


Abstract
ToppGene Suite (http://toppgene.cchmc.org; this web site is free and open to all users and does not require a login to access) is a one-stop portal for (i) gene list functional enrichment, (ii) candidate gene prioritization using either functional annotations or network analysis and (iii) identification and prioritization of novel disease candidate genes in the interactome. Functional annotation-based disease candidate gene prioritization uses a fuzzy-based similarity measure to compute the similarity between any two genes based on semantic annotations. The similarity scores from individual features are combined into an overall score using statistical meta-analysis. A P-value of each annotation of a test gene is derived by random sampling of the whole genome. The protein–protein interaction network (PPIN)-based disease candidate gene prioritization uses social and Web networks analysis algorithms (extended versions of the PageRank and HITS algorithms, and the K-Step Markov method). We demonstrate the utility of ToppGene Suite using 20 recently reported GWAS-based gene–disease associations (including novel disease genes) representing five diseases. ToppGene ranked 19 of 20 (95%) candidate genes within the top 20%, while ToppNet ranked 12 of 16 (75%) candidate genes among the top 20%.
INTRODUCTION
High-throughput genome-wide studies like linkage analysis and gene expression profiling, although useful for classification and characterization, do not provide sufficient information to identify specific disease causal genes. Both of these approaches typically result in hundreds of potential candidate genes, often failing to help researchers in reducing the target genes to a manageable number for further validation. To overcome these limitations, several gene prioritization methods have been developed (1–10). While all of these tools are based on the assumption that similar phenotypes are caused by genes with similar or related functions (2,11–13), they differ by the strategy they adopt in calculating similarity and by the data sources they use (14). Except for ENDEAVOUR (5,14) and ToppGene (10), most of the existing approaches mainly focus on the combination of few data sources. Interestingly, none of these approaches utilize mouse phenotype data explicitly in their prioritization approaches even though the mouse is the key model organism for the analysis of mammalian developmental, physiological and disease processes (15). Additionally, previous reports (16,17) have shown that a direct comparison of human and mouse phenotypes allowed rapid recognition of disease causal genes. In an earlier study (10), we have demonstrated that employing mouse phenotype data in fact improves candidate gene prioritization. Through various examples, we also demonstrated (10) that ToppGene performs better than SUSPECTS (9), PROSPECTR (3) and ENDEAVOUR (5), three commonly used methods in candidate gene prioritization.
Most of the current computational disease candidate gene prioritization methods (1–10) rely on functional annotations, gene-expression data or sequence-based features. The coverage of the gene functional annotations, however, is a limiting factor. Currently, only a fraction of the genome is annotated with pathways and phenotypes (10). While two-thirds of all the genes are annotated by at least one functional annotation, the remaining one-third is yet to be annotated. Recent biotechnological advances such as the high-throughput yeast two-hybrid screen have facilitated building proteome-wide protein–protein interaction networks (PPINs) or ‘interactome’ maps in humans (18,19). The shift in focus to systems biology in the post-genomic era has generated further interest in PPINs and biological pathways. While protein–protein interactions (PPI) have been used widely to identify novel disease candidate genes (20–24), several recent studies (22,23,25–27) report also using them for candidate gene prioritization.
Since biological networks have been found to be comparable to communication and social networks (28) through commonalities such as scale-freeness and small-world properties, we reasoned that the algorithms used for social and Web networks should be equally applicable to biological networks and developed ToppNet (27). One of the earliest efforts (24) uses a classifier based on several topological features, including degree (number of links to the protein), 1N index (proportion of links to disease-related proteins), 2N index (average 1N index in the neighbors), average distance to disease genes and positive topology coefficient (average neighborhood overlapping with disease genes). A more recent application, Genes2Networks (29), identifies important genes based on a list of ‘seed’ genes. It generates a Z-score for each ‘intermediate’ gene from a binomial proportions test to represent its specificity or significance to the ‘seed’ genes. The former method, independent of known disease-related genes, is used for disease candidate gene identification, especially in cases where there is little or no prior knowledge about the disease. The latter application, on the other hand, uses a ‘seed’ list as training to score the neighboring genes. It avoids bias toward highly connected ‘hub’ genes, but the candidate gene is searched in a local network region unlike ToppNet, and the user has to provide the size of the neighborhood region in the network.
