


Abstract
Outer membrane proteins (OMPs) are the transmembrane proteins found in the outer membranes of Gram-negative bacteria, mitochondria and plastids. Most prediction methods have focused on analogous features, such as alternating hydrophobicity patterns. Here, we start from the observation that almost all β-barrel OMPs are related by common ancestry. We identify proteins as OMPs by detecting their homologous relationships to known OMPs using sequence similarity. Given an input sequence, HHomp builds a profile hidden Markov model (HMM) and compares it with an OMP database by pairwise HMM comparison, integrating OMP predictions by PROFtmb. A crucial ingredient is the OMP database, which contains profile HMMs for over 20 000 putative OMP sequences. These were collected with the exhaustive, transitive homology detection method HHsenser, starting from 23 representative OMPs in the PDB database. In a benchmark on TransportDB, HHomp detects 63.5% of the true positives before including the first false positive. This is 70% more than PROFtmb, four times more than BOMP and 10 times more than TMB-Hunt. In Escherichia coli, HHomp identifies 57 out of 59 known OMPs and correctly assigns them to their functional subgroups. HHomp can be accessed at http://toolkit.tuebingen.mpg.de/hhomp.
INTRODUCTION
Outer membrane proteins (OMPs) occur in Gram-negative bacteria as well as in eukaryotic organelles of endosymbiotic origin, such as mitochondria and plastids (1). Except for the recently discovered α-helical OMP Wza from Escherichia coli (2), bacterial OMPs belong to the functionally diverse group of outer membrane β-barrels (OMBBs). Their transmembrane (TM) domains consist of ββ-hairpins in a barrel-shaped arrangement, forming a closed β-sheet around a central pore. OMBBs vary greatly in size, the β-barrels consisting of between 8 and 24 β-strands (3,4). They generally have an even number of β-strands, with the exception of VDAC-1, a mitochondrial OMBB with 19 β-strands (5,6). OMPs are involved in a broad range of biological functions such as active and passive transport, enzymatic activity, cell adhesion and structural anchoring. Sometimes, their very extended surface-exposed loops are prominent epitopes, which are exploited in vaccine development and strain typing with immunological methods.
Previous methods to predict the occurrence and topology (i.e. number and location of strands) of OMBBs from sequence data have mostly used analogous features such as amino acid composition or alternating hydrophobicity patterns (7,8), either implicitly, such as in neural network and SVM-based methods (9–11), or explicitly, such as in TMB-Hunt, which applies a k-nearest neighbour algorithm to the whole-sequence amino acid composition (12,13), or in BOMP, which employs C-terminal pattern recognition combined with a sliding window analysis of amino acids frequencies in alternating positions (14). TransFold is a topology prediction method that employs statistical pair potentials to predict inter-β-strand contacts (15,16). Various topology prediction methods were benchmarked in (17). The best-performing OMBB predictors, such as PROFtmb (18), have specially designed hidden Markov models (HMMs). These possess a circular topology containing states for upward and downward β-strands and two groups of states for the inner and outer loops (18–21). None of the existing methods can classify OMP sequences according to their functional subgroup.
In contrast to existing methods, our OMP prediction and classification server explicitly makes use of the fact that almost all OMBBs, spanning sizes from 8 to 24 strands, are homologous to each other (M. Remmert et al., submitted for publication) (22). Their structural similarity is not sufficient to demonstrate homology, since structure space is limited by a finite number of arrangements of secondary structure elements and hence structural convergence cannot be excluded (23). A tried and reliable indication for homology is significant sequence similarity, because sequence space is vast and sequence convergence therefore unlikely (24,25). We use profile–profile comparison methods, since sequence profiles conserve the signature of proteins’ past for much longer than their sequences. We assemble a database of putative OMPs by exhaustive, transitive homology detection starting from known OMPs. The database is likely to be nearly complete, as indicated by the 97% coverage of known OMPs in the E. coli genome (Table S1) and our other benchmark results (Figure 1, Table 1). HHomp searches this database using an OMP-specific extension of HHsearch, a method for pairwise profile HMM comparison (26) that integrates the OMP predictions from PROFtmb (18). We achieve considerably better sensitivity in predicting OMPs than the other tested methods. In addition, HHomp shows excellent performance in assigning OMP sequences to the correct functional subgroups.
Figure 1.

