


Abstract
FASTR3D is a web-based search tool that allows the user to fast and accurately search the PDB database for structurally similar RNAs. Currently, it allows the user to input three types of queries: (i) a PDB code of an RNA tertiary structure (default), optionally with specified residue range, (ii) an RNA secondary structure, optionally with primary sequence, in the dot-bracket notation and (iii) an RNA primary sequence in the FASTA format. In addition, the user can run FASTR3D with specifying additional filtering options: (i) the released date of RNA structures in the PDB database, and (ii) the experimental methods used to determine RNA structures and their least resolutions. In the output page, FASTR3D will show the user-queried RNA molecule, as well as user-specified options, followed by a detailed list of identified structurally similar RNAs. Particularly, when queried with RNA tertiary structures, FASTR3D provides a graphical display to show the structural superposition of the query structure and each of identified structures. FASTR3D is now available online at http://bioalgorithm.life.nctu.edu.tw/FASTR3D/.
INTRODUCTION
In recent years, there is a fast growing interest in non-coding RNAs (ncRNAs) because, although their transcripts are not translated into proteins, they play essential roles in many cellular processes, including gene regulation, RNA modification and chromosome replication (1–4). However, the function of most ncRNAs has yet to be determined. Likewise to proteins, a common and useful approach for annotating the function of an ncRNA is by searching databases for similar RNA molecules whose functions are already known. For this purpose, several databases of ncRNAs have been proposed, such as NONCODE (5), RNAdb (6), miRBase (7), fRNAdb (8) and ncRNAdb (9). For these databases, however, the search is performed solely by querying keywords, accession numbers, transcript/organism names and/or sequences. Compared with the 20-letter protein alphabet, the 4-letter RNA alphabet is smaller and less informative, leading to that searching for similar RNA molecules based on sequence comparison/alignment is not as accurate and powerful as it does for proteins.
Actually, a more reliable way for determining the functions of ncRNAs is from the analysis on the structure level, since structures of molecules are typically more evolutionarily conserved than their sequences. In this regard, a series of recent efforts and studies has led to a substantial increase in both the number and the size of solved RNA structures deposited in the PDB and NDB databases (10,11). Therefore, it has become more and more crucial to develop automatic tools that are able to efficiently and accurately search for structurally similar RNA substructures and motifs against the PDB/NDB database. Basically, detecting structural similarities in two RNA molecules at secondary structure level is an easy job, whereas it is intractable at tertiary structure level, because it has been shown to be an nondeterministic polynomial time (NP)-hard problem even to find a constant ratio approximation algorithm for computing a pair of maximal substructures from two RNA (or protein) tertiary [three-dimensional (3D)] structures with exhibiting the highest degree of similarity (12). Therefore, currently available tools, such as ARTS (13,14), DIAL (15), SARSA (16) and SARA (17), are all based on some heuristic approaches for comparing the similarities of two RNA tertiary structures. All these methods, however, have at least quadratic-time complexity and hence are impractical for searching ever-increasing databases of RNA tertiary structures. Currently, there are several tools that can be used to search motifs in RNA structures, including FR3D (18), PRIMOS (19) and RNAMotif (20). FR3D uses a base-centred method to perform a geometric search of RNA local/composite 3D motifs. PRIMOS searches for locally structural similarities of consecutive RNA fragments by comparing their pseudotorsion angles. RNAMotif finds the fragments of an RNA sequence that conform to a predefined descriptor of defining a particular motif of secondary structure.
In this study, we have developed a web server, called FASTR3D (‘Fast and Accurate Search Tool for RNA 3D structures’), based on a hashing algorithm that is able to fast and accurately find structural similarities for a query of RNA molecule in the PDB database. In principle, this hashing algorithm consists of three main procedures as follows. The first procedure is to derive the primary sequence, secondary structure and tertiary structure information of all RNA molecules currently deposited in the PDB database and then store the derived second structures in a hash table. The secondary procedure is to derive some possible secondary structures of the query RNA if it is a primary sequence or tertiary structure. The third procedure is to search the hash table for all candidate RNAs whose secondary structures exactly match that of the query RNA, followed by primary sequence filter and/or tertiary structure filter to screen out those candidates whose primary sequences and/or tertiary structures are not equal to that of the query RNA. The FASTR3D web server is now available online at http://bioalgorithm.life.nctu.edu.tw/FASTR3D/ for public access.