Here, we describe a unique, one-stop online assembly of computational software tools (summarized in Table 1 and Figure 1) that enables biomedical researchers to (i) perform gene list enrichment analysis (ToppFun), (ii) perform candidate gene prioritization based on functional annotations (ToppGene), (iii) perform candidate gene prioritization based on protein interactions network analysis (ToppNet) and (iv) identify and rank candidate genes in the interactome based on both functional annotations and PPIN analysis (ToppGeNet). Instructions and ‘help’ for each of these modules can be accessed from the homepage. The database is updated periodically, and the current status of the data (versions and coverage) can also be accessed from the homepage (‘Database details’). Additionally, several examples with stepwise instructions are provided to demonstrate the utility of these applications (see ‘Supplementary’ section from ToppGene homepage).
Table 1.
Summary of ToppGene suite applications
| Application | Description | Input | Output |
|---|---|---|---|
| ToppFun | Detects functional enrichment of input gene list based on Transcriptome (gene expression), Proteome (protein domains and interactions), Regulome (TFBS and miRNA), Ontologies (GO, Pathway), Phenotype (human disease and mouse phenotype), Pharmacome (Drug–Gene associations) and Bibliome (literature co-citation). | Supported identifiers include NCBI Entrez gene IDs, approved human gene symbols, NCBI Reference Sequence accession numbers; single gene list. | HTML output; Tab-delimited downloadable text file; graphical charts |
| ToppGene | Prioritize or rank candidate genes based on functional similarity to training gene list. | Same as above but with two gene lists (training and test) | HTML output |
| ToppNet | Prioritize or rank candidate genes based on topological features in protein–protein interaction network. | Same as above | HTML output; Cytoscape-compatible input file; graphical networks |
| ToppGeNet | Identify and prioritize the neighboring genes of the ‘seeds’ in protein–protein interaction network based on functional similarity to the ‘seed’ list (ToppGene) or topological features in protein–protein interaction network (ToppNet). | Single gene list | Same as above |
Figure 1.

Schematic representation of workflow and methodology in ToppGene Suite applications. (A) Genes in the training set are selected based on their attributes or current gene annotations (genes associated with a disease, phenotype, pathway or a GO term). (B) The test gene source can be candidate genes from linkage analysis studies or genes differentially expressed in a particular disease or phenotype or genes from the interactome. (C) ToppFunEnriched terms of the gene annotations and sequence features, namely, GO: Molecular Function, GO: Biological Process, Mouse Phenotype, Pathways, Protein Interactions, Protein Domains, transcription factor-binding sites, miRNA-target genes, disease-gene associations, drug-gene interactions, and Gene Expression, compiled from various data sources and also used to build the training set gene profile. (C and D) ToppGene—a similarity score is generated for each annotation of each test gene by comparing to the enriched terms in the training set of genes. The final prioritized gene list is then computed based on the aggregated values of the 14 similarity scores. (E and F) ToppNet—Training and test set genes are mapped to a protein–protein interaction network. Scoring and ranking of test set genes are based on the relative location to all of the training set genes using global network-distance measures in the PPIN.
TOPPFUN: GENE LIST FUNCTIONAL ENRICHMENT
ToppFun can be used for gene list functional enrichment analysis. It uses as many as 14 annotation categories including GO terms, pathways, protein–protein interactions, protein functional domains, transcription factor-binding sites, microRNAs, gene tissue expressions and literatures. Flexible options are provided to either download results as a tab-delimited file or display as a chart. Hypergeometric distribution with Bonferroni correction is used as the standard method for determining statistical significance.
