


Abstract
Isotope labeling combined with liquid chromatography–mass spectrometry (LC–MS) provides a robust platform for analyzing differential protein expression in proteomics research. We present a web service, called MaXIC-Q Web (http://ms.iis.sinica.edu.tw/MaXIC-Q_Web/), for quantitation analysis of large-scale datasets generated from proteomics experiments using various stable isotope-labeling techniques, e.g. SILAC, ICAT and user-developed labeling methods. It accepts spectral files in the standard mzXML format and search results from SEQUEST, Mascot and ProteinProphet as input. Furthermore, MaXIC-Q Web uses statistical and computational methods to construct two kinds of elution profiles for each ion, namely, PIMS (projected ion mass spectrum) and XIC (extracted ion chromatogram) from MS data. Toward accurate quantitation, a stringent validation procedure is performed on PIMSs to filter out peptide ions interfered with co-eluting peptides or noise. The areas of XICs determine ion abundances, which are used to calculate peptide and protein ratios. Since MaXIC-Q Web adopts stringent validation on spectral data, it achieves high accuracy so that manual validation effort can be substantially reduced. Furthermore, it provides various visualization diagrams and comprehensive quantitation reports so that users can conveniently inspect quantitation results. In summary, MaXIC-Q Web is a user-friendly, interactive, robust, generic web service for quantitation based on ICAT and SILAC labeling techniques.
INTRODUCTION
Recent advances in incorporating isotopic-labeling strategies into mass spectrometry (MS)-based proteomics have facilitated quantitation studies of differently expressed levels of proteins in complex biological samples (1). Stable isotope labeling for quantitation based on MS survey scans can be divided into three major categories: chemical labeling, e.g. ICAT (2); enzymatic labeling, e.g. 18O-labeling (3); and metabolic labeling, e.g. SILAC (4,5). To calculate the relative peptide ratio, the corresponding extracted ion chromatograms (XICs) are constructed and their areas are used to calculate peptide and protein ratios. Several tools have been developed to facilitate quantitation analysis, for example, XPRESS (6), ASAPRatio (7), RelEx (8), MSQuant (9), MFPaQ (10) and MaxQuant (11). The pioneering XPRESS tool utilizes signals of precursor ions to reconstruct XICs. ASAPRatio adopts several numerical and statistical methods for smoothing XICs, detecting ratio outliers, and error analysis for assessing the quantitation results. In addition, ASAPRatio uses signal-to-noise (S/N) ratios to filter out unquantifiable XICs before ratio determination. RelEx uses the least-squares regression technique to align paired XICs and determine quantifiable areas of the aligned XICs. The ion abundance ratio is determined by the regression slope, and the maximum correlation coefficient serves as a confidence measure of the quantitation results. MSQuant is another widely used quantitation tool. It is based on the Mascot search output and utilizes liquid chromatography (LC) profiles to compute quantitation ratios. MFPaQ is also specifically designed for Mascot users and utilizes the parsed identification results to extract quantitation data. The program selects MS survey scans to construct the elution peaks of each pair of ions, and the elution profile intensities are used to compute the corresponding ratio. MaxQuant provides a platform that sequentially performs 3D feature detection, SILAC pairs finding, mass recalibration, peptide identification and protein quantitation for high-resolution MS data. After the powerful mass recalibration, the number of identified peptides can be significantly increased. For protein quantitation, it also provides a significance factor to indicate the confidence of the calculated protein ratio.
Compared with MS/MS quantitation based on signature ions, constructing an XIC requires a larger retention time range and consequently may include co-eluted signal of other peptide ions or noise. Most of the aforementioned tools evaluate the data validity for quantitation, i.e. determining whether the peptide ion is quantifiable, based on the shapes of XICs. As observed, to detect the interference of co-eluted peptides and noise, both elution time and m/z of the underlying peptide ion should be taken into account. Therefore, ion mass spectra over the elution time range are accumulated to generate the projected ion mass spectrum (PIMS, whose concept is shown in Figure 2), which is an important indicator to determine data quality. Interference from co-eluted peptides and noise is detected by a validation procedure on the PIMS to ensure data quality of quantifiable peptides. In this article, we present a web service called MaXIC-Q Web, which provides a generic platform for quantitation analysis in cICAT, SILAC or user-defined stable isotope labeling proteomic experiments. It can automatically process signals, remove noise, validate data quality for quantitation, select appropriate quantitation signals and calculate abundance ratios.