In addition, our FASTR3D was tested with a number of RNA primary sequences, secondary structures and tertiary structures, and its experimental results on querying RNA primary sequences and secondary structures were also compared with those obtained by the search tool of RNA FRABASE (http://rnafrabase.ibch.poznan.pl/), which was developed by Popenda et al. (21) on the basis of RNA primary sequences and/or secondary structures using the methods of regular expression and pattern recognition. The comparison of experimental results on querying secondary structures reveals that FASTR3D has a comparable performance as RNA FRABASE, both with returning the search results in a short time. However, our FASTR3D is able to find more structurally similar RNAs for a query of RNA primary sequence, when compared with RNA FRABASE, because FASTR3D searches for structurally similar RNAs using the secondary structure derived from the query sequence, while RNA FRABASE searches them solely based on the primary sequence. In addition, the function of querying RNA tertiary structures in FASTR3D, as well as the online graphical display of showing the structural superposition of the query and identified structures, is not available in RNA FRABASE.
METHODS
Our FASTR3D was implemented based on a hashing algorithm whose procedure flowchart, as shown in Figure 1, consists of three major procedures. The first procedure is a preprocessing job that is to derive the primary sequence, secondary structure and tertiary structure information of all RNAs in the PDB database and particularly store the derived secondary structures (i.e. standard Watson–Crick and wobble base pairs) in a hash table. Note that the secondary structure information was derived using the RNAView program (22), while the tertiary structure information of pseudotorsion angles η and θ values was derived using the AMIGOS program (23). The second procedure is to derive the secondary structure information for the RNA queried by the user. Currently, the user can input any of the following three types of queries: (i) a PDB code of an RNA tertiary structure optionally with specified residue range, (ii) an RNA secondary structure, optionally with primary sequence, in the dot-bracket notation, and (iii) an RNA primary sequence in the FASTA format. If the query is a PDB code of an RNA tertiary structure, then its secondary structure is derived from its PDB file, which is downloaded from the PDB database, using the RNAView program (22). If the query is an RNA primary sequence, then a set of at most X suboptimal secondary structures is derived using the RNAsubopt program (24), where the default value of X is 16. It is often observed that the suboptimal secondary structure predicted by RNAsubopt for an RNA molecule may not be the true secondary structure. Therefore, we design an alternative approach as follows to derive a set of at most X most frequently occurring true secondary structures for the query RNA sequence. First, we search the PDB database for all the RNAs whose primary sequences are equal to the query sequence. Then, we use RNAView to derive all the secondary structures from the PDB files of these RNAs and from them we finally select at most X most frequently occurring secondary structures. The third procedure is to use the hash table to quickly search for all candidate RNAs whose secondary structures exactly match that of the query RNA (or any of X predicted/true secondary structures for the query RNA), followed by primary sequence filter (if the query RNA has primary sequence information) and/or tertiary structure filter (if the query is an RNA tertiary structure) to screen out those candidates whose primary sequences and/or tertiary structures are not equal to that of the query RNA.
Figure 1.

The procedure flowchart of FASTR3D, where 1D, 2D and 3D refer to primary, secondary and tertiary, respectively.
In the following, we describe the details of the significant steps in the above procedures, including how to prepare the hash table of the secondary structures of all RNA molecules currently deposited in the PDB database, how to use this hash table to search for RNA structural similarities and how to utilize the η and θ values to efficiently screen out structurally non-similar candidates. For simplicity, we let D = {S1, S2, …, Sm} denote the database of the secondary structures derived from the PDB database using the RNAView program (22), and let Q be the secondary structure of the query RNA. Note that in the structural database D, each structure Si is labelled with an integer i, to which we refer as the index of Si. Moreover, we denote by the k-tuple a consecutive sequence of k nt (residues) within an RNA molecule. Clearly, there are (|S| − k + 1) overlapping k-tuples for a given RNA secondary structure S with |S| residues. The offset of a k-tuple within S is defined to be the position of its first residue with respect to the first residue of S. For convenience, we use the letter j to denote offset and use the notation wj(S) to denote the k-tuple of S that has offset j. Therefore, the position of each occurrence of each k-tuple within a structure Si of D can be represented by an (i, j) pair.
