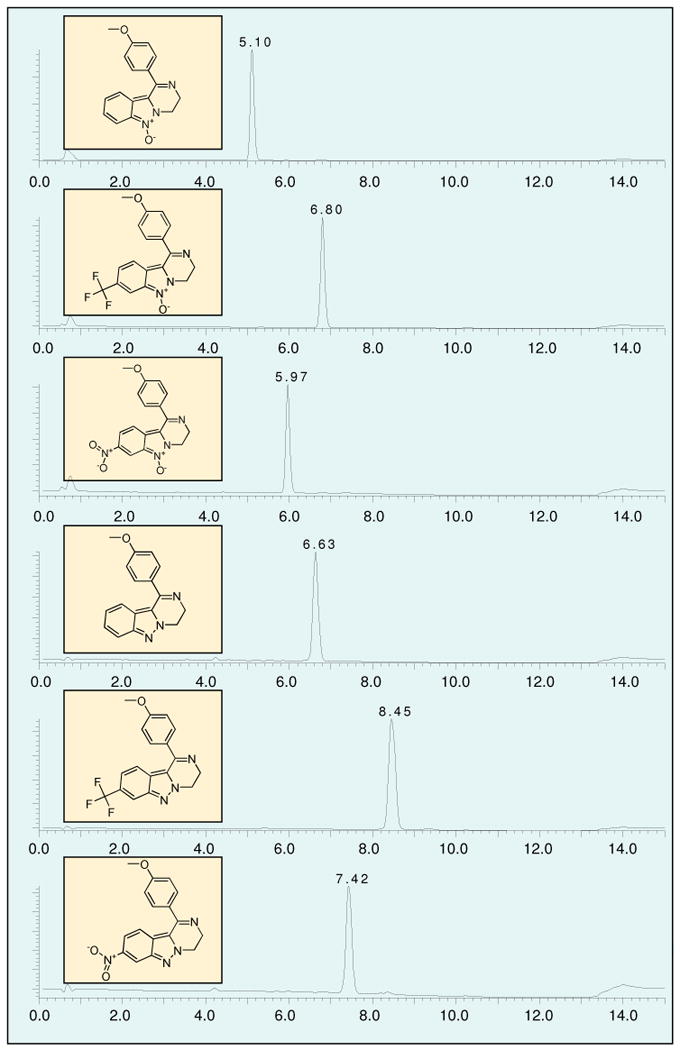
LCMS of crude compounds 7 and 8
The structure of compound 7(2,1,1) was determined by analysis of 1D and 2D NMR spectra.
Conclusion
We reported a very efficient traceless synthesis of 3,4-dihydropyrazino[1,2-b]indazoles and their 6-oxides by using commercially available and easily accessible building blocks. The synthesis provided high-purity crude compounds under very mild conditions. The synthesis tolerated diversely substituted building blocks without compromising on the purity of crude products. A small combinatorial library was synthesized.
Experimental Section
Syntheses were carried out on Domino Blocks17 (www.torviq.com) in disposable polypropylene reaction vessels.
Reaction with CDI and alkyldiamine (resin 1)
Wang resin (Advanced ChemTech, 1 mmol/g, 8 g) was washed three times with DCM. Solution of CDI (40 mmol, 6.48 g) and pyridine (40 mmol, 3.2 mL) in 80 mL DCM was added and the resin slurry was shaken at ambient temperature for 3 h. Resin was washed three times with DCM and solution of alkyldiamine (40 mmol) in 80 mL DCM was added. Resin slurry was shaken at ambient temperature for 2 h and washed three times with DCM.
Reaction with 2-Nos-Cl (resins 2)
Resins 1, ∼2 g, were washed three times with DCM, a solution of 2-Nos-Cl (6 mmol) and lutidine (6.6 mmol, 764 μL) in 20 mL DCM was added to the resin and the slurry was shaken at ambient temperature overnight. The resin was washed three times with DMF and three times with DCM.
Reaction with bromoketones/bromoacetates (resins 3)
Resins 2, ∼500 mg, were washed three times with DCM and five times with DMF. A solution of 0.5 M bromoketone/bromoacetate (3 mmol) and 1 M DIEA (6 mmol, 1.04 mL) in 6 mL DMF was added. The slurry was shaken at ambient temperature overnight. The resin was washed three times with DMF and five times with DCM.
Cyclization to indazole oxides (resins 4)
Resins 3, ∼500 mg, were washed five times with DMF. A solution of 0.2 M DBU (2 mmol, 300 μL) in 10 mL DMF was added and the resin slurry was shaken for 30 min. Resins were washed three times with DMF, three times with DCM, one time with TEA, three times with DCM, one time with 5% AcOH in DCM, three times with MeOH and three times with DCM.