Figure 2.

Construction of the PIMS and XIC: (A) PIMS and (B) XIC.
As a generic computation platform for high-throughput quantitative proteomics, MaXIC-Q Web offers the following features: (i) it accepts the mzXML (12) spectral format, which can be converted from raw files of various mass spectrometers by existing tools, as well as search results from commonly used search engines, including Mascot, SEQUEST and ProteinProphet (13). (ii) It allows user-defined isotope codes, which cover a very broad range of quantitation strategies for various in vivo and in vitro labeling techniques, and even user-developed labeling methods. (iii) MaXIC-Q Web constructs the PIMS for each peptide ion from MS survey scans and performs stringent data validation on the PIMS using three criteria to validate whether the peptide ion has good spectral data without interference from noise or co-eluted peptides for correct quantitation in an unattended manner. However, to add flexibility, MaXIC-Q Web allows users to determine whether partially use or not to use the stringent validation procedure. (iv) MaXIC-Q Web provides visualization of quantitation data, including Elution3D, XIC and PIMS diagrams.
As MaXIC-Q Web is designed as a generic platform, it has been applied to datasets from different instruments. Here, we report the performance of MaXIC-Q Web on three datasets generated from cICAT-labeling experiments and a SILAC experiment. The first dataset is a model dataset of nine standard protein mixtures published by Moulder et al. (14), which was analyzed by an LC–MS/MS system consisting of a nano-LC coupled with a QSTAR Pulsar ESI-hybrid quadrupole-TOF instrument. The second dataset is a large-scale dataset containing 33 SCX fractions from cICAT-labeled proteins of endothelial cells in response to nitric oxide stimulation, which were analyzed by μLC–MS/MS analysis with the HP 1100 solvent delivery system (Hewlett-Packard, Palo Alto, CA, USA) and quadrupole/time-of-flight mass spectrometer (QSTAR Pulsar i, Applied Biosystems, Foster City, CA, USA). The dataset is used to assess quantitation accuracy and robustness and computation speed of the web service. The computation results show that MaXIC-Q Web can efficiently generate accurate and reliable quantitation results. Furthermore, the quantitation accuracy on the second dataset is 80%. The third dataset is a large-scale dataset of SILAC-labeled proteins from EAhy926 cell lysate, and the light- and heavy-labeled samples were mixed at ratio 1:1 and analyzed by LTQ-Orbitrap. In the SILAC experiment, MaXIC-Q Web reported a convincible mean protein ratio of 0.987 ± 0.278 for 253 quantifiable proteins; details of quantitation are available in Supplementary Table.
METHOD
The pipeline for quantitation analysis using MaXIC-Q Web is shown in Figure 1, where MaXIC-Q Web's workflow is also shown in the quantitation stage. Toward automatic quantitation with minimum manual inspection, MaXIC-Q Web is equipped with modules for reconstruction of unbiased mass spectra and XICs, stringent spectra feature validation, and accurate ion, peptide and protein level calculation. The workflow of MaXIC-Q Web comprises five steps: filtering, ion level processing, peptide ion validation, peptide level processing and protein level processing.
Figure 1.

Pipeline of quantitation analysis using MaXIC-Q Web.
In the first step, i.e. filtering, MaXIC-Q Web filters out low-confidence peptides according to a user-defined threshold to ensure accurate quantitation from confidently assigned peptides. In addition, MaXIC-Q Web provides an option for users to filter out degenerate peptides. As peptide ion level processing and peptide ion validation are crucial steps for quantitation accuracy, we briefly described these two steps and more details are provided in Supplementary Data.
Peptide ion level processing
In addition to quantifying an identified peptide ion with a specified charge state (CS), MaXIC-Q Web aims to detect the peptide ion unidentified in other CSs (range: from +1 to +4) in order to quantify peptide ions as many as possible to reduce quantitation bias. For example, if a peptide ion is identified as +2 charged, MaXIC-Q Web also detects and quantifies +1, +3 and +4 peptide ions.
For each isotope-labeled, confidently identified peptide ion, MaXIC-Q constructs PIMS for each specific CS and reconstructs the corresponding XIC (details described in Supplementary Data), as shown in Figure 2. Then the area of XIC is used to represent peptide ion abundance.