Hash table construction for a structural database
Here, we reorganize the structural database D by using a hash table to store the position of each occurrence of each k-tuple. Note that each RNA tertiary structure Si in the structural database D is represented by its secondary structure in the dot-bracket format, where an unpaired nucleotide is denoted by a dot and a Watson–Crick (e.g. AU, UA, CG, GC) or wobble (e.g. GU and UG) base pair by a pair of opening and closing round brackets (e.g. ‘(’ and ‘)’). Moreover, to correctly represent complicated secondary structures in RNA molecules, the bracket notation used in this study is extended by allowing the user to use additional squared brackets (e.g. ‘[’ and ‘]’) to represent simple pseudoknots and kissing loops, and curly brackets (e.g. ‘{’ and ‘}’) to represent high-order pseudoknotted structures.
To simplify our implementation, all the brackets appearing in an RNA secondary structure are transformed into the round brackets, since their exact pairing relationships between the opening and closing brackets are already recorded in advance using a data structure of 1D array. For each secondary structure Si with |Si| residues, we break it into 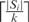 non-overlapping k-tuples and store the position of each occurrence of each k-tuple in the hash table. Recall that for any k-tuple w = r1r2 … rk, each residue rx, where 1 ≤ x ≤ k, can be either a dot, opening bracket or closing bracket. Therefore, each of these three possible symbols is then encoded as a base-3 digit as follows: e(·) = 03, e(() = 13 and e()) = 23. Using this encoding, w can be represented uniquely by a decimal integer
non-overlapping k-tuples and store the position of each occurrence of each k-tuple in the hash table. Recall that for any k-tuple w = r1r2 … rk, each residue rx, where 1 ≤ x ≤ k, can be either a dot, opening bracket or closing bracket. Therefore, each of these three possible symbols is then encoded as a base-3 digit as follows: e(·) = 03, e(() = 13 and e()) = 23. Using this encoding, w can be represented uniquely by a decimal integer 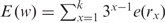 . Finally, the hash table of the structural database D is represented by two data structures, a list of positions L and an array A of pointers into L. Basically, there are 3k pointers in A, with one pointer corresponding to each of the 3k possible k-tuples. More clearly, the pointer at position E(w) of A points to the entry of L that describes the positions of the first occurrence of the k-tuple w in the database D. Then we can obtain the positions of all occurrences of w in D by traversing L from this position until we reach the location pointed by the pointer located at position E(w) + 1 of A. Below, we illustrate the above hash table construction with a simple example. For simplicity, we let k = 2 and D consist of two RNAs S1 and S2 whose secondary structures are S1 = ‘(((···)·))’ and S2 = ‘·((····)) ·’, respectively. In Table 1, each row contains the list of the positions of all occurrences for each of the nine possible 2-tuples, denoted by w. Then the pointer at E(w) of A points to the beginning of the position list corresponding to w and the concatenation of the nine position lists in the order from top to bottom forms L.
. Finally, the hash table of the structural database D is represented by two data structures, a list of positions L and an array A of pointers into L. Basically, there are 3k pointers in A, with one pointer corresponding to each of the 3k possible k-tuples. More clearly, the pointer at position E(w) of A points to the entry of L that describes the positions of the first occurrence of the k-tuple w in the database D. Then we can obtain the positions of all occurrences of w in D by traversing L from this position until we reach the location pointed by the pointer located at position E(w) + 1 of A. Below, we illustrate the above hash table construction with a simple example. For simplicity, we let k = 2 and D consist of two RNAs S1 and S2 whose secondary structures are S1 = ‘(((···)·))’ and S2 = ‘·((····)) ·’, respectively. In Table 1, each row contains the list of the positions of all occurrences for each of the nine possible 2-tuples, denoted by w. Then the pointer at E(w) of A points to the beginning of the position list corresponding to w and the concatenation of the nine position lists in the order from top to bottom forms L.
Table 1.
