

Abstract
AIM: To examine the effect of troglitazone, a peroxisome proliferator-activated receptor γ (PPARγ) ligand, on the proliferation and apoptosis of human liver cancer cells.
METHODS: Liver cancer cell line HepG2 was cultured and treated with troglitazone. Cell proliferation was detected by 3-(4-,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay; apoptosis was detected by flow cytometry and terminal deoxynucleotidyl transferase-mediated nick end labeling of DNA fragmentation sites (TUNEL) assay; and apoptosis-related protein was detected by immunocytochemistry and Western blotting.
RESULTS: Troglitazone inhibited growth and induced apoptosis of HepG2 cells in a dose-dependent manner, and induced activation of caspase-3 expression. Troglitazone not only drove apoptosis-inhibiting factor survivin to translocate incompletely from the nucleus to the cytoplasm, but also inhibited expression of survivin, while it did not affect expression of apoptosis-promoting factor Bax.
CONCLUSION: PPARγ ligands inhibit growth and induce apoptosis of liver cancer cells, and may have applications for the prevention and treatment of liver cancer.
Keywords: Peroxisome proliferator-activated receptor γ, Troglitazone, Liver neoplasms, Apoptosis
INTRODUCTION
Primary hepatocellular carcinoma (HCC) is the fifth most common malignant tumor worldwide, and causes more than 500 000 deaths annually[1]. Dissemination of Hepatitis B virus (HBV) and Hepatitis C virus (HCV) causes the incidence of HCC to increase[2]. Surgical resection is the treatment of choice for HCC, but only 10%-20% of HCC patients are resectable at the time of diagnosis[3]. Liver transplantation is only indicated for early HCC (Milan criteria 1 tumor, < 5 cm in diameter, or up to three tumors, all < 3 cm in diameter), as far as outcome is concerned[4]. In addition, recurrence remains high in patients who receive radical treatment[5,6]. It is therefore urgent to seek a new approach for the treatment of HCC.
Peroxisome proliferator-activated receptor (PPAR) belongs to the nuclear hormone receptor superfamily[7]. Three subtypes of PPAR (PPARα, PPARγ and PPARδ) have been identified, among which PPARγ has been studied most extensively. PPAR-γ exerts its effects by forming heterodimers with the 9-cis-retinoid X receptor (RXR) after ligand activation. The PPAR/RXR heterodimer acts as a regulatory transcription factor with peroxisome proliferator responsive element (PPRE) to regulate the expression of target genes which participate in the physiological and pathological processes of lipid metabolism, glucose metabolism, adipocyte differentiation, energy balance, inflammatory reactions and atherosclerosis[8,9]. PPARγ ligands are classified as natural and synthetic. The former include long-chain polyunsaturated fatty acid and eicosanoids, and the latter include thiazolidinediones (TZDs), the most commonly used drugs for diabetes, such as troglitazone, pioglitazone, rosiglitazone and LY171.833. Several recent studies have shown most human tumors express PPARγ, and there is evidence that PPARγ ligands have anti-tumor activity[10–20]. One of the important effects of these drugs is induction of apoptosis, although the exact mechanism remains elusive.
The present study used liver cancer cell line HepG2 that endogenously expresses PPARγ as an experimental model[15] to study the effects of PPARγ ligand troglitazone on proliferation and apoptosis of liver cancer cells. It also analyzed the molecular mechanisms of these effects in an attempt to provide experimental clues for the use of PPARγ ligands in the treatment of liver cancer.
MATERIALS AND METHODS
Chemicals
Troglitazone (Cayman Chemical Industry, USA) was dissolved in DMSO and then diluted to appropriate concentrations with culture medium. The final concentration of DMSO in the culture medium did not exceed 1 mL/L.
Cell culture
HepG2 cells were cultured in Dulbecco’s modified Eagle’s medium (GIBCO Laboratories, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS, GIBCO Laboratories), 100 U/mL penicillin, and 100 g/mL streptomycin. Cells were grown at 37°C in an atmosphere of 95% air and 5% CO2.
MTT assay
Cell proliferation was detected by 3-(4-,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (Sigma, St Louis, MO, USA) assay. When cells were cultured to the log phase, they were seeded on a 96-well plate (2 × 104 cells/100 μL/well) for 24 h until confluence. Cells were divided into a control (DMSO) group and a troglitazone group. The concentration of troglitazone was 5, 10, 20, 40, 80 and 100 μmol/L. After 120 h, 10 μL MTT (5 g/L) was added and incubated at 37°C for 4 h. DMSO (75 μL) was added to each well, which was oscillated for 10 min until the crystals were dissolved completely. Absorbance (A) was detected with an enzyme calibrator at 560 nm. Cell viability = (A of study group/A of control group) × 100%. The experiment was repeated twice. There were six wells for each concentration.