TOPPGENE: FUNCTIONAL ANNOTATIONS-BASED CANDIDATE GENE PRIORITIZATION
ToppGene works by generating a representative profile of the training genes using as many as 14 features and identifies over-representative terms from the training genes. This forms the first step and is done by using ToppFun (see previous section). The test set genes are compared to this representative profile of the training set or the overrepresented terms from the training genes for all categorical annotations and the average vector for the expression values (Figure 1). For a test gene, a similarity score to the training profile for each of the 14 features is derived and summarized by the 14 similarity scores. In the case of a missing value (for instance, lack of one or more annotations for a test gene), the score is set to −1. Otherwise, it is a real value in [0, 1]. Different methods are used for similarity measures of categorical (e.g. GO annotations) and numeric (i.e. gene expression) annotations. While a fuzzy-based similarity measure is applied for categorical terms [see Popescu et al. (30) for additional details], for numeric annotation, i.e. the microarray expression values, the similarity score is calculated as the Pearson correlation of the two expression vectors of the two genes. The 14 similarity scores are combined into an overall score using statistical meta-analysis. A P-value of each annotation of a test gene G is derived by random sampling of the whole genome. The P-value of similarity score Si is defined as:
 |
Fisher's inverse chi-square method, which states that 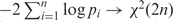 (assuming pi values come from independent tests) is then applied to combine the P-values from multiple annotations into an overall P-value. The final similarity score of the test gene is then obtained by 1 minus the combined P-value. For more details, validation and comparison with other related applications; the readers are referred to our previous study (10).
(assuming pi values come from independent tests) is then applied to combine the P-values from multiple annotations into an overall P-value. The final similarity score of the test gene is then obtained by 1 minus the combined P-value. For more details, validation and comparison with other related applications; the readers are referred to our previous study (10).
TOPPNET: NETWORK ANALYSIS-BASED CANDIDATE GENE PRIORITIZATION
ToppNet gene prioritization is based on protein–protein interaction network (PPIN) analyses. Based on the observation that biological networks share many properties with Web and social networks (28), ToppNet uses extended versions of three algorithms from White and Smyth (31)—PageRank with Priors, HITS with Priors and K-step Markov—to prioritize disease candidate genes by estimating their relative importance in the PPIN to the disease-related genes. For more details about the protein interaction datasets used, algorithmic details and validation, see our recently published study (27).
TOPPGENET: PRIORITIZATION OF NEIGHBORING GENES IN PPIN
ToppGeNet differs from ToppGene and ToppNet in that the test set is derived from the protein interactome. In other words, for a training set of known disease genes, the test set is generated by mining the protein interactome and compiling the genes either directly or indirectly interacting (based on user input) with the training set. After any overlapping or common genes between test and training sets are removed, interactome-based test set genes can be prioritized using either a functional annotation-based method (ToppGene) or PPIN-based method (ToppNet). The human protein interaction dataset (file ‘interactions.gz’), a compilation of PPIs from BIND (32), BioGRID (33) and HPRD (34), is downloaded from NCBI Entrez Gene FTP site (ftp://ftp.ncbi.nih.gov/gene/).
TOPPGENE SUITE IMPLEMENTATION AND ACCESS
The programs of our enrichment and prioritization methods are implemented in JAVA. An open-source JAVA package, FtpBean by Calvin Tai (http://www.geocities.com/SiliconValley/Code/9129), is used to automatically download data and annotation files from FTP servers. BioJava packages are used to process UniProt records and extract related protein domain information. GOLEM
(http://function.princeton.edu/GOLEM/download.html) source code is adapted and modified for dealing with ontology annotations. Colt (http://dsd.lbl.gov/~hoschek/colt) and Jakarta Commons-Math libraries
(http://jakarta.apache.org/commons/math) are used for statistical analysis. The fuzzy similarity measure and related functions are implemented locally. The user front end of ToppGene Suite is a web application written in JAVA. The application server is Sun GlassFish Enterprise Server v2.1 running on OpenSUSE 10.3 Linux. Speed is a key consideration in the design choices of the ToppGene Suite front end. When the web server is started, most of the data is loaded from a relational database and kept in memory.