Peptide ion validation
After constructing PIMS, MaXIC-Q Web sequentially checks the three criteria, S/N ratio, CS and isotope pattern (IP), on each PIMS to determine whether each individual labeled ion has spectral data of good quality for quantitation. The S/N criterion checks whether there exists a valid peak, i.e. a peak with an S/N ≥2.5, and the peak's m/z range covers the precursor m/z. The CS criterion checks whether there exist at least three isotope peaks that have correct charge state as the targeted peptide ion. The IP criterion checks whether the peak distribution is similar to the theoretical isotope distribution of the targeted ion, as measured by chi-squared goodness-of-fit test. When the PIMS fails in one criterion, the validation procedure stops and the labeled ion is determined as no expression or unquantifiable. Only when the PIMS pass the three criteria, the labeled ion is quantifiable by its XIC area. When both light- and heavy-paired [shortened as (L,H)-paired] ions pass the validation, the ion ratio is calculated.
Peptide and protein ratio determination
After the peptide validation for all CSs (from +1 to +4), a peptide ratio is calculated by weighted average of all the ratios of quantifiable ions. In the final step, before calculating protein ratios, the ratios of non-degenerate peptides are normalized using Central Tendency Normalization (15) to eliminate systematic errors. Then the protein ratio is calculated based on the normalized peptide ratios.
RESULTS
MaXIC-Q Web has been tested on a mixture of nine standard proteins using ICAT labeling. We compared quantitation results of MaXIC-Q Web with existing web-based tools, XPRESS and ASAPRatio. The complete results are provided in the Supplementary.xls file and an example of MaXIC-Q's result is shown in Supplementary Data. For the four proteins commonly quantified by the three tools, we calculated the average error of protein and peptide ratios as follows:
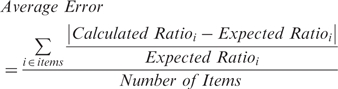 |
1 |
For XPRESS, ASAPRatio and MaXIC-Q Web, the average errors of the four proteins were 0.675, 0.821 and 0.339, respectively. Overall speaking, the MaXIC-Q Web's results are comparable with those of the other two programs, and it demonstrates more stable and accurate computation capability.
Large-scale quantitation for a comparative study of Nitric oxide (NO)-treated endothelial cells by cICAT strategy and 2D LC–MS/MS
MaXIC-Q Web was applied to a large-scale quantitative proteomic dataset generated from the comparative study of NO-treated EA.hy926 endothelial cells. The control and NO-treated cells were labeled with light and heavy cICAT, respectively; and a total of 33 fractions were generated by SCX fractionation and analyzed by LC–MS/MS. The search result files and 66 mzXML files (∼6 GB) were input to MaXIC-Q Web. To process the input data, MaXIC-Q Web only took 110 min on Microsoft Windows Server 2003 R2 × 64 edition service pack 2 with 64-bit AMD Opteron Processor 2210 CPU, 1.8 GHz processors, 7200 r.p.m. SATA hard disk 500 GB and 8 G RAM (only 2 G was used for this operation). In contrast, ASAPRatio took 10 h and 33 min on the same computer. Thus, MaXIC-Q Web provides an efficient platform to expedite quantitation analysis.
Using the SEQUEST search engine, the dataset generated 7109 identified peptides and 1259 corresponding proteins without restriction on the peptide and protein probability settings. After filtering low-confidence peptides and proteins, we obtained 5488 non-degenerate peptides (peptide probability > 0.7) and 770 proteins (protein probability > 0.6). Partial output reports of the peptide and protein quantitation results are presented in Supplementary Data. Moreover, in this experiment, MaXIC-Q Web reported a mean peptide ratio of 0.945 ± 0.245 for 3948 peptides. The result was reasonably accurate. The mean of the protein ratios was 0.956 ± 0.235 after peptide ratio normalization, and 0.924 ± 0.227 without peptide ratio normalization. The SDs for the mean peptide ratio and the mean protein ratio were approximately within the 20% range. We assume that this can be attributed to the c-ICAT labeling method combined with MS analysis (10).