Flow cytometry
Cells were seeded onto a six-well plate and allowed to grow to 40% confluence. After treatment with or without the drug, both floating and adherent cells were collected. Cells were suspended and then fixed with ice-cold 70% alcohol at -20°C, followed by washing and staining with 50 μg/mL propidium iodide in the presence of 100 μg/mL ribonuclease A for 30 min at 37°C in the dark. DNA content was analyzed by flow cytometry using Cell Quest software (Becton Dickinson and Beckman-Coulter, San Jose, CA, USA).
TUNEL assay
HepG2 cells were grown on poly-L-lysine-coated slides in a six-well plate. After treatment with or without the drug, the slides were gently washed three times in 0.1 mol/L PBS (pH 7.4), fixed with 80% ice-cold ethanol, and immediately transferred to a freezer until use. To study apoptosis of cultured cells, terminal deoxynucleotidyl transferase-mediated nick end labeling of DNA fragmentation sites (TUNEL) assay was performed using Fluorescein FragEL DNA Fragmentation Detection Kit (Oncogene Research Products, San Diego, CA, USA) according to the manufacturer’s instructions. Ten high-power areas with even distribution of positive cells were used for calculation of the percentage of apoptotic cells under a fluorescence microscope.
Immunocytochemistry
HepG2 cells were grown on poly-L-lysine-coated slides in a six-well plate. After treatment with the drug, the slides were washed twice with PBS and fixed in 4% paraformaldehyde for 30 min at room temperature. Immunostaining was performed using the streptavidin-biotin complex method with UltraSensitive S-P Kit (Fuzhou Maxim Biotech, Fuzhou, Fujian, China). The slides were pretreated first with 0.3% hydrogen peroxide in PBS for 10 min to inactivate endogenous peroxidase, and then microwave antigen retrieval was performed with 0.01 mol/L citrate buffer at pH 6.0 for 20 min, followed by incubation with polyclonal rabbit anti-human antibody against the active caspase-3 (1:25; R&D Systems, Wiesbaden, Germany), rabbit anti-human survivin polyclonal antibody (1:50; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C overnight, biotinylated secondary antibody for 30 min, and peroxidase-labeled streptavidin for 30 min. Color reaction was developed with 3, 3’-diaminobenzidine as a chromogen. Finally, the slides were counterstained with hematoxylin, and dehydrated through graded alcohol. For negative controls, slides were processed as above but treated with PBS instead of the primary antibody.
Western blotting
Troglitazone-treated HepG2 cells were prepared in a cell-lysis solution. The protein concentration was determined by BCA Protein assay kit (Perbio Science Deutschland, Germany). Protein samples (50 μg) were separated by SDS-PAGE and transferred to nitrocellulose membranes, to which rabbit anti-human survivin polyclonal antibody (1:1000), mouse anti-human Bax antibody (1:1000), mouse anti-human β-actin antibody (1:1000), and horseradish-peroxidase-labeled secondary antibody (all from Santa Cruz Biotechnology) were added. The signals were detected by enhanced chemiluminescence according to the protocol supplied with the kit.
Statistical analysis
Data were expressed as mean ± SD. Statistical correlation of data was checked for significance by ANOVA and Student’s t test. Differences with P < 0.05 were considered significant. These analyses were performed using SPSS 11.0 software (SPSS, Chicago, IL, USA).
RESULTS
Troglitazone inhibits cell growth
The MTT assay showed that cell viability was 96.8% ± 1.22%, 53.4% ± 1.2%, 42.3% ± 1.2%, 31.4% ± 1%, 13.6% ± 0.8% and 9.6% ± 0.7% at 5, 10, 20, 40, 80 and 100 μmol/L troglitazone for 120 h, respectively (Figure 1), which indicated that troglitazone inhibited the growth of liver cells in a dose-dependent manner.
Figure 1.
