


Abstract
The CentroidFold web server (http://www.ncrna.org/centroidfold/) is a web application for RNA secondary structure prediction powered by one of the most accurate prediction engine. The server accepts two kinds of sequence data: a single RNA sequence and a multiple alignment of RNA sequences. It responses with a prediction result shown as a popular base-pair notation and a graph representation. PDF version of the graph representation is also available. For a multiple alignment sequence, the server predicts a common secondary structure. Usage of the server is quite simple. You can paste a single RNA sequence (FASTA or plain sequence text) or a multiple alignment (CLUSTAL-W format) into the textarea then click on the ‘execute CentroidFold’ button. The server quickly responses with a prediction result. The major advantage of this server is that it employs our original CentroidFold software as its prediction engine which scores the best accuracy in our benchmark results. Our web server is freely available with no login requirement.
INTRODUCTION
Recent research has discovered that functional noncoding RNAs (ncRNAs) play essential roles in cells. It is well-known that functions of ncRNAs are deeply related to their secondary structures rather than primary sequence structures (e.g. hairpin structures for miRNA precursors and cloverleaf structures for tRNAs). Therefore, the importance of accurate secondary structure predictions has increased. The most successful approach for predicting RNA secondary structures is based on the free energy minimization such as Mfold (1) and RNAfold in the Vienna RNA package (2). Alternative approach is based on probabilistic frameworks, including stochastic context-free grammars (SCFGs), which can model RNA secondary structures without pseudoknots (3). These approaches employ a dynamic programming technique called the Cocke–Younger–Kasami (CYK) algorithm for calculating the minimum free energy (MFE) or maximum likelihood (ML) structure (4). However, several studies have pointed out a drawback of the MFE/ML estimators that the MFE/ML structure generally has an extremely low probability and is even not optimal with respect to the number of corrected predicted base pairs (5–8). Hence, alternative estimators which consider the ensemble of all possible solutions, instead of only the solution with the highest probability, have been developed. These include the centroid estimator employed by Sfold (6,7) and the maximum expected accuracy (MEA) estimator employed by CONTRAfold (9). These estimators maximize the expectation of an object function related to the accuracy of the prediction.
We have recently proposed a generalized centroid estimator, called a γ-centroid estimator, which can be more appropriate for the accuracy measure of RNA secondary structure prediction than the MEA estimator, and have furthermore shown that the γ-centroid estimator is theoretically and experimentally superior to the MEA estimator (10).
CentroidFold is an implementation of the γ-centroid estimator for predicting RNA secondary structures, and is distributed as a free software from http://www.ncrna.org/software/centroidfold/. In this article, we introduce a web application of CentroidFold with a very simple interface. It takes an individual RNA sequence or a multiple alignment of RNA sequences, and returns its predicted (common) secondary structure with a graphical representation. Our web application is available at http://www.ncrna.org/centroidfold/ for unrestricted use.
METHODS
Algorithm
CentroidFold predicts RNA secondary structures with the γ-centroid estimator (10) which is a kind of posterior decoding method based on statistical decision theory. We define a gain function between a true structure y and a candidate structure ŷ by
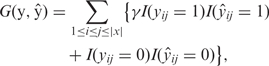 |
1 |
where γ is a weight for base pairs, yij is 1 if the i-th and the j-th nucleotides form a base pair in y, or 0 otherwise, and I(condition) is an indicator function which takes a value of 1 or 0 depending on whether the condition is true or false. The gain function (1) is equal to the weighted sum of the number of true positives and the number of true negatives of base pairs.
The expectation of the gain function (1) with respect to an ensemble of all possible secondary structures under a given posterior distribution p(y|x) is
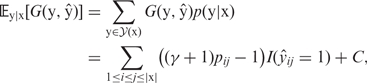 |
2 |
where 𝒴(x) is a set of all possible secondary structures for x, |x| is the length of x and C is a constant independent of ŷ. The base-pairing probability pij = 𝔼y|x[yij] is the probability that the i-th and j-th nucleotides form a base pair in y, which can be interpreted as confidence measure of predicted base pairs. The posterior distribution p(y|x) for calculating base-pairing probabilities can be chosen from various implementations including the McCaskill model (11) and the CONTRAfold model (9). We employ the CONTRAfold model as the default setting of CentroidFold in accordance with our benchmark (10).
Then, we can find ŷ which maximizes the expected gain (2) using the recursive equations:
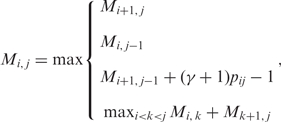 |
3 |
and tracing back from M1,|x|.
We can control the trade-off between specificity and sensitivity by γ. If γ = 1, our estimator is equivalent to the centroid estimator (7,8). The γ-centroid estimator is similar to the MEA estimator (9). The difference between them is only in the gain functions: the gain function of the γ-centroid is more suitable for evaluation measures for RNA secondary structure prediction than that of the MEA estimator. See (10) for more details.
Web server
The CentroidFold web server can be accessed on http://www.ncrna.org/centroidfold/ providing a very simple form for inputs. The server can accept two types of sequence formats: the FASTA format for predicting secondary structures of a single RNA sequence, and the CLUSTAL-W format for predicting common secondary structures of a multiple alignment of RNA sequences. The format of entered sequences can be automatically detected, and the appropriate prediction method is executed after the ‘execute CentroidFold’ button is clicked (Figure 1). The result of prediction is shown as a standard base-pair notation (Figure 2A) and a graphical representation (Figure 2B). Each predicted base pair is colored with the heat color gradation from blue to red corresponding to the base-pairing probability from 0 to 1. You can see the PDF version of the graphical presentation from a link given below the Figure 2.
Figure 1.