ROC plot comparing HHomp with the β-barrel prediction methods PROFtmb, TMB-Hunt and BOMP. The true positives (TPs) set consists of the 2164 proteins annotated as OMPs in TransportDB without a BLAST E-value < 0.01 to any of the 23 proteins in our trainings set. The FPs are 5000 randomly selected non-OMP proteins from SCOP. HHomp detects 63.5% of all TPs before the first FP. The effective error rate is the number of effective FPs divided by the sum of TPs and effective FPs are defined as FPs multiplied with 21 to obtain the same fraction of OMPs in the benchmark set as in Gram-negative genomes (2%) (1).
Table 1.
Number of proteins predicted as OMPs by HHomp and PROFtmb for various genomes
| Organism class | Organism | Proteins | HHomp hits with prob |
PROFtmb hits with score |
||
|---|---|---|---|---|---|---|
| 100 (%) | >90 (%) | >10 | >7 | |||
| Archaea | Aeropilum pernix | 1841 | 0 | 0 | 0 | 4 (0.2%) |
| Methanocaldococcus jan. | 1784 | 0 | 0 | 1 (0.1%) | 2 (0.1%) | |
| Gram-positive bacteria | Staphylococcus aureus | 2618 | 0 | 1 (0.04) | 6 (0.2%) | 13 (0.5%) |
| Bacillus subtilis | 4102 | 0 | 0 | 0 | 8 (0.2%) | |
| Corynebacterium diphtheriae | 2272 | 0 | 1 (0.04) | 3 (0.1%) | 20 (0.9%) | |
| Lactobacillus casei | 2771 | 0 | 0 | 4 (0.1%) | 17 (0.6%) | |
| Gram-negative bacteria | Escherichia coli | 4240 | 71 (1.7) | 77 (1.8) | 29 (0.7%) | 82 (1.9%) |
| Neisseria meningitidis | 2063 | 34 (1.6) | 36 (1.7) | 10 (0.5%) | 26 (1.2%) | |
| Agrobacterium tumefaciens | 5288 | 36 (0.7) | 39 (0.8) | 26 (0.5%) | 98 (1.8%) | |
| Bartonella henselae | 1488 | 29 (1.9) | 31 (2.1) | 7 (0.5%) | 21 (1.4%) | |
| Cyanobacteria | Synechococcus | 2892 | 20 (0.7) | 26 (0.9) | 2 (0.1%) | 32 (1.1%) |
| Eucarya | Saccharomyces cerevisiae | 5869 | 1 (0.02) | 4 (0.07) | 20 (0.3%) | 64 (1.0%) |
| Homo sapiens | 34 143 | 1 (∼0) | 20 (0.1) | 74 (0.2%) | 362 (1.0%) | |
Gram-positive bacteria and archaea do not have an outer membrane and should therefore not possess OMPs. Note that the error rate of HHomp at 90% probability is significant lower than the error rate of PROFtmb at a score of 10. In most Gram-negative bacteria, HHomp detects >1.5% OMPs with 100%, over twice more than PROFtmb at a score of 10. In yeast, HHomp correctly predicts the major mitochondrial OMBBs (35,36).
METHOD
HHomp is based on a database of precomputed profile HMMs of putative OMPs. The underlying sequences were identified by the exhaustive, transitive sequence search method HHsenser (27). To obtain representative bona fide OMPs as starting points for the transitive searches, we filtered the sequences of all bacterial OMPs in the Protein Data Bank (PDB) (28) for a maximum of 25% pairwise sequence identity. For each of these 23 proteins (Table S2), we performed an HHsenser run, searching through all bacterial sequences in the NCBI non-redundant (nr) database, and all sequences in the NCBI environmental database (both filtered for 90% maximum pairwise sequence identity). We pooled the largely overlapping results from these 23 searches into a nr database of over 20 000 proteins. Note that, to collect the sequences in the OMP database, the only information about OMPs we used was the identity of the 23 OMPs of known structure. We did not use any annotation such as that contained in SwissProt or TransportDB for filtering or complementing this sequence set.
Because the exhaustive HHsenser searches produce only sequence fragments that can be aligned reliably with one of the 23 starting OMPs in a transitive chain, we extracted the corresponding full-length sequences from the nr and environmental database for each of the fragments. The full-length sequences were then clustered and visualized with CLANS (29). In CLANS, sequences attract each other with a strength proportional to their pairwise BLAST (30) log E-values, so that similar sequences come to lie closely together. Clusters in this cluster map were defined by visual analysis. The obtained 474 clusters were manually annotated using the annotation of member proteins. Fifty five percent of the clusters contained only hypothetical proteins and thus likely represent as yet unknown groups of OMPs. Four clusters were identified as false positives (FPs). These were not removed for the benchmarks to permit a realistic estimation of the FP rate. A profile HMM was built for each cluster from a multiple alignment of its sequences (31). In addition to amino acid and gap frequencies, the database HMMs contain the secondary structure predicted by PSIPRED (32) and the β-barrel structure predicted by PROFtmb (18).