Handling co-elution of peptide ions
In large-scale experiments, noisy data present a critical challenge for accurate quantification. Various levels of noise inevitably occur depending on the separation capability, resolution and mass accuracy of LC–MS/MS. Data noise includes background signal and co-elution of multiple peptides, i.e. the signals of a target ion are interfered by co-eluted peptides. Most quantitation tools, including MaXIC-Q Web, handle background noise by background subtraction. However, the co-elution problem cannot be resolved by simply examining the XIC or ion mass spectrum alone. For example, in Figure 3, a peptide VTCPNHPDAILVEDYR is identified by the SEQUEST search engine with a high peptide probability 0.9985 reported by PeptideProphet. By examining the Elution3D diagram, it can be seen that there is a peptide ion with close precursor m/z, co-eluting with the heavy peptide ion of VTCPNHPDAILVEDYR. However, validation based on XICs or MS/MS spectra may not detect this circumstance, because both heavy and light ions have good bell-shaped XICs, and the MS/MS spectrum has very good quality as evidenced by the high peptide probability 0.9985 reported by PeptideProphet. In contrast, using peptide ion validation on PIMS, the heavy-labeled ion failed at the IP criterion and the co-elution can be detected. The peptide ion is reported as unquantifiable to prevent inaccurate ion ratio interfering the peptide and protein ratios.
Figure 3.

An example of peptide co-elution from the dataset of NO-treated endothelial cells: (A) Elution3D diagram, PIMS and XIC; (B) heavy-labeled peptide ion; and (C) light-labeled peptide ion of the peptide VTCPNHPDAILVEDYR.
Effect of peptide ion validation
To evaluate the accuracy of MaXIC-Q Web and the effect of peptide ion validation, we also investigated the standard errors of the calculated peptide ratios for each protein. Figure 4 shows the cumulative percentage of proteins versus the standard error of peptide ion ratios. We observe that 95.50% proteins have standard errors of ion ratios less than 0.225 when we applied full peptide ion validation (S/N, CS and IP). If the CS and IP validation criteria are not applied, the results drop to 69.28%, which indicates that the CS and IP criteria detect interference from co-elution peaks and incorrectly identified peptides; thus, most unquantifiable ions are filtered out prior to the IP validation step. The result shows that the peptide ion validation can prevent the poor quality quantitation and assure the quantitation accuracy.
Figure 4.

Cumulative distribution of proteins versus the standard error of ion ratios in the comparative study of NO-treated endothelial cells.
Accuracy of MaXIC-Q Web quantitation by manual validation
To further evaluate the accuracy of MaXIC-Q Web, we manually validated the quantitation results. Of the 603 quantified proteins, 102 proteins (1188 ions) were manually validated in the evaluation analysis. They are classified as differentially expressed—defined as having down-regulated expression (ratio < 0.73) or up-regulated expression (ratio > 1.37), unchanged expression, or unquantifiable. The manual validation procedure for the 1188 ions proceeds as follows. For each ion, we first use Elution3D image to determine whether an ion co-elutes with other peptide ions or contains noise. If the ion is categorized as unquantifiable according to the ion validation criteria, we use the PIMS image to check whether another suitable ion peak can be selected for reconstructing an XIC. If no suitable ion peak can be selected, the labeled ion is deemed unquantifiable. If a suitable ion peak can be chosen, we reconstruct the XIC and use the XIC viewer to select the elution time ranges to calculate the ion ratio. The manually validated results of each ion are classified into four categories: unchanged, down-regulated, up-regulated and unquantifiable. For each ion, if the categories assigned by manual validation and MaXIC-Q Web are the same, we say that it is correctly quantified by MaXIC-Q Web. The accuracy and the rates of different error types of MaXIC-Q Web quantitation for the dataset are shown in Table 1. MaXIC-Q Web achieves an accuracy of 80.05%.
Table 1.
Accuracy of MaXIC-Q Web on the manually validated 1188 ions in the NO-treated endothelial cells experiment
| Performance | Rate (%) |
|---|---|
| Accuracy | 80.05 |
| False positive rate | 5.05 |
| False negative rate | 9.34 |
| Serious errors | 0.17 |
| Other errors | 5.39 |
False positive rate = unquantifiable peptide ions falsely quantified by MaXIC-Q Web; False negative rate = quantifiable peptide ions incorrectly analyzed by MaXIC-Q Web as unquantifiable; Serious errors = up-regulated peptide ions incorrectly recognized by MaXIC-Q Web as down-regulated, or vice versa); Other errors = peptide ions with unchanged expressions incorrectly quantified by MaXIC-Q Web as having changed expressions, and vice versa).