A 2-tuple hash table for S1 = ‘(((···)·))’ and S2 = ‘·((····)) ·’
| 2-tuple w | E(w) | Position lists |
|---|---|---|
| ·· | 0 | (1, 5), (2, 5) |
| ·( | 1 | (2, 1) |
| ·) | 2 | (2, 7) |
| (· | 3 | (1, 3), (2, 3) |
| (( | 4 | (1, 1) |
| () | 5 | |
| )· | 6 | (1, 7), (2, 9) |
| )( | 7 | |
| )) | 8 | (1, 9) |
Query substructure search
In the following, we describe how to use the hash table of the structural database D as constructed above to search for all occurrences of a query Q of an RNA secondary structure. Suppose that the length of Q is n. Then we can proceed position-by-position along Q from position 1 to n − k + 1. At position p, where 1 ≤ p ≤ n − k + 1, we obtain the list of the positions of all the occurrences of the k-tuple wp(Q) from the hash table of D via the pointer of E(wp(Q)). Let this list contain q positions, say (i1, j1), (i2, j2), …, (iq, jq). From this list, we derive a list of hits H1 = (i1, j1 − p, j1), H2 = (i2, j2 − p, j2), …, Hq = (iq, jq − p, jq). This list of hits is then added to a master list M of hits that accumulates all the hits we derived when p runs from 1 to n − k + 1. For convenience, the elements of a hit are referred to as the index, shift and offset. Next, we sort all the elements in M first by index and then by shift. Finally, we scan through M by looking for runs of hits for which the index and shift are identical. Clearly, by further sorting each of these runs by offset, we can determine the region of some structure in D that exactly matches the query structure Q. For example, we search for the query of an RNA secondary structure Q = ‘(···) ·’ within the hash table of D as constructed in Table 1. In Table 2, column 3 displays the occurrence positions in D for each 2-tuple of Q, with corresponding hits shown in column 4, and column 5 shows the sorted M in which the run of three hits highlighted in bold indicates that there is a match between Q and S1 that starts at the third nucleotide and ends at the eighth nucleotide. Basically, the search speed of the above hashing algorithm is proportional to the size of the master list M, which falls off rapidly with increasing the value of k. Although a greater k increases the search speed, the condition |Q| ≥ 2k − 1 should be satisfied to guarantee that the hashing algorithm will find a hit at some point in the matching region. For example, suppose that S = ‘((····))’ and Q = ‘(····)’. If k = 4, then none of three overlapping 4-tuples in Q is able to match any of two non-overlapping 4-tuples in S. In addition, the hash table is generated in advance for a fixed k in our algorithm. Therefore, to achieve the best search speed and reduce the storage requirement, we set the value of k as 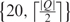 .
.
Table 2.
The search of the query secondary structure Q = ‘(···)·’
| p | wp(Q) | Positions | H | M |
|---|---|---|---|---|
| 1 | (· | (1, 3) | (1, 2, 3) | (1, 2, 3) |
| (2, 3) | (2, 2, 3) | (1, 2, 5) | ||
| 2 | ·· | (1, 5) | (1, 3, 5) | (1, 2, 7) |
| (2, 5) | (2, 3, 5) | (1, 3, 5) | ||
| 3 | ·· | (1, 5) | (1, 2, 5) | (2, 2, 3) |
| (2, 5) | (2, 2, 5) | (2, 2, 5) | ||
| 4 | ·) | (2, 7) | (2, 3, 7) | (2, 3, 5) |
| 5 | )· | (1, 7) | (1, 2, 7) | (2, 3, 7) |
| (2, 9) | (2, 4, 9) | (2, 4, 9) |
Tertiary structure filter using pseudotorsion angles
Basically, the comparison of RNA conformation is a high-dimensional problem, because six standard torsion angles (α, β, γ, δ, ɛ and ζ) are needed to specify the backbone conformation of a single nucleotide. Duarte and Pyle (23), however, pointed out that the pseudotorsion angles η (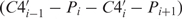 ) and θ (
) and θ (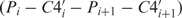 ) are at least as descriptive of backbone morphology as standard torsion angles and they may be even superior in terms of specifying the backbone conformation of an individual nucleotide. This suggests that the η–θ plot can provide us a 2D representation of the conformation properties of an entire RNA molecule, so that we can carry out the rapid and accurate comparison of RNA conformations. Duarte et al. (19) further called such an ordered set of η–θ coordinates as an RNA worm. As was used by Duarte et al. (19), we can detect the conformation difference of two RNAs by comparing their worms based on a Euclidean metric as follows. Let Q′ denote an identified candidate RNA whose secondary structure matches that of the query RNA Q with n residues, and let the worms of Q and Q′ denoted by {(η1,1, θ1,1), …, (η1,n, θ1,n)} and {(η2,1, θ2,1), …, (η2,n, θ2,n)}, respectively. The conformational difference between two residues (η1,i, θ1,i) and (η2,i, θ2,i) is defined to be