Given a sequence as input, HHomp builds a profile HMM by searching homologous sequences with buildali.pl from the HHsearch package (26) with default parameters. We predict the secondary structure and the β-barrel strands using PSIPRED and PROFtmb in the same way as for the database HMMs. This profile HMM is compared with the database of precomputed putative OMP HMMs (see below). The result is a list of OMP clusters, ranked by probability of a correct match. In the following, we explain how we have adapted HHsearch to the special case of OMP prediction.
- We combine the score between pairs of HMM columns in HHsearch with a score that measures how well the OMP β-strand predictions from PROFtmb match between query and database profiles. This β-barrel score SBB is added to the amino acid match score in the Viterbi algorithm of HHsearch with a weight factor wBB in the same way as is done for the secondary structure score (26). For each column in the profile HMM, PROFtmb predicts one of four β-barrel states, ρ ∈ {I, O, U, D} (inner and outer loop, upward and downward strand), together with a confidence value c ∈ {1, 3, 5, 7, 9}. To compare a predicted β-barrel state (ρ, c) with a β-barrel state σ of known structure, we construct five 4 × 4 substitution matrices, one for each value of c. As training set, we used 17 OMBB sequences with manually identified β-barrel states and 500 random non-OMP sequences. For all 517 proteins, we performed a PROFtmb prediction and estimated the probabilities P(σ; ρ, c) from the observed counts between predicted state (ρ, c) and manually annotated state σ. For simplicity, HHomp compares predicted states with each other, even when one of the structures is known. The log-odds score between predicted states (ρq, cq) and (ρp, cp) is
where the model probability in the numerator is the probability that both predicted states are obtained from an ancestral state σ, summed over all possible states σ.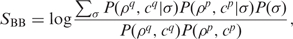
To avoid FP matches with non-TM domains, HHomp requires that a certain minimum number of query sequence residues is aligned to the predicted TM region of the database HMM. We use a minimum coverage of 50 amino acids and a minimum raw score of 50 in the predicted TM region. To predict the TM regions in the database HMMs, we searched through all OMPs in the SCOP database with each database HMM. The predicted TM region in the database HMM was defined as comprising all residues aligned to the TM domain of at least one of the best five OMP matches in the SCOP database.
- Not all profile HMMs in the OMP database are equally likely to represent OMPs. For each database match X, HHomp reports the corresponding probability that the query protein is an OMP, calculated according to the following formula:
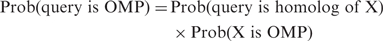
The probabilies Prob(X is OMP) are estimated for the entire database by searching with each database HMM X through all OMPs in the PDB and setting these probabilities to the match probability of the best-matching OMP.
RESULTS
We first compare HHomp with the prediction methods PROFtmb (18), BOMP (14) and TMB-Hunt (12) on a test set of annotated OMPs from TransportDB (33) and 5000 negative, non-OMP sequences randomly selected from the SCOP database (version 1.69) (34). The TransportDB contains 4494 outer membrane channels from fully sequenced organisms, which were annotated by experimental and bioinformatic evidence. To avoid testing HHomp on proteins that are similar to one of the 23 proteins with which it was trained (Table S2), we exclude all sequences from the test set that have a BLAST E-value better than 0.01 or a sequence identity larger than 20% to one of the 23 proteins. This yields a test set of 2164 OMBBs.
HHomp detects 63.5% of the true positives (TPs) before including the first FP. This is 70% more than PROFtmb, four times more than BOMP and 10 times more than TMB-Hunt (Figure 1). Similar improvements are observed at 10% effective error rate. Note that, in contrast to the other methods, the performance of PROFtmb is impaired when we use negative sequences with the same mean length as in the positive set (Figure S1).