Usage
Step 1: data preparation
To accommodate spectral data files generated by different mass spectrometers, MaXIC-Q Web adopts the generic mzXML format. As shown in Figure 1, spectral data files from the major MS manufacturers are converted into the mzXML format by existing tools, e.g. mzStar for.wiff files from Applied Biosystems, ReAdW (http://tools.proteomecenter.org/ReAdW.php) for.raw files from Thermal Finnigan, and MassWolf (http://tools.proteomecenter.org/MassWolf.php) for.raw directories from Waters. Each converted mzXML file contains all the necessary attributes, including MS and MS/MS peak lists and the scan number index for subsequent quantitation. For input of search results, MaXIC-Q Web accepts search result files from Mascot and SEQUEST. SEQUEST users need to input pepXML and protXML files generated by PeptideProphet and ProteinProphet, respectively. For Mascot users, MaXIC-Q Web accepts the XML format exported directly from the Mascot web interface.
Step 2: data upload and assignment
MaXIC-Q Web provides a user-friendly interface to upload all the mzXML files and search result files. By using the interface shown in Figure 5, users can easily upload the raw data and search result files.
Figure 5.

Interface for file assignment.
Step 3: light- and heavy-label configuration
(Figure 6) In this step, users have to define how to differentiate light- and heavy-labeled peptides, either by using modification code or amino acid combination. According to the definition of light- and heavy-labeling code or combination, and the mass difference, MaXIC-Q Web automatically detects and quantifies the paired light/heavy peptide ion.
Figure 6.

Interface for light- and heavy-label configuration.
Step 4: parameters setting
The final step before quantifying is to input the user-defined parameters, such as peptide score threshold, protein score threshold, etc.
Quantitation results
After finishing quantitation, MaXIC-Q Web automatically redirects to a quantitation result page, as shown in Figure 7. MaXIC-Q Web also allows user to download all quantitation results in the html format, which is portable and executable on any computer platform. The result page contains protein and peptide list with quantitation information and for each peptide ion three types of visualization images, i.e. Elution3D, XIC and PIMS images, for users to conveniently browse or inspect quantitation results.
Figure 7.

Quantitation result page.
Elution3D diagram
Elution3D shows the three dimensional elution profile of the targeted peptide ion near its precursor m/z and elution time. An example is shown in Figure 8A, in which x-axis denotes elution time, y-axis represents m/z, and color stands for different intensity in raw data. In this example, the extracted data have elution time in the range from 26.84 to 29 min and m/z from 824.37 to 842 Da.
Figure 8.

Example of (A) Elution3D, (B) XIC and (C) PIMS diagrams.
XIC
In the quantitation result report, MaXIC-Q Web shows the constructed XICs for a targeted peptide ion with light label and heavy label. An example is shown in Figure 8B, in which x-axis denotes elution time, and y-axis represents intensity, and the region marked by yellow color is the area that we use to calculate the relative expression ratio.
PIMS
As shown in Figure 8C, x-axis denotes m/z, and y-axis represents ion intensity. Peptide ion validation is performed on PIMS. Validation results on the three criteria, S/N, CS and IP, are also shown. Blue color represents passing the criterion and red represents failing.
CONCLUSION
In contrast to many quantitation tools that only process search results derived by Mascot or raw data from a specific instrument, MaXIC-Q Web is generic that can accept both SEQUEST and Mascot search results and the mzXML files easily converted from raw data generated by major mass spectrometers. Moreover, MaXIC-Q Web is designed to accommodate ICAT, SILAC and user-defined modifications.