) are at least as descriptive of backbone morphology as standard torsion angles and they may be even superior in terms of specifying the backbone conformation of an individual nucleotide. This suggests that the η–θ plot can provide us a 2D representation of the conformation properties of an entire RNA molecule, so that we can carry out the rapid and accurate comparison of RNA conformations. Duarte et al. (19) further called such an ordered set of η–θ coordinates as an RNA worm. As was used by Duarte et al. (19), we can detect the conformation difference of two RNAs by comparing their worms based on a Euclidean metric as follows. Let Q′ denote an identified candidate RNA whose secondary structure matches that of the query RNA Q with n residues, and let the worms of Q and Q′ denoted by {(η1,1, θ1,1), …, (η1,n, θ1,n)} and {(η2,1, θ2,1), …, (η2,n, θ2,n)}, respectively. The conformational difference between two residues (η1,i, θ1,i) and (η2,i, θ2,i) is defined to be 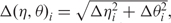 where Δηi = min{|η1,i − η2,i|, 360 − |η1,i − η2,i|} and Δθi = min{|θ1,i − θ2,i|, 360 − |θ1,i − θ2,i|} (since 0° and 360° are the same). As was also pointed out by Duarte et al. (19), two residues (η1,i, θ1,i) and (η2,i, θ2,i) can be considered structurally identical if Δ(η, θ)i < 25°. Therefore, based on this property, we design our tertiary structure filter to discard the identified RNA Q′ from consideration if the average conformation difference
where Δηi = min{|η1,i − η2,i|, 360 − |η1,i − η2,i|} and Δθi = min{|θ1,i − θ2,i|, 360 − |θ1,i − θ2,i|} (since 0° and 360° are the same). As was also pointed out by Duarte et al. (19), two residues (η1,i, θ1,i) and (η2,i, θ2,i) can be considered structurally identical if Δ(η, θ)i < 25°. Therefore, based on this property, we design our tertiary structure filter to discard the identified RNA Q′ from consideration if the average conformation difference 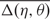 between Q and Q′ is greater than or equal to a predefined cutoff, where
between Q and Q′ is greater than or equal to a predefined cutoff, where 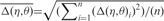 and for our purpose, the cutoff value is set as 55°.
and for our purpose, the cutoff value is set as 55°.
USAGE OF FASTR3D
Input
FASTR3D provides an intuitive user interface as illustrated in Figure 2. In basic search, the user can submit a job by entering or pasting one of the following three types of queries to search for structurally similar RNA structures: (i) a PDB code of an RNA tertiary structure (default), optionally with specified residue range, (ii) an RNA secondary structure, optionally with primary sequence, in the RNA FRABASE format (i.e. a kind of dot-bracket notation) and (iii) an RNA primary sequence in the FASTA format. In addition, the user can further restrict FASTR3D to return those RNAs whose primary sequences exactly match that of the query RNA if the query RNA contains the information of its primary sequence. If the query is an RNA tertiary structure, then the user can determine whether to calculate the RMSD between the query RNA and identified candidate RNAs with the considerations of computational performance. If the query is a primary sequence, then the user can choose to use either at most X true, frequently occurring secondary structures or predicted suboptimal secondary structures to perform the PDB database search. The default value of X is 16 and can be changed by the user. In advanced search, the user can run FASTR3D with specifying additional filtering options: (i) the released date of identified RNA structures in the PDB database, and (ii) the experimental methods used to determine identified RNA structures and their least resolutions.
Figure 2.

The web interface of FASTR3D.
Output
In the output page, FASTR3D will first show the user-queried RNA molecule, as well as user-specified options. Next, it will show a detailed list of identified structurally similar RNAs (Figure 3 for an example), including corresponding PDB ID, primary sequence, secondary structure, tertiary structure, RMSD between the query and identified structures, chain ID, starting and ending nucleotide numbers, experimental method used to determine the structure, classification of RNA molecule (based on function, metabolic role, molecule type, cellular location and so on), released date in the PDB database and solved resolution. Particularly, if the query RNA is a tertiary structure, then FASTR3D allows the user to visually view, rotate and enlarge the superposition of the query RNA and each of identified RNA (Figure 4). If the query RNA is a primary sequence or secondary structure, then the user still can visually view, rotate and enlarge the tertiary structure of each identified RNA.
Figure 3.

The output of FASTR3D on querying an RNA tertiary structure.
Figure 4.

The visual display of query RNA (top left panel) and an identified RNA (top right panel) and their superposition (bottom panel).