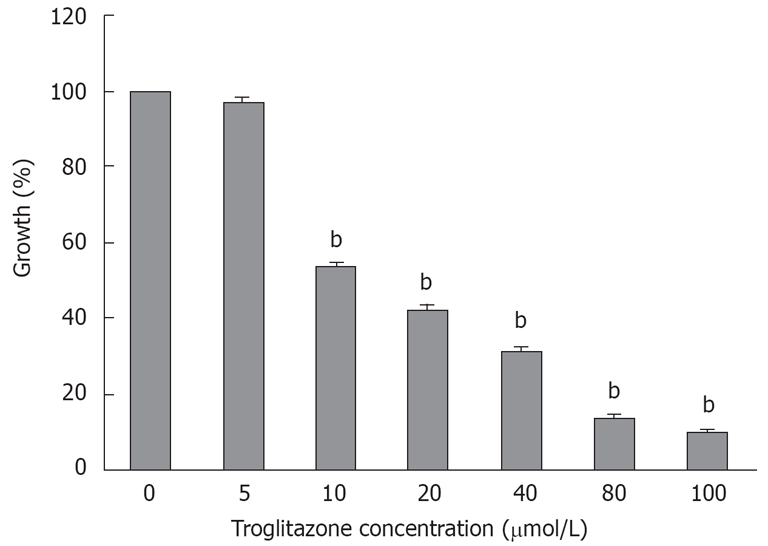
Effect of troglitazone on growth of HepG2 cells. Cells were incubated with different concentrations of troglitazone for 120 h. Troglitazone inhibited growth of HepG2 cells in a dose-dependent manner, as demonstrated by MTT assay. Data represent the mean ± SD from six wells. bP < 0.01 vs corresponding control group (unpaired Student’s t test).
Troglitazone induces apoptosis
HepG2 cells were treated with troglitazone (20 and 30 μmol/L) for 24 h, and phase-contrast microscopy revealed that some cells became round, blunt and smaller in size, accompanied by karyopyknosis; light refraction was increased; and cells became detached and suspended in the medium, especially with 30 μmol/L troglitazone. In the control group, cells were regular in morphology and grew fully in patches and confluently, rarely sloughing off (Figure 2).
Figure 2.
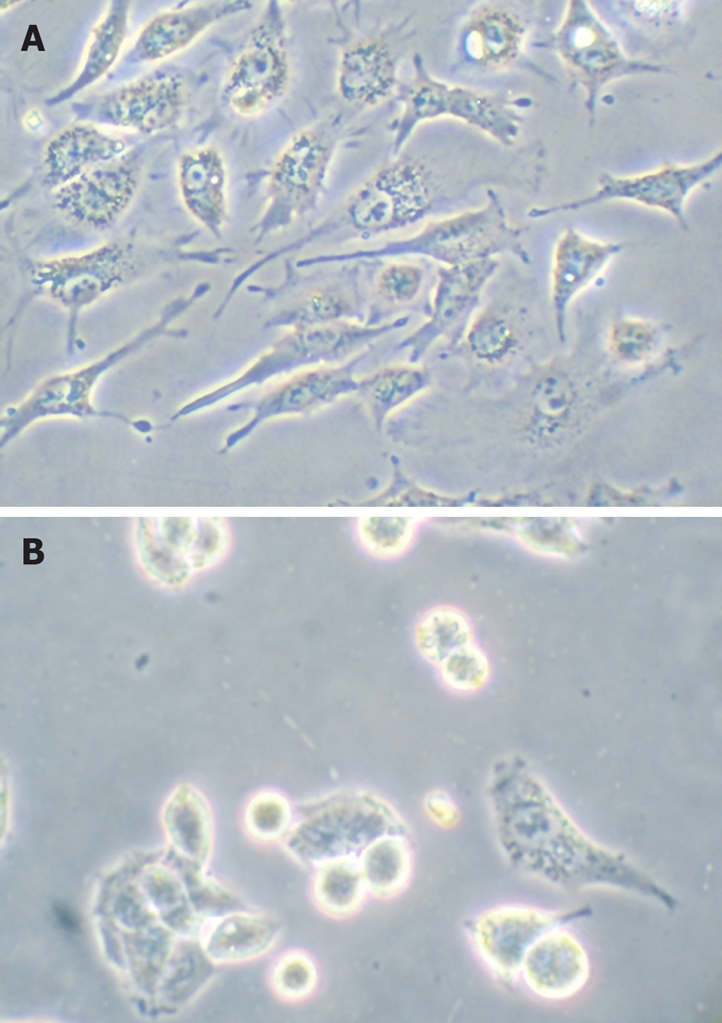
Morphological changes were examined by phase-contrast microscopy. A:Controls; B: 30 μmol/L troglitazone.