In a second benchmark, we count the number of proteins predicted as OMPs by HHomp and PROFtmb in various genomes (Table 1). Gram-positive bacteria and archaea do not have an outer membrane and should therefore not possess OMPs. We use probability cut-offs of 100% and 90% for HHomp and score cut-offs of 10 and 7 for PROFtmb [these cut-offs should correspond roughly to 100% and 90% accuracy (18)]. The error rate at these cut-offs can be estimated by dividing the FP hits in Gram-positive bacteria and archaea by the total number of proteins in their genomes. In this way, we estimate an error rate of ∼2×10−4 for HHomp at 90% cut-off and of ∼10−3 for PROFtmb at a score of 10. In Gram-negative bacteria, ∼1.5–3% of the proteins is assumed to be OMPs (1). HHomp predicts >1.5% OMPs in most Gram-negative bacteria with a probability of 100%, over twice more than PROFtmb at a score of 10. HHomp detects 57 out of the 59 known E. coli OMPs in TransportDB with >94% probability, corresponding to an estimated error rate <2 × 10−4, whereas PROFtmb identifies 23 at an error rate of ∼10−3 (Table S1). Furthermore, HHomp is able to detect OMPs in eukarya even though its database is built from bacterial protein sequences. In the genome of Saccharomyces cerevisiae, HHomp yields fours matches with >90% probability. All of them are TPs, comprising the major known mitochondrial OMBBs (35,36)—two VDAC isoforms (GI 6322077 and 6324273), one component of the TOM complex (TOM40, GI 6323859) and one component of the SAM complex (SAM50, GI 6324302). None of these proteins can be identified by PROFtmb with a score >7 (Table S3), and none of the 64 proteins predicted by PROFtmb is annotated as known or putative mitochondrial OMP. In human, HHomp identifies a known mitochondrial OMP (SAM 50 homolog) with 100% probability and a further 17 proteins >90% probability. Of these, seven are known mitochondrial OMPs and 10 are FP hits (Table S4). Eight FP had P-values >0.05 but received high HHomp probabilities through their elevated PROFtmb scores. Two FPs (ladinin 1) matched the N-terminal α-helix of the autotransporters, which is part of the OMBB domain in the SCOP domain definition.
In contrast to other OMP predictions, HHomp is able to assign proteins to OMP families with high reliability. In E. coli, all annotated OMPs are classified correctly when the best-ranked hit is used for the family assignment (Table S1).
A method based on homology detection like HHomp might be favoured by the presented benchmarks: the proteins in TransportDB, which were annotated by inference from homologous, experimentally validated OMPs, might be more easily predicted as OMBBs by such a method. However, this also applies to some extent to TMB-Hunt and PROFtmb, which use homology information by constructing a profile from the query sequence. Note that the two described benchmarks are fair in the sense that HHomp was not given more information than the other OMBB predictors. All four methods merely used the identity of bona fide OMBBs in the PDB. HHomp's database of putative OMBBs was used as obtained from our automatic searches without modifications (e.g. adding annotated OMBBs from Swissprot or TransportDB). Only after the benchmarks we removed four obvious FP clusters. Therefore, the benchmarks provide a rather conservative estimate for HHomp's prediction performance. One obvious limitation of HHomp is that it can only detect OMPs that are homologous to known groups with which it was trained. If a still undiscovered group of OMBBs exists that is not homologous to the known OMBBs, tools relying on analogous features instead of homology will likely be at an advantage.
WEBSERVER
The HHomp webserver (available at http://toolkit.tuebingen.mpg.de/hhomp) consists of an OMP prediction and searching interface and a browsing interface for the underlying OMP database. With the prediction interface, the user can search the database with a query protein sequence. Various parameters for alignment building and searching can be modified (explained in the online help). After a few minutes, the server returns a graphical overview of the matched regions, a detailed and annotated list of matched OMP database HMMs and the corresponding alignments (Figure 2). An alternative view of the alignments can be selected, in which the columns of the aligned profile HMMs are represented as coloured histograms. The detected OMP HMMs are linked to the browsing interface with detailed description pages for the OMP clusters, containing alignments and 3D models for the TM domain if available (Figure 2, upper insert). Detailed help pages explain input parameters and output formats. HHomp is integrated into the MPI Bioinformatics Toolkit (37), which provides a user-friendly framework for job control (e.g. for running jobs in parallel) and offers many other tools for sequence analysis. In particular, a 3D model can be built with the MODELLER software (38) based on the HHomp results.
Figure 2.

HHomp results page with graphical overview of regions matched to OMPs from the HHomp database, summary results list and detailed alignments.
Executables for Linux (32/64bit) and the HHomp database are freely available for academic users and can be downloaded from ftp://ftp.tuebingen.mpg.de/pub/protevo/HHomp.