EXPERIMENTAL RESULTS
For the purpose of evaluation, our FASTR3D was tested with a number of RNA primary sequences and secondary/tertiary structures, and its experimental results on querying RNA primary sequences and secondary structures were also compared with those obtained by RNA FRABASE. Basically, our FASTR3D has a comparable performance as RNA FRABASE on querying RNA secondary structures, because the basic principles behind these two tools are the same, even though they were implemented based on different algorithms. As to the queries of RNA primary sequences, the search result of our FASTR3D is greatly different from those obtained by RNA FRABASE. Recall that, when queried with an RNA primary sequence, our FASTR3D searches for query-matching substructures (fragments) within RNA molecules using the secondary structure information of the query sequence, while RNA FRABASE searches them solely based on the query sequence. As mentioned before, RNA structures are more evolutionarily conserved than their sequences and, therefore, it can be commonly observed that different RNA sequences have the same/similar structures. This indicates that our FASTR3D may be able to find more structurally similar RNA fragments, when compared with RNA FRABASE. For the purpose of demonstration, we selected a fragment from the large subunit of the ribosome in Haloarcula marismortui (PDB ID: 1FFK, chain: 0, nucleotide number: 2558–2575) and applied its sequence (GGGGCUGAAGAAGGUCCC) to RNA FRABASE (with default parameters) and our FASTR3D (with searching frequently occurring true secondary structures and without matching the query sequence). Consequently, RNA FRABASE found 51 candidate RNAs that have the same primary sequence as the query, while our FASTR3D found 304 candidates that have the same secondary structure as that of the query derived by the program RNAsubopt. By further verification, we found that 94 out of the 304 tertiary substructures returned by our FASTR3D are highly similar to that of the query. This experiment demonstrates that the number of structurally similar substructures identified by our FASTR3D is greater than that by RNA FRABASE.
In the following, we demonstrate the utility of our FASTR3D on querying RNA tertiary structures, which is currently not available in RNA FRABASE. First of all, we used the tertiary substructure of a riboswitch (PDB ID: 1Y27, chain: X, nucleotide numbers: 27–43 and 54–72), as shown in Figure 5, to test our FASTR3D for its capability of searching the PDB database for structurally similar riboswitches. The so-called riboswitches are genetic regulatory elements typically found in the non-coding regions of various bacterial mRNAs. They are to regulate the expression of the genes encoded by their downstream mRNAs, via the binding of small metabolites that do not require the assistance of any protein factor (25). More importantly, it has been suggested by recent studies that riboswitches can serve as antibacterial drug targets, due to their importance to the control of genes in many bacteria (26). Basically, riboswitches are composed of a ligand binding aptamer domain and an expression platform that interfaces with RNA elements involved in gene expression. Particularly, the aptamer domain for guanine-responsive riboswitches consists of three stems and two hairpin loops. It has been reported that the interaction between these two hairpin loops, as was illustrated in Figure 5, is required for the biological function of the guanine-responsive riboswitches (27). In this experiment, FASTR3D quickly found other nine riboswitches (PDB IDs: 2G9C, 2B57, 2EEW, 2EEU, 2EES, 1U8D, 2EET, 2EEV and 3DS7) that possess substructures highly similar to the query, where their RMSDs to the query range from 0.98 Å to 1.04 Å (Figure 3 for other details). The superposition of the query and the identified substructure in 2EEV is shown in the bottom panel in Figure 4.
Figure 5.

The interaction between two hairpin loops from the guanine-responsive riboswitch (PDB ID: 1Y27, chain: X, nucleotide numbers: 27–43 and 54–72). One loop is in cyan and the other is in magenta, with interacting residues in the loops colored yellow and green. Helical stems of the hairpin loops are in blue. This figure was prepared using the program PyMoL (http://www.pymol.org/).
Next, we tested our FASTR3D using a frameshifting pseudoknot (PDB ID: 1YG3, chain: A, nucleotide numbers: 3–30) from sugarcane yellow leaf virus (ScYLV), as shown in Figure 6a. Programmed −1 ribosomal frameshifting (−1 PRF) is a recoding mechanism by which the translational ribosome switches from the zero reading frame to the −1 reading frame at a specific position and continues its translation in the new frame. The recording of −1 PRF leads to an expression of an alternative protein, which is different from that produced by standard translation. To date, this recoding mechanism has been found to occur in many viruses, as well as a few cellular genes (28,29). The mechanism allows viruses to produce different proteins from the same mRNA and hence increases the diversity of their proteins. In most cases (but not all), the −1 PRF is commonly stimulated by an RNA pseudoknot located downstream from a heptanucleotide slip site where the −1 PRF event takes place. It has been shown that the absence or destabilization of a stable pseudoknot can eliminate efficient stimulation of −1 PRF in ScYLV (30). In this experiment, FASTR3D quickly found other three RNA pseudoknots (PDB IDs: 1YG4, 2AP0 and 2AP5) in the PDB database whose 3D structures are very similar to that of the query, where their RMSDs to the query is between 1.71 Å and 2.97 Å. Figure 6b displays the superposition of the query and the identified pseudoknot 2AP5 whose RMSD is 2.97 Å.