Flow cytometry showed that after treatment of HepG2 cells with 20 and 30 μmol/L troglitazone for 24 h, the percentage of sub-G0/G1 peak apoptotic cells was 25.5% ± 1.1% and 43.9% ± 1.7%, respectively. In the untreated controls, none of the cells was found in the sub-G0/G1 peak (Figure 3). TUNEL staining also showed that treatment with troglitazone for 24 h caused apoptosis of HepG2 cells. The apoptotic rate increased with concentration of troglitazone: 22.4% ± 1.2% for 20 μmol/L, and 38.6% ± 0.8% for 30 μmol/L troglitazone. In the control group, there were only background cells, without the presence of TUNEL positive cells (Figure 4, P < 0.001).
Figure 3.

Cell cycle analysis by flow cytometry. A: Control cells; B: 20 μmol/L troglitazone; C: 30 μmol/L troglitazone.
Figure 4.

Troglitazone significantly increased the number of TUNEL-positive cells in a dose-dependent manner. A: Control cells; B: 20 μmol/L troglitazone; C: 30 μmol/L troglitazone.
Troglitazone induces activation of caspase-3 expression
The caspase family plays a very important role in mediating apoptosis, among which caspase-3 is the key executive molecule, and its activated form has multiple functions in signal transmission of apoptosis[21,22]. Troglitazone activated caspase-3 expression. Positive staining was signified by a yellow-brown color, present in smaller cells whose plasma was condensed. After treatment of HepG2 cells with 20 and 30 μmol/L troglitazone, the positive cell rate was 21.2% ± 1.4% and 35.8% ± 2.4%, respectively (Figure 5). No positive cells were found in the controls.
Figure 5.
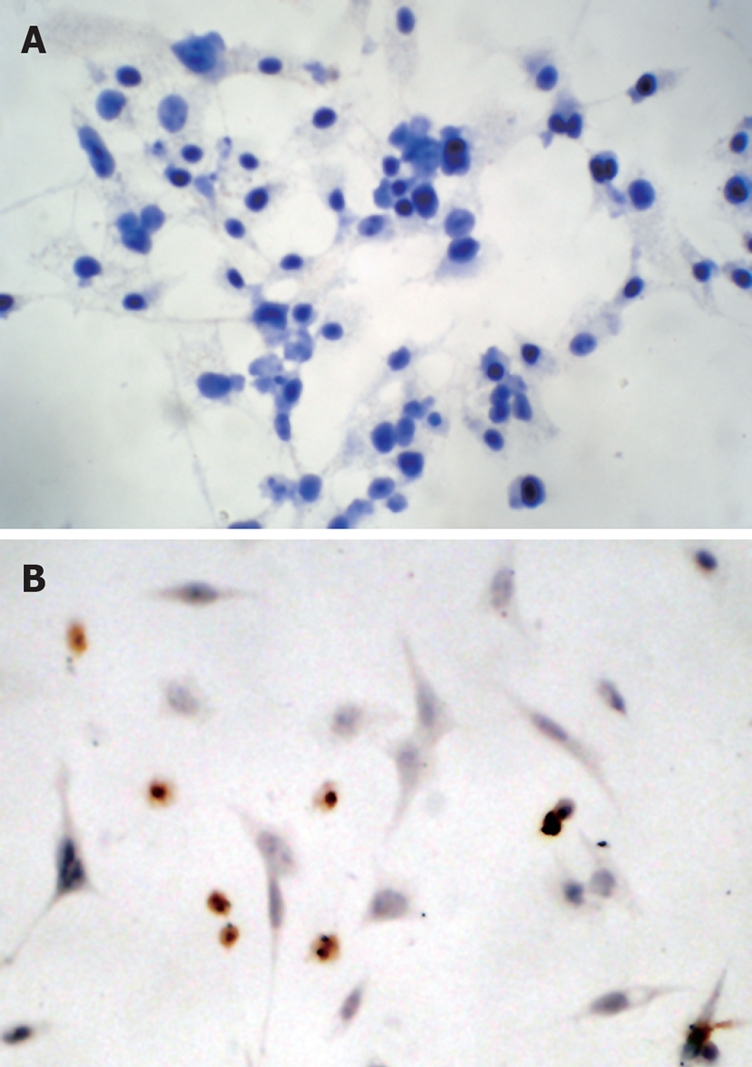
Immunocytochemistry showed that caspase-3 was activated. The shrunken cells were positively stained for activated caspase-3. No positive cell was detectable in the control group ( A: original magnification, × 200). Whereas large numbers of positive cells labeled for activated caspase-3 were observed in HepG2 cells treated with 30 μmol/L troglitazone for 24 h ( B: original magnification, × 400).