CONCLUSION
The finding that almost all OMBBs are homologous to each other irrespective of their number of β-strands and hence that they can be predicted and classified using homology detection methods has proved very fruitful. Our OMP database, constructed with exhaustive, transitive homology searches, contains only few non-OMP sequences, as indicated by the very low number of FP matches in Gram-positive bacteria and archaea. Regarding the completeness of our OMP database, we note that (i) a large number of database clusters contain only hypothetical proteins, (ii) we predict nearly all known OMPs in the E. coli genome and (iii) we correctly predict the major mitochondrial OMBBs. This shows that HHomp is able to identify even very distant relatives of the 23 bacterial OMPs used for training the OMP database. In summary, although slower than most other methods, HHomp offers excellent sensitivity at very low error rate for the detection of OMPs among bacterial and eukaryotic sequences. For the application to entire genomes, a downloadable version of the software is available.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Ludwig-Maximilians-University Munich.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Oliver Kohlbacher for support during MR's diploma thesis and all users who helped to improve our webserver with their questions, feedback and bug reports.
REFERENCES
- 1.Wimley WC. The versatile β-barrel membrane protein. Curr. Opin. Struct. Biol. 2003;13:404–411. doi: 10.1016/s0959-440x(03)00099-x. [DOI] [PubMed] [Google Scholar]
- 2.Dong C, Beis K, Nesper J, Brunkan-LaMontagne AL, Clarke BR, Whitfield C, Naismith JH. Wza the translocon for E. coli capsular polysaccharides defines a new class of membrane proteins. Nature. 2006;444:226–229. doi: 10.1038/nature05267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koebnik R, Locher KP, Gelder PV. Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol. Microbiol. 2000;37:239–253. doi: 10.1046/j.1365-2958.2000.01983.x. [DOI] [PubMed] [Google Scholar]
- 4.Remaut H, Tang C, Henderson NS, Pinkner JS, Wang T, Hultgren SJ, Thanassi DG, Waksman G, Li H. Fiber formation across the bacterial outer membrane by the chaperone/usher pathway. Cell. 2008;133:640–652. doi: 10.1016/j.cell.2008.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bayrhuber M, Meins T, Habeck M, Becker S, Giller K, Villinger S, Vonrhein C, Griesinger C, Zweckstetter M, Zeth K. Structure of the human voltage-dependentanion channel. Proc. Natl Acad. Sci. USA. 2008;105:15370–15375. doi: 10.1073/pnas.0808115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hiller S, Garces RG, Malia TJ, Orekhov VY, Colombini M, Wagner G. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science. 2008;321:1206–1210. doi: 10.1126/science.1161302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Punta M, Forrest LR, Bigelow H, Kernytsky A, Liu J, Rost B. Membrane protein prediction methods. Methods. 2007;41:460–474. doi: 10.1016/j.ymeth.2006.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neuwald AF, Liu JS, Lawrence CE. Gibbs motif sampling: detection of bacterial outer membrane protein repeats. Protein Sci. 1995;4:1618–1632. doi: 10.1002/pro.5560040820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gromiha MM, Ahmad S, Suwa M. Neural network-based prediction of transmembrane β-strand segments in outer membrane proteins. J. Comp. Chem. 2004;25:762–767. doi: 10.1002/jcc.10386. [DOI] [PubMed] [Google Scholar]
- 10.Jacoboni I, Martelli PL, Fariselli P, De Pinto V, Casadio R. Prediction of the transmembrane regions of β-barrel membrane proteins with a neural network-based predictor. Protein Sci. 2001;10:779–787. doi: 10.1110/ps.37201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Natt NK, Kaur H, Raghava GP. Prediction of transmembrane regions of β-barrel proteins using ANN- and SVM-based methods. Proteins. 2004;56:11–18. doi: 10.1002/prot.20092. [DOI] [PubMed] [Google Scholar]
- 12.Garrow AG, Agnew A, Westhead DR. TMB-Hunt: a web server to screen sequence sets for transmembrane β-barrel proteins. Nucleic Acids Res. 2005;33:W188–W192. doi: 10.1093/nar/gki384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garrow AG, Westhead DR. A consensus algorithm to screen genomes for novel families of transmembrane β-barrel proteins. Proteins. 2007;69:8–18. doi: 10.1002/prot.21439. [DOI] [PubMed] [Google Scholar]