Since limited accuracy is a serious problem in current quantitation tools, MaXIC-Q Web adopts several strategies to achieve better accuracy. First, MaXIC-Q Web contains a filtering module to filter out low-confidence peptides and proteins, as defined by users. Second, MaXIC-Q Web constructs PIMSs and defines validation criteria to automatically determine whether the corresponding XICs are valid for calculating the ion ratio. The stringent validation criteria ensure detection of inevitable co-elution that frequently occurs in proteomics experiments. It also provides various visualizations of spectral data, including Elution3D, XIC and PIMS. In addition, MaXIC-Q Web generates a very comprehensive HTML output report, which is portable and executable on most browsers, to facilitate manual inspection of quantitation results or spectral data. In summary, MaXIC-Q Web is a fully automated and easy to use quantitation web service suitable for large-scale proteomic quantitation using stable isotope-labeling techniques.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Thematic Program of Academia Sinica (Grant AS94B003 and AS95ASIA02); National Science Council of Taiwan (Grant NSC 95-3114-P-002-005-Y). Funding for open access charge: Thematic Program of Academia Sinica (Grant AS94B003 and AS95ASIA02).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr Robert Moulder at Turku Centre for Biotechnology, Finland, for providing the dataset of standard protein mixture experiment, and Dr Kay-Hooi Khoo and Mr Ying-Che Chang at NRPGM Core Facilities for Proteomics Research, Academia Sinica, for providing the dataset of SILAC experiments. The website http://ms.iis.sinica.edu.tw/MaXIC-Q_Web/ is free and open to all users and there is no login requirement.
REFERENCES
- 1.Tao WA, Aebersold R. Advances in quantitative proteomics via stable isotope tagging and mass spectrometry. Curr. Opin. Biotechnol. 2003;14:110–118. doi: 10.1016/s0958-1669(02)00018-6. [DOI] [PubMed] [Google Scholar]
- 2.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 3.Yao X, Freas A, Ramirez J, Demirev PA, Fenselau C. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal. Chem. 2001;73:2836–2842. doi: 10.1021/ac001404c. [DOI] [PubMed] [Google Scholar]
- 4.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 5.Ong SE, Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC) Nat. Protoc. 2006;1:2650–2660. doi: 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]
- 6.Han DK, Eng J, Zhou H, Aebersold R. Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat. Biotechnol. 2001;19:946–951. doi: 10.1038/nbt1001-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li XJ, Zhang H, Ranish JA, Aebersold R. Automated statistical analysis of protein abundance ratios from data generated by stable-isotope dilution and tandem mass spectrometry. Anal. Chem. 2003;75:6648–6657. doi: 10.1021/ac034633i. [DOI] [PubMed] [Google Scholar]
- 8.MacCoss MJ, Wu CC, Liu H, Sadygov R, Yates JR., 3rd A correlation algorithm for the automated quantitative analysis of shotgun proteomics data. Anal. Chem. 2003;75:6912–6921. doi: 10.1021/ac034790h. [DOI] [PubMed] [Google Scholar]
- 9.Schulze W, Mann M. A novel proteomic screen for peptide-protein interactions. J. Biol. Chem. 2004;11:10756–10764. doi: 10.1074/jbc.M309909200. [DOI] [PubMed] [Google Scholar]
- 10.Bouyssie D, de Peredo AG, Mouton E, Albigot R, Roussel L, Ortega N, Cayrol C, Burlet-Schiltz O, Girard JP, Monsarrat B. Mascot file parsing and quantification (MFPaQ), a new software to parse, validate, and quantify proteomics data generated by ICAT and SILAC mass spectrometric analyses: application to the proteomics study of membrane proteins from primary human endothelial cells. Mol. Cell Proteomics. 2007;6:1621–1637. doi: 10.1074/mcp.T600069-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 12.Pedrioli PG, Eng JK, Hubley R, Vogelzang M, Deutsch EW, Raught B, Pratt B, Nilsson E, Angeletti RH, Apweiler R, et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 2004;22:1459–1466. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- 13.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 14.Moulder R, Filen JJ, Salmi J, Katajamaa M, Nevalainen OS, Oresic M, Aittokallio T, Lahesmaa R, Nyman TA. A comparative evaluation of software for the analysis of liquid chromatography-tandem mass spectrometry data from isotope coded affinity tag experiments. Proteomics. 2005;5:2748–2760. doi: 10.1002/pmic.200401187. [DOI] [PubMed] [Google Scholar]
- 15.Callister SJ, Barry RC, Adkins JN, Johnson ET, Qian WJ, Webb-Robertson BJM, Smith RD, Lipton MS. Normalization approaches for removing systematic biases associated with mass spectrometry and label-free proteomics. J. Proteome Res. 2006;5:277–286. doi: 10.1021/pr050300l. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.