Troglitazone drives survivin protein to translocate incompletely from the nucleus to the cytoplasm and inhibits its expression
Immunocytochemical staining showed that apoptosis-inhibiting factor survivin was mainly expressed in the nucleus and weakly in the cytoplasm of untreated HepG2 cells. After treatment with 20 and 30 μmol/L troglitazone for 24 h, survivin translocated partly from the nucleus to the cytoplasm, and the intensity of its expression decreased markedly. Also, apoptotic cells were seen in the treatment group, and presented with a smaller cell size and condensed cytoplasm (Figure 6). Western blotting revealed that, in HepG2 cells treated with troglitazone for 24 h, expression of HepG2 survivin was inhibited in a dose-dependent manner, but the treatment did not significantly affect expression of apoptosis-promoting factor Bax (Figure 7).
Figure 6.
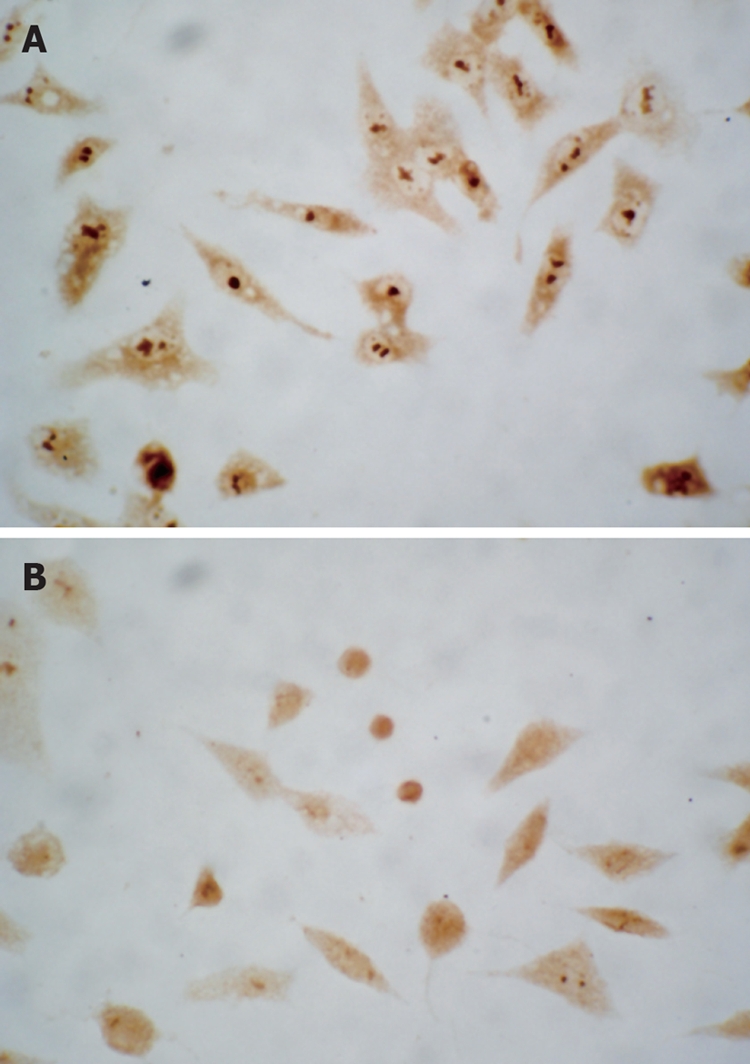
Survivin was present predominantly in the nucleus in untreated cells ( A: original magnification, × 400) as demonstrated by immunocytochemistry. After exposure of HepG2 cells to 30 μmol/L troglitazone for 24 h, survivin incompletely translocated from the nucleus to the cytoplasma ( B: original magnification, × 400).
Figure 7.

Effect of troglitazone on the expression of Bax and survivin. Lane 1: Control cells; lane 2: 20 μmol/L troglitazone; lane 3: 30 μmol/L troglitazone.
DISCUSSION
Apoptosis is a normal phenomenon in the process of embryonic growth of all organisms and human development. Disturbance of this process is associated with the development of many severe diseases and disorders including tumors. Selective induction of tumor-cell apoptosis may become the basic strategy in the treatment of malignant tumors. Most of the available chemotherapy drugs work through destroying tumor cells by inducing apoptosis[23]. In our study, the MTT assay showed troglitazone inhibited growth of liver cancer cells in a dose-dependent manner. To determine whether the inhibitory effect of troglitazone on the proliferation of liver cancer cells was associated with induction of apoptosis, flow cytometry and TUNEL staining were conducted. The results confirmed troglitazone induced apoptosis of HepG2 cells in a dose-dependent manner. Thus, clarifying the apoptosis-related gene protein that is associated with troglitazone-induced apoptosis is of primary importance in explaining the mechanism of action of troglitazone.