ToppGene Suite uses two different relational databases for persistence of data: (i) Oracle Database 10g Enterprise Edition Release 10.2.0.3.0 – 64 bit; and (ii) Apache Derby Server - 10.4.2.0. The two databases are used differently. The ‘production data’ are stored in a Derby database on the same computer as the web server, which gives Derby the advantage that it does not have to fetch large data sets across a network and therefore eliminates network latency for small queries. The Oracle Database, on the other hand, is used for data collection and refresh. The data schemas in Oracle are highly structured according to the generally accepted database practice of Third Normal Form.
ToppGene Suite uses Hibernate (http://www.hibernate.org/) for updating and retrieving data to and from the databases. The back end of ToppGene Suite is a scripted process that automatically downloads data from publicly available data sources [see (10,27) for more details]. The process, also written in JAVA, is launched using a common JAVA utility called Ant (provided by the Apache Foundation).
The gene information, annotation and the interactions data is updated automatically except for pathways (see the ‘Database details’ section from the homepage of ToppGene Suite for a list of data resources, coverage and version details and dates of last updates). The ‘Database details’ is a dynamic web page that reads the in-memory data structures and displays the counts and statistics of the live data. As the data are refreshed, the counts and statistics are automatically updated. Users can enter the training and test sets of genes of interest as queries from the interface, and the application will display enriched themes in the training set genes along with annotated prioritized test genes. Alternately, users can enter training sets and use the extended gene list from the PPIN as a test set to rank the genes in the interactome using either functional annotations or network features.
UTILITY OF THE TOPPGENE SUITE
For a more detailed validation study using ToppGene, the readers are referred to our previous study (10). In the present study, to demonstrate the utility of ToppGene Suite, we focused on recently reported GWAS. The aim was to test whether ToppGene and ToppNet are capable of retrieving or prioritizing the GWAS-discovered novel disease genes in a training-test type of analysis. We used 20 gene–disease associations (including novel disease genes) representing five diseases (Bipolar Disorder, Cardiomyopathy, Celiac Disease, Crohns Disease and Obesity; Table 2). For each of these five disorders, we built a training set containing all the genes already known to play a role in that disorder according to the OMIM and NCBI's Entrez Gene records (limiting the search field to ‘Disease/Phenotype’ and organism ‘Homo sapiens’) (See ‘Supplementary’ section from ToppGene homepage). The test set consisted of the GWAS-reported disease gene plus 99 nearest neighboring genes based on their location on the same chromosome. ToppGene and ToppNet prioritization results are presented in Table 2. ToppGene ranked 19 of 20 (95%) candidate genes within the top 20%, while ToppNet ranked 12 of 16 (75%) candidate genes among the top 20%. The mean ranks for ToppGene- and ToppNet-based prioritization were 6.8 and 11.75, respectively (excluding four disease genes that lacked interaction data).
Table 2.