Deoxygenation (resins 5)
Resins 4, ∼250 mg, in 10-mL plastic reaction vessels were washed five times with DCM and a solution of 2.5 mL of 0.7 M TEA (1.8 mmol, 245 μL) in DCM was added into each reaction vessel. Plastic 3-mL syringes were charged with 2.5 mL 0.5 M methanesulfonyl chloride (1.2 mmol, 96 μL) in DCM. These syringes were connected with the plastic reaction vessels containing the resins 4. Connected syringes were left in a freezer for 30 min and the methanesulfonyl chloride solution was drawn into the syringe with a resin. The resin slurry was shaken at ambient temperature (for reaction time, cf., Table 1).
Cleavage, cyclization and isolation (resins 6-10)
Resins 4 and 5, ∼250 mg, were treated with 50% TFA for 1 h. TFA solution was collected, resin was washed three times with 50% TFA and combined extracts were evaporated by a stream of nitrogen, the residual material was analyzed by LCMS. Compounds that did not spontaneously cyclized (for structures cf., Table 1) were dissolved in a neat acetic acid and shaken for 2 h. Compound 9 was dissolved in MeOH with TEA and shaken overnight. Solvent was evaporated in a stream of nitrogen. Products were isolated and purified according to the purity of crude product. Products with purity >90% were either precipitated by diethylether and collected precipitate was freeze dried (method A) or dissolved in 5% MeCN in water and passed through C18 cartridge. The cartridge was washed with water and products eluted by MeCN. The solution was evaporated by a stream of nitrogen and the residue freeze dried (method B). Products with purity <90% were purified by semi-preparative HPLC (method C). All products were analyzed by LCMS and HRMS.
Supplementary Material
Acknowledgments
The work was supported by the Department of Chemistry and Biochemistry University of Notre Dame and the NIH (GM079576). We gratefully appreciate the use of the NMR facility at the University of Notre Dame.
Footnotes
Supporting Information Available. Details of experimental procedures, spectroscopic data and NMR spectra for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Soural M, Krchňák V. J Comb Chem. 2007;9:793–796. doi: 10.1021/cc070071v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bouillon I, Krchňák V. J Comb Chem. 2007;9:912–915. doi: 10.1021/cc700122a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soural M, Bouillon I, Krchňák V. J Comb Chem. 2008;10:923–933. doi: 10.1021/cc8001074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bouillon I, Zajíček J, Pudelová N, Krchňák V. J Org Chem. 2008;73:9027–9032. doi: 10.1021/jo8018895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fukuyama T, Jow CK, Cheung M. Tetrahedron Lett. 1995;36:6377–6374. [Google Scholar]
- 6.Tapolcsányi P, Krajsovszky G, Andó R, Lipcsey P, Horváth G, Mátyus P, Riedl Z, Hajós G, Maes BUW, Lemiére GLF. Tetrahedron. 2002;58:10137–10143. [Google Scholar]
- 7.Timári G, Soós T, Hajós G, Messmer A, Nacsa J, Molnár J. Bioorg Med Chem Lett. 1996;6:2831–2836. [Google Scholar]
- 8.Csányi D, Timári G, Hajós G. Synth Commun. 1999;29:3959–3969. [Google Scholar]
- 9.Cimanga K, DeBruyne T, Lasure A, VanPoel B, Pieters L, Claeys M, VandenBerghe D, Kambu K, Tona L, Vlietinck AJ. Planta Medica. 1996;62:22–27. doi: 10.1055/s-2006-957789. [DOI] [PubMed] [Google Scholar]
- 10.Noamesi BK, Bamgbose SOA. Planta Medica. 1983;47:100–102. doi: 10.1055/s-2007-969962. [DOI] [PubMed] [Google Scholar]
- 11.Cimanga K, DeBruyne T, Pieters L, Vlietinck AJ. J Nat Prod. 1997;60:688–691. doi: 10.1021/np9605246. [DOI] [PubMed] [Google Scholar]
- 12.Cerecetto H, Gerpe A, Gonzalez M, Aran VJ, de Ocariz CO. Mini-Reviews in Medicinal Chemistry. 2005;5:869–878. doi: 10.2174/138955705774329564. [DOI] [PubMed] [Google Scholar]
- 13.Wang SS. J Am Chem Soc. 1973;95:1328–1333. doi: 10.1021/ja00785a602. [DOI] [PubMed] [Google Scholar]
- 14.Hauske JR, Dorff P. Tetrahedron Lett. 1995;36:1589–1592. [Google Scholar]
- 15.Munson MC, Cook AW, Josey JA, Rao C. Tetrahedron Lett. 1998;39:7223–7226. [Google Scholar]
- 16.Morimoto Y, Kurihara H, Yokoe C, Kinoshita T. Chem Lett. 1998:829–830. [Google Scholar]
- 17.Krchňák V, Paděra V. Bioorg Med Chem Lett. 1998;22:3261–3264. doi: 10.1016/s0960-894x(98)00594-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.