Figure 6.

(a) Tertiary structure of a frameshifting pseudoknot (PDB ID: 1YG3, chain: A, nucleotide numbers: 3–30). Stem 1 is in yellow, stem 2 is in blue, loop 1 is in red, loop 2 is in green and the nucleotide (A13) between the two stems is in violet. (b) The superposition between the query pseudoknot (1YG3) colored orange and an identified pseudoknot (2AP5) colored green with an RMSD of 2.97 Å.
For more details on the above experiments, as well as other experiments, we refer the reader to help page of our FASTR3D at http://bioalgorithm.life.nctu.edu.tw/FASTR3D/help.html. Basically, when queried with RNA primary sequences, our FASTR3D can provide more unintended structures than RNA FRABASE as the query sequences are not as conserved as their secondary structures. On the other hand, the search results by our FASTR3D using RNA tertiary structures have the intended structures with more various sequences than those by RNA FRABASE using their primary sequences and secondary structures as the input.
SUMMARY
FASTR3D is a web-based search tool that allows the user to quickly and accurately search the PDB database for structural similarities of a query RNA. The user can query this tool by using either an RNA tertiary structure, an RNA secondary structure or an RNA primary sequence. Since the hashing algorithm, as well as tertiary structure filter, behind our FASTR3D is highly efficient, a typical query can be done in a short time. It is worth mentioning again that the function of querying RNA tertiary structures in our FASTR3D, as well as the online graphical display of showing structural superposition, is not available in RNA FRABASE. Therefore, we believe that our FASTR3D can serve as a useful tool in the study of structural biology.
FUNDING
National Science Council of Republic of China (NSC97-2221-E-009-081-MY3 in part). Funding for open access charge: ATU plan of MOE.
Conflict of interest statement. None declared.
REFERENCES
- 1.Doudna JA. Structural genomics of RNA. Nat. Struct. Biol. 2000;7:954–956. doi: 10.1038/80729. [DOI] [PubMed] [Google Scholar]
- 2.Eddy SR. Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2001;2:919–929. doi: 10.1038/35103511. [DOI] [PubMed] [Google Scholar]
- 3.Mattick JS, Makunin IV. Non-coding RNA. Hum. Mol. Genet. 2006;15:R17–R29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- 4.Storz G. An expanding universe of noncoding RNAs. Science. 2002;296:1260–1263. doi: 10.1126/science.1072249. [DOI] [PubMed] [Google Scholar]
- 5.He S, Liu C, Skogerbo G, Zhao H, Wang J, Liu T, Bai B, Zhao Y, Chen R. NONCODE v2.0: decoding the non-coding. Nucleic Acids Res. 2008;36:D170–D172. doi: 10.1093/nar/gkm1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pang KC, Stephen S, Dinger ME, Engstrom PG, Lenhard B, Mattick JS. RNAdb 2.0–an expanded database of mammalian non-coding RNAs. Nucleic Acids Res. 2007;35:D178–D182. doi: 10.1093/nar/gkl926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kin T, Yamada K, Terai G, Okida H, Yoshinari Y, Ono Y, Kojima A, Kimura Y, Komori T, Asai K. fRNAdb: a platform for mining/annotating functional RNA candidates from non-coding RNA sequences. Nucleic Acids Res. 2007;35:D145–D148. doi: 10.1093/nar/gkl837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szymanski M, Erdmann VA, Barciszewski J. Noncoding RNAs database (ncRNAdb) Nucleic Acids Res. 2007;35:D162–D164. doi: 10.1093/nar/gkl994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berman HM, Westbrook J, Feng Z, Iype L, Schneider B, Zardecki C. The nucleic acid database. Acta Crystallogr. D Biol. Crystallogr. 2002;58:889–898. doi: 10.1107/s0907444902003487. [DOI] [PubMed] [Google Scholar]
- 12.Kolodny R, Linial N. Approximate protein structural alignment in polynomial time. Proc. Natl Acad. Sci. USA. 2004;101:12201–12206. doi: 10.1073/pnas.0404383101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dror O, Nussinov R, Wolfson H. ARTS: alignment of RNA tertiary structures. Bioinformatics. 2005;21 (Suppl. 2):47–53. doi: 10.1093/bioinformatics/bti1108. [DOI] [PubMed] [Google Scholar]