Caspase is a group of cysteine hydrolytic proteinases that are able to specifically cleave the peptide chain behind the residue base of the target protein aspartate. It is the key molecule in the regulation of apoptosis. Caspase can trigger a cascade reaction under the control of apoptosis signals. According to their method of entering the apoptotic site, caspases are classified as initiator and effector caspases[21], among which, caspase-3 is the most important terminal effector caspase in apoptosis, and plays an irreplaceable role. After initiation of the apoptotic process, caspase-3 transforms from the zymogen form to the activated form and functions by hydrolyzing proteins essential for survival of many kinds of cells[22].
Survivin is a member of the inhibitor of apoptosis (IAP) family of negative regulators of programmed cell death, and is undetectable in normal adult tissue, but is abundantly expressed in fetal tissue and a variety of human cancers including HCC. High levels of survivin in tumors are frequently correlated with malignant parameters, which suggest a role in tumorigenesis[24–26]. Survivin can inhibit terminal effector caspase-3 and caspase-7, and interfere with caspase-9 activity and processing. Therefore, approaches designed to counteract antiapoptotic activity may inhibit tumor growth[27].
There has long been controversy over the subcellular location of survivin. Some researchers have found survivin expression is located in the nucleus in HCC, esophageal squamous cell carcinoma, ovarian carcinoma, mantle cell lymphoma, cholangiocarcinoma, and non-small cell lung cancer, and is associated with poor prognosis. Other researchers have pointed out that survivin expression is located in the nucleus in gastric cancer, bladder mucosa and transitional cell carcinoma, and breast cancer, and is a predictor of good prognosis[28]. The results of our immunohistochemical staining confirmed that survivin was expressed mainly in the nucleus of HepG2 cells, and weakly in the cytoplasm. Fortugno et al[29] have found two survivin pools using immunofluorescence, one in the cytoplasm and the other in the nucleus. Suzuki et al[30] have found survivin protein can translocate from the cytoplasm to the nucleus. Here, it forms a complex with CDK4, which enables p21 to combine with a precursor released from the CDK4 complex and mitochondrial caspase-3, and inhibits apoptosis by inhibiting the activity of caspase-3. The results of our study showed troglitazone induced apoptosis in a dose-dependent manner, during which survivin translocated incompletely from the nucleus to the cytoplasm, with a decrease in the intensity of expression; at the same time caspase-3 expression was activated. We presume troglitazone dissociated the survivin/CDK4 complex by inducing survivin expression and driving survivin to translocate incompletely from the nucleus to the cytoplasm. This forced p21 to form a complex with CDK4 by dissociating the combination of p21 and the precursor of mitochondrial caspase-3 and activating caspase-3. This led to cleavage of the corresponding nuclear and cytoplasmic substrates and finally caused apoptosis.
TZD induces massive apoptosis in renal cancer cells, with increased Bax expression and decreased Bcl-2 expression[18]. However, troglitazone significantly increases the expression of c-myc mRNA, but has no effect on expression of Bcl-2 and Bax in thyroid carcinoma cells[13]. In the current study, we also found Bax may not participate in troglitazone-induced apoptosis, suggesting that the mechanisms by which the PPARγ ligands cause tumor cell apoptosis are different depending on the type of cancer.
In conclusion, PPARγ ligands have the effect of inhibiting growth and inducing the apoptosis of liver cancer cells, and may have applications for the prevention and treatment of liver cancer.
COMMENTS
Background
Peroxisome proliferator-activated receptor (PPAR) belongs to the nuclear hormone receptor superfamily. Three subtypes of PPAR (PPARα, PPARγ and PPARδ) have been identified, among which PPARγ has been studied most extensively. Recent data have shown that ligands for PPARγ exhibit growth-inhibitory effects on many types of human cancer. One of the important effects of these drugs is inducing apoptosis, although the exact mechanism remains elusive.
Research frontiers
We used liver cancer cell line HepG2 that endogenously expresses PPARγ as an experimental model to study the effects of PPARγ ligand troglitazone on the proliferation and apoptosis of liver cancer cells, and analyzed the molecular mechanisms of these effects.