Results of the 20 genetic disease prioritizations using ToppGene and ToppNet
| Disease | Reference | Gene | ToppGene rank | ToppNet rank |
|---|---|---|---|---|
| Bipolar disorder | Le-Niculescu et al. (35) | KLF12 | 2 | 15 |
| Bipolar disorder | Le-Niculescu et al. (35) | RORB | 4 | 18 |
| Bipolar disorder | Le-Niculescu et al. (35) | RORA | 7 | 13 |
| Bipolar disorder | Le-Niculescu et al. (35) | ALDH1A1 | 10 | No interaction data |
| Bipolar disorder | Le-Niculescu et al. (35) | AK3L1 | 11 | No interaction data |
| Cardiomyopathy | Dhandapany et al. (36) | MYBPC3 | 1 | 2 |
| Celiac disease | Hunt et al. (37) | SH2B3 | 1 | 8 |
| Celiac disease | Hunt et al. (37) | CCR3 | 2 | 3 |
| Celiac disease | Hunt et al. (37) | IL18R1 | 3 | 29 |
| Celiac disease | Hunt et al. (37) | RGS1 | 9 | 26 |
| Celiac disease | Hunt et al. (37) | TAGAP | 14 | No interaction data |
| Celiac disease | Hunt et al. (37) | IL12A | 14 | 10 |
| Crohns disease | Fisher et al. (38) | MST1 | 1 | 27 |
| Crohns disease | Fisher et al. (38) | NKX2-3 | 1 | 27 |
| Crohns disease | Fisher et al. (38) | IRGM | 2 | No interaction data |
| Crohns disease | Villani et al. (39) | NLRP3 | 5 | 1 |
| Crohns disease | Fisher et al. (38) | IL12B | 7 | 1 |
| Crohns disease | Barrett et al. (40) Franke et al. (41) | STAT3 | 11 | 1 |
| Crohns disease | Franke et al. (41) | PTPN2 | 30 | 6 |
| Obesity | Renstrom et al. (42) | MC4R | 1 | 1 |
| Mean | 6.8 | 11.75 |
The gene-disease associations were from recently reported GWAS and include novel disease gene associations. The training sets were compiled using ‘phenotype/disease’ annotations in NCBI's Entrez Gene records and OMIM. To build the test set genes, we defined the artificial linkage interval to be the set of genes containing the 99 nearest neighboring genes to the novel disease gene based on their genomic distance on the same chromosome.
LIMITATIONS
ToppGene or any functional annotation-based prioritization method has some limitations. First, when using a training set of genes, the assumption is that the disease genes we have yet to discover will be consistent with what is already known about a disease and/or its genetic basis, which may not always be the case. Second, the annotations and analyses, as well as the prioritization, can only be as accurate as the underlying online sources from which the annotations are retrieved. Similar to functional annotation-based methods, the performance of network-based prioritization methods (ToppNet) is also dependent on the quality of interaction data, which currently suffers from incompleteness and unreliability with missing interactions and false positives.
CONCLUSIONS
Existing disease candidate gene prioritization methodologies mine biological and functional information about candidate genes, and we believe that our ToppGene Suite can complement these existing approaches by applying novel methods that mine mouse phenotype data and PPIN. Through various examples, we demonstrate that ToppGene Suite is capable of identifying true candidate genes. However, it needs to be emphasized that our aim is not to prove that ToppGene Suite-prioritized genes are true disease genes but rather to aid in selection of a subset of most likely disease gene candidates from larger sets of disease-implicated genes identified by high-throughput genome-wide techniques like linkage analysis and microarray analysis. As the functional annotations of human and mouse genes and the quality of PPIN improves, we envisage a proportional increase in the performance of ToppGene Suite and strongly believe that it will be a valuable adjunct to wet lab experiments in human genetics and disease research. We further hypothesize that integrating the rankings obtained using functional annotations and PPIN-based approaches may improve the prioritization of disease genes.
FUNDING
State of Ohio Computational Medicine Center (ODD TECH 04-042); National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases (NIH/NIDDK) 1U01 DK70219 (Murine Atlas of a Genitourinary Smooth Muscle Development); PHS Grant P30 DK078392 (Cincinnati Digestive Health Center). Funding for open access charge: CCHMC, Cincinnati, OH, USA.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We acknowledge the help of Ron Bryson, Technical Writer, Division of Biomedical Informatics, CCHMC, Ohio, USA, in editing the manuscript.