- 14.Dror O, Nussinov R, Wolfson HJ. The ARTS web server for aligning RNA tertiary structures. Nucleic Acids Res. 2006;34:W412–W415. doi: 10.1093/nar/gkl312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrè F, Ponty Y, Lorenz WA, Clote P. DIAL: a web server for the pairwise alignment of two RNA three-dimensional structures using nucleotide, dihedral angle and base-pairing similarities. Nucleic Acids Res. 2007;35:W659–W668. doi: 10.1093/nar/gkm334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang YF, Huang YL, Lu CL. SARSA: a web tool for structural alignment of RNA using a structural alphabet. Nucleic Acids Res. 2008;36:W19–W24. doi: 10.1093/nar/gkn327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Capriotti E, Marti-Renom MA. RNA structure alignment by a unit-vector approach. Bioinformatics. 2008;24:i112–i118. doi: 10.1093/bioinformatics/btn288. [DOI] [PubMed] [Google Scholar]
- 18.Sarver M, Zirbel CL, Stombaugh J, Mokdad A, Leontis NB. FR3D: finding local and composite recurrent structural motifs in RNA 3D structures. J. Mol. Biol. 2008;56:215–252. doi: 10.1007/s00285-007-0110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duarte CM, Wadley LM, Pyle AM. RNA structure comparison, motif search and discovery using a reduced representation of RNA conformational space. Nucleic Acids Res. 2003;31:4755–4761. doi: 10.1093/nar/gkg682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macke TJ, Ecker DJ, Gutell RR, Gautheret D, Case DA, Sampath R. RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res. 2001;29:4724–4735. doi: 10.1093/nar/29.22.4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Popenda M, Blazewicz M, Szachniuk M, Adamiak RW. RNA FRABASE version 1.0: an engine with a database to search for the three-dimensional fragments within RNA structures. Nucleic Acids Res. 2008;36:D386–D391. doi: 10.1093/nar/gkm786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang H, Jossinet F, Leontis N, Chen L, Westbrook J, Berman H, Westhof E. Tools for the automatic identification and classification of RNA base pairs. Nucleic Acids Res. 2003;31:3450–3460. doi: 10.1093/nar/gkg529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duarte CM, Pyle AM. Stepping through an RNA structure: a novel approach to conformational analysis. J. Mol. Biol. 1998;284:1465–1478. doi: 10.1006/jmbi.1998.2233. [DOI] [PubMed] [Google Scholar]
- 24.Wuchty S, Fontana W, Hofacker IL, Schuster P. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers. 1999;49:145–165. doi: 10.1002/(SICI)1097-0282(199902)49:2<145::AID-BIP4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 25.Mandal M, Breaker RR. Gene regulation by riboswitches. Nat. Rev. Mol. Cell Biol. 2004;5:451–463. doi: 10.1038/nrm1403. [DOI] [PubMed] [Google Scholar]
- 26.Blount KF, Breaker RR. Riboswitches as antibacterial drug targets. Nat. Biotechnol. 2006;24:1558–1564. doi: 10.1038/nbt1268. [DOI] [PubMed] [Google Scholar]
- 27.Batey RT, Gilbert SD, Montange RK. Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature. 2004;432:411–415. doi: 10.1038/nature03037. [DOI] [PubMed] [Google Scholar]
- 28.Farabaugh PJ. Programmed translational frameshifting. Microbiol. Rev. 1996;60:103–134. doi: 10.1128/mr.60.1.103-134.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Namy O, Rousset JP, Napthine S, Brierley I. Reprogrammed genetic decoding in cellular gene expression. Mol. Cell. 2004;13:157–168. doi: 10.1016/s1097-2765(04)00031-0. [DOI] [PubMed] [Google Scholar]
- 30.Cornish PV, Hennig M, Giedroc DP. A loop 2 cytidine-stem 1 minor groove interaction as a positive determinant for pseudoknot-stimulated -1 ribosomal frameshifting. Proc. Natl Acad. Sci. USA. 2005;102:12694–12699. doi: 10.1073/pnas.0506166102. [DOI] [PMC free article] [PubMed] [Google Scholar]