Innovations and breakthroughs
Troglitazone inhibited growth and induced apoptosis of HepG2 cells in a dose-dependent manner, and induced activation of caspase-3 expression. Troglitazone not only drove apoptosis-inhibiting factor survivin to translocate incompletely from the nucleus to the cytoplasm, but also inhibited expression of survivin.
Applications
PPARγ ligand has the effect of inhibiting growth and inducing apoptosis of liver cancer cells, and may have potential as a drug against liver cancer.
Peer review
This study should arouse interest in readers. It provides important information for the investigation of the anti-cancer mechanism of troglitazone and other PPARγ ligands, and for the development of anti-cancer therapy using PPARγ ligands.
Supported by Grants from the State Key Basic Research Program, No. 2002CB513100
Peer reviewers: Frank A Anania, Professor, Emory University School of Medicine, Division of Division Digestive Diseases, 615 Michael Street, Room 255 Whitehead Biomedical Research Building, Atlanta, GA 30322, United States; Dusan M Jovanovic, Professor, Institute of Oncology, Institutski Put 4, Sremska Kamenica 21204, Serbia
S- Editor Yang RH L- Editor Kerr C E- Editor Ma WH
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: Globocan 2000. Int J Cancer. 2001;94:153–156. doi: 10.1002/ijc.1440. [DOI] [PubMed] [Google Scholar]
- 2.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 3.Pergolizzi JV Jr, Auster M, Conaway GL, Sardi A. Cryosurgery for unresectable primary hepatocellular carcinoma: a case report and review of literature. Am Surg. 1999;65:402–405. [PubMed] [Google Scholar]
- 4.Mazzaferro V, Regalia E, Doci R, Andreola S, Pulvirenti A, Bozzetti F, Montalto F, Ammatuna M, Morabito A, Gennari L. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med. 1996;334:693–699. doi: 10.1056/NEJM199603143341104. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto J, Kosuge T, Takayama T, Shimada K, Yamasaki S, Ozaki H, Yamaguchi N, Makuuchi M. Recurrence of hepatocellular carcinoma after surgery. Br J Surg. 1996;83:1219–1222. [PubMed] [Google Scholar]
- 6.Otto G, Heuschen U, Hofmann WJ, Krumm G, Hinz U, Herfarth C. Survival and recurrence after liver transplantation versus liver resection for hepatocellular carcinoma: a retrospective analysis. Ann Surg. 1998;227:424–432. doi: 10.1097/00000658-199803000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schoonjans K, Staels B, Auwerx J. The peroxisome proliferator activated receptors (PPARS) and their effects on lipid metabolism and adipocyte differentiation. Biochim Biophys Acta. 1996;1302:93–109. doi: 10.1016/0005-2760(96)00066-5. [DOI] [PubMed] [Google Scholar]
- 9.Fajas L, Auboeuf D, Raspe E, Schoonjans K, Lefebvre AM, Saladin R, Najib J, Laville M, Fruchart JC, Deeb S, et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J Biol Chem. 1997;272:18779–19789. doi: 10.1074/jbc.272.30.18779. [DOI] [PubMed] [Google Scholar]
- 10.Sato H, Ishihara S, Kawashima K, Moriyama N, Suetsugu H, Kazumori H, Okuyama T, Rumi MA, Fukuda R, Nagasue N, et al. Expression of peroxisome proliferator-activated receptor (PPAR)gamma in gastric cancer and inhibitory effects of PPARgamma agonists. Br J Cancer. 2000;83:1394–1400. doi: 10.1054/bjoc.2000.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clay CE, Atsumi GI, High KP, Chilton FH. Early de novo gene expression is required for 15-deoxy-Delta 12,14-prostaglandin J2-induced apoptosis in breast cancer cells. J Biol Chem. 2001;276:47131–47135. doi: 10.1074/jbc.C100339200. [DOI] [PubMed] [Google Scholar]
- 12.Debrock G, Vanhentenrijk V, Sciot R, Debiec-Rychter M, Oyen R, Van Oosterom A. A phase II trial with rosiglitazone in liposarcoma patients. Br J Cancer. 2003;89:1409–1412. doi: 10.1038/sj.bjc.6601306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohta K, Endo T, Haraguchi K, Hershman JM, Onaya T. Ligands for peroxisome proliferator-activated receptor gamma inhibit growth and induce apoptosis of human papillary thyroid carcinoma cells. J Clin Endocrinol Metab. 2001;86:2170–2177. doi: 10.1210/jcem.86.5.7493. [DOI] [PubMed] [Google Scholar]