The CentroidFold web server.
Figure 2.

The result of predicting a common secondary structure for an example multiple alignment of Qrr RNAs. (A) A standard base-pair notation. (B) A graphical representation.
DISCUSSION AND CONCLUSIONS
The CentroidFold web server allows biologists to predict RNA (common) secondary structures with the most accurate prediction engine which scores the best accuracy in our benchmark results. For example, RNAfold based on MFE fails to predict a secondary structure of a typical tRNA sequence (Rfam id: M19341.1/98-169), whereas CentroidFold almost successfully predicts its secondary structure as shown in Figure 3. This result suggests that several ncRNA sequences do not always form MFE secondary structures, and posterior decoding methods including the γ-centroid estimator can provide more reliable predictions.
Figure 3.

Comparison of secondary structures of a tRNA sequence (Rfam id: M19341.1/98–169) between RNAfold (left), CentroidFold (center) and the reference structure (right).
The most recent CentroidFold software has implemented the stochastic suboptimal folding algorithm like Sfold (7) with the stochastic traceback algorithm for the CONTRAfold model instead of the McCaskill model. We are planing to provide its web interface for easy use.
FUNDING
This work was supported in part by a grant from ‘Functional RNA Project’ funded by the New Energy and Industrial Technology Development Organization (NEDO) of Japan, and was also supported in part by Grant-in-Aid for Scientific Research on Priority Area ‘Comparative Genomics’ from the Ministry of Education, Culture, Sports, Science and Technology of Japan. Funding for open access charge: Internal fund of Computational Biology Research Center.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Hisanori Kiryu and our colleagues from the RNA Informatics Team at the Computational Biology Research Center (CBRC) for fruitful discussions.
REFERENCES
- 1.Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981;9:133–148. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dowell RD, Eddy SR. Evaluation of several lightweight stochastic context-free grammars for RNA secondary structure prediction. BMC Bioinformatics. 2004;5:71. doi: 10.1186/1471-2105-5-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Durbin R, Eddy S, Krogh A, Mitchison G. Biological Sequence Analysis. Cambridge, England: Cambridge University Press; 1998. [Google Scholar]
- 5.Knudsen B, Hein J. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res. 2003;31:3423–3428. doi: 10.1093/nar/gkg614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding Y, Lawrence CE. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res. 2003;31:7280–7301. doi: 10.1093/nar/gkg938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding Y, Chan CY, Lawrence CE. RNA secondary structure prediction by centroids in a Boltzmann weighted ensemble. RNA. 2005;11:1157–1166. doi: 10.1261/rna.2500605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carvalho LE, Lawrence CE. Centroid estimation in discrete high-dimensional spaces with applications in biology. Proc. Natl Acad. Sci. USA. 2008;105:3209–3214. doi: 10.1073/pnas.0712329105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Do CB, Woods DA, Batzoglou S. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics. 2006;22:e90–e98. doi: 10.1093/bioinformatics/btl246. [DOI] [PubMed] [Google Scholar]
- 10.Hamada M, Kiryu H, Sato K, Mituyama T, Asai K. Prediction of RNA secondary structure using generalized centroid estimators. Bioinformatics. 2009;25:465–473. doi: 10.1093/bioinformatics/btn601. [DOI] [PubMed] [Google Scholar]
- 11.McCaskill JS. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990;29:1105–1119. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]