- 14.Berven FS, Flikka K, Jensen HB, Eidhammer I. BOMP: a program to predict integral β-barrel outer membrane proteins encoded within genomes of Gram-negative bacteria. Nucleic Acids Res. 2004;32:W394–W399. doi: 10.1093/nar/gkh351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waldispühl J, Berger B, Clote P, Steyaert JM. trandFold: a web server for predicting the structure and residue contacts of transmembrane beta-barrels. Nucleic Acids Res. 2006;34:189–193. doi: 10.1093/nar/gkl205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waldispühl J, O'Donnell CW, Devadas S, Clote P, Berger B. Modeling ensembles of transmembrane beta-barrel proteins. Proteins. 2008;71:1097–1112. doi: 10.1002/prot.21788. [DOI] [PubMed] [Google Scholar]
- 17.Bagos PG, Liakopoulos TD, Hamodrakas SJ. Evaluation of methods for predicting the topology of beta-barrel outer membrane proteins and a consensus prediction method. BMC Bioinformatics. 2005;6:7. doi: 10.1186/1471-2105-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bigelow HR, Petrey DS, Liu J, Przybylski D, Rost B. Predicting transmembrane β-barrels in proteomes. Nucleic Acids Res. 2004;32:2566–2577. doi: 10.1093/nar/gkh580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martelli PL, Fariselli P, Krogh A, Casadio R. A sequence-profile-based HMM for predicting and discriminating β-barrel membrane proteins. Bioinformatics. 2002;18:46–53. doi: 10.1093/bioinformatics/18.suppl_1.s46. [DOI] [PubMed] [Google Scholar]
- 20.Liu Q, Zhu YS, Wang BH, Li YX. A HMM-based method to predict the transmembrane regions of β-barrel membrane proteins. Comput. Biol. Chem. 2003;27:69–76. doi: 10.1016/s0097-8485(02)00051-7. [DOI] [PubMed] [Google Scholar]
- 21.Bagos PG, Liakopoulos TD, Spyropoulos IC, Hamodrakas SJ. PREDTMBB: a web server for predicting the topology of β-barrel outer membrane proteins. Nucleic Acids Res. 2004;32:W400–W404. doi: 10.1093/nar/gkh417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnold T, Poynor M, Nussberger S, Lupas AN, Linke D. Gene duplication of the eight-stranded beta-barrel OmpX produces a functional pore: a scenario for the evolution of transmembrane beta-barrels. J. Mol. Biol. 2007;366:1174–1184. doi: 10.1016/j.jmb.2006.12.029. [DOI] [PubMed] [Google Scholar]
- 23.Krishna SS, Grishin NV. Structurally analogous proteins do exist! Structure. 2004;12:1125–1127. doi: 10.1016/j.str.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 24.Murzin AG. How far divergent evolution goes in proteins. Curr. Opin. Struct. Biol. 1998;8:380–387. doi: 10.1016/s0959-440x(98)80073-0. [DOI] [PubMed] [Google Scholar]
- 25.Doolittle RF. Convergent evolution: the need to be explicit. Trends Biochem. Sci. 1994;19:15–18. doi: 10.1016/0968-0004(94)90167-8. [DOI] [PubMed] [Google Scholar]
- 26.Söding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 27.Söding J, Remmert M, Biegert A, Lupas AN. HHsenser: exhaustive transitive profile search using HMM-HMM comparison. Nucleic Acids Res. 2006;34:W374–W378. doi: 10.1093/nar/gkl195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frickey T, Lupas AN. CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics. 2004;20:3702–3704. doi: 10.1093/bioinformatics/bth444. [DOI] [PubMed] [Google Scholar]
- 30.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 31.Lassmann T, Sonnhammer EL. Kalign - an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics. 2005;6:298–298. doi: 10.1186/1471-2105-6-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 33.Ren Q, Kang KH, Paulsen IT. TransportDB: a relational database of cellular membrane transport systems. Nucleic Acids Res. 2004;32:D284–D288. doi: 10.1093/nar/gkh016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol.Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 35.Paschen SA, Waizenegger T, Stan T, Preuss M, Cyrklaff M, Hell K, Rapaport D, Neupert W. Evolutionary conservation of biogenesis of beta-barrel membrane proteins. Nature. 2003;426:862–866. doi: 10.1038/nature02208. [DOI] [PubMed] [Google Scholar]
- 36.Pfanner N, Wiedemann N, Meisinger C, Lithgow T. Assembling the mitochondrial outer membrane. Nat. Struct. Mol. Biol. 2004;11:1044–1048. doi: 10.1038/nsmb852. [DOI] [PubMed] [Google Scholar]
- 37.Biegert A, Mayer C, Remmert M, Söding J, Lupas A. The MPI Bioinformatics Toolkit for protein sequence analysis. Nucleic Acids Res. 2006;34:W335–W339. doi: 10.1093/nar/gkl217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.