- 14.Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome proliferator-activated receptor gamma agonists. Lancet Oncol. 2004;5:419–429. doi: 10.1016/S1470-2045(04)01509-8. [DOI] [PubMed] [Google Scholar]
- 15.Date M, Fukuchi K, Morita S, Takahashi H, Ohura K. 15-Deoxy-delta12,14-prostaglandin J2, a ligand for peroxisome proliferators-activated receptor-gamma, induces apoptosis in human hepatoma cells. Liver Int. 2003;23:460–466. doi: 10.1111/j.1478-3231.2003.00877.x. [DOI] [PubMed] [Google Scholar]
- 16.Han S, Sidell N, Fisher PB, Roman J. Up-regulation of p21 gene expression by peroxisome proliferator-activated receptor gamma in human lung carcinoma cells. Clin Cancer Res. 2004;10:1911–1919. doi: 10.1158/1078-0432.ccr-03-0985. [DOI] [PubMed] [Google Scholar]
- 17.Ceni E, Mello T, Tarocchi M, Crabb DW, Caldini A, Invernizzi P, Surrenti C, Milani S, Galli A. Antidiabetic thiazolidinediones induce ductal differentiation but not apoptosis in pancreatic cancer cells. World J Gastroenterol. 2005;11:1122–1130. doi: 10.3748/wjg.v11.i8.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang FG, Zhang ZW, Xin DQ, Shi CJ, Wu JP, Guo YL, Guan YF. Peroxisome proliferator-activated receptor gamma ligands induce cell cycle arrest and apoptosis in human renal carcinoma cell lines. Acta Pharmacol Sin. 2005;26:753–761. doi: 10.1111/j.1745-7254.2005.00753.x. [DOI] [PubMed] [Google Scholar]
- 19.Shigeto T, Yokoyama Y, Xin B, Mizunuma H. Peroxisome proliferator-activated receptor alpha and gamma ligands inhibit the growth of human ovarian cancer. Oncol Rep. 2007;18:833–840. [PubMed] [Google Scholar]
- 20.Lin MS, Chen WC, Bai X, Wang YD. Activation of peroxisome proliferator-activated receptor gamma inhibits cell growth via apoptosis and arrest of the cell cycle in human colorectal cancer. J Dig Dis. 2007;8:82–88. doi: 10.1111/j.1443-9573.2007.00290.x. [DOI] [PubMed] [Google Scholar]
- 21.Nicholson DW. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999;6:1028–1042. doi: 10.1038/sj.cdd.4400598. [DOI] [PubMed] [Google Scholar]
- 22.Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- 23.Bremer E, van Dam G, Kroesen BJ, de Leij L, Helfrich W. Targeted induction of apoptosis for cancer therapy: current progress and prospects. Trends Mol Med. 2006;12:382–393. doi: 10.1016/j.molmed.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 24.Li F. Role of survivin and its splice variants in tumorigenesis. Br J Cancer. 2005;92:212–216. doi: 10.1038/sj.bjc.6602340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T, Reed JC. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 1998;58:5315–5320. [PubMed] [Google Scholar]
- 26.Shin S, Sung BJ, Cho YS, Kim HJ, Ha NC, Hwang JI, Chung CW, Jung YK, Oh BH. An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and -7. Biochemistry. 2001;40:1117–1123. doi: 10.1021/bi001603q. [DOI] [PubMed] [Google Scholar]
- 27.Zaffaroni N, Pennati M, Daidone MG. Survivin as a target for new anticancer interventions. J Cell Mol Med. 2005;9:360–372. doi: 10.1111/j.1582-4934.2005.tb00361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li F, Yang J, Ramnath N, Javle MM, Tan D. Nuclear or cytoplasmic expression of survivin: what is the significance? Int J Cancer. 2005;114:509–512. doi: 10.1002/ijc.20768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fortugno P, Wall NR, Giodini A, O’Connor DS, Plescia J, Padgett KM, Tognin S, Marchisio PC, Altieri DC. Survivin exists in immunochemically distinct subcellular pools and is involved in spindle microtubule function. J Cell Sci. 2002;115:575–585. doi: 10.1242/jcs.115.3.575. [DOI] [PubMed] [Google Scholar]
- 30.Suzuki A, Ito T, Kawano H, Hayashida M, Hayasaki Y, Tsutomi Y, Akahane K, Nakano T, Miura M, Shiraki K. Survivin initiates procaspase 3/p21 complex formation as a result of interaction with Cdk4 to resist Fas-mediated cell death. Oncogene. 2000;19:1346–1353. doi: 10.1038/sj.onc.1203429. [DOI] [PubMed] [Google Scholar]