REFERENCES
- 1.Freudenberg J, Propping P. A similarity-based method for genome-wide prediction of disease-relevant human genes. Bioinformatics. 2002;18(Suppl. 2):S110–S115. doi: 10.1093/bioinformatics/18.suppl_2.s110. [DOI] [PubMed] [Google Scholar]
- 2.Turner FS, Clutterbuck DR, Semple CA. POCUS: mining genomic sequence annotation to predict disease genes. Genome Biol. 2003;4:R75. doi: 10.1186/gb-2003-4-11-r75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tiffin N, Kelso JF, Powell AR, Pan H, Bajic VB, Hide WA. Integration of text- and data-mining using ontologies successfully selects disease gene candidates. Nucleic Acids Res. 2005;33:1544–1552. doi: 10.1093/nar/gki296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adie EA, Adams RR, Evans KL, Porteous DJ, Pickard BS. Speeding disease gene discovery by sequence based candidate prioritization. BMC Bioinformatics. 2005;6:55. doi: 10.1186/1471-2105-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aerts S, Lambrechts D, Maity S, Van Loo P, Coessens B, De Smet F, Tranchevent LC, De Moor B, Marynen P, Hassan B, et al. Gene prioritization through genomic data fusion. Nat. Biotechnol. 2006;24:537–544. doi: 10.1038/nbt1203. [DOI] [PubMed] [Google Scholar]
- 6.Thornblad TA, Elliott KS, Jowett J, Visscher PM. Prioritization of positional candidate genes using multiple web-based software tools. Twin Res. Hum. Genet. 2007;10:861–870. doi: 10.1375/twin.10.6.861. [DOI] [PubMed] [Google Scholar]
- 7.Zhu M, Zhao S. Candidate gene identification approach: progress and challenges. Int. J. Biol. Sci. 2007;3:420–427. doi: 10.7150/ijbs.3.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tiffin N, Adie E, Turner F, Brunner HG, van Driel MA, Oti M, Lopez-Bigas N, Ouzounis C, Perez-Iratxeta C, Andrade-Navarro MA, et al. Computational disease gene identification: a concert of methods prioritizes type 2 diabetes and obesity candidate genes. Nucleic Acids Res. 2006;34:3067–3081. doi: 10.1093/nar/gkl381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adie EA, Adams RR, Evans KL, Porteous DJ, Pickard BS. SUSPECTS: enabling fast and effective prioritization of positional candidates. Bioinformatics. 2006;22:773–774. doi: 10.1093/bioinformatics/btk031. [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Xu H, Aronow BJ, Jegga AG. Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinformatics. 2007;8:392. doi: 10.1186/1471-2105-8-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc. Natl Acad. Sci. USA. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jimenez-Sanchez G, Childs B, Valle D. Human disease genes. Nature. 2001;409:853–855. doi: 10.1038/35057050. [DOI] [PubMed] [Google Scholar]
- 13.Smith NG, Eyre-Walker A. Human disease genes: patterns and predictions. Gene. 2003;318:169–175. doi: 10.1016/s0378-1119(03)00772-8. [DOI] [PubMed] [Google Scholar]
- 14.Tranchevent LC, Barriot R, Yu S, Van Vooren S, Van Loo P, Coessens B, De Moor B, Aerts S, Moreau Y. ENDEAVOUR update: a web resource for gene prioritization in multiple species. Nucleic Acids Res. 2008;36:W377–W384. doi: 10.1093/nar/gkn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clarke AR. Murine genetic models of human disease. Curr. Opin. Genet, Dev. 1994;4:453–460. doi: 10.1016/0959-437x(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 16.Gorgels TG, Hu X, Scheffer GL, van der Wal AC, Toonstra J, de Jong PT, van Kuppevelt TH, Levelt CN, de Wolf A, Loves WJ, et al. Disruption of Abcc6 in the mouse: novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum. Mol. Genet. 2005;14:1763–1773. doi: 10.1093/hmg/ddi183. [DOI] [PubMed] [Google Scholar]
- 17.van Bokhoven H, Celli J, Kayserili H, van Beusekom E, Balci S, Brussel W, Skovby F, Kerr B, Percin EF, Akarsu N, et al. Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nat. Genet. 2000;25:423–426. doi: 10.1038/78113. [DOI] [PubMed] [Google Scholar]
- 18.Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 19.Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 20.George RA, Liu JY, Feng LL, Bryson-Richardson RJ, Fatkin D, Wouters MA. Analysis of protein sequence and interaction data for candidate disease gene prediction. Nucleic Acids Res. 2006;34:e130. doi: 10.1093/nar/gkl707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kann MG. Protein interactions and disease: computational approaches to uncover the etiology of diseases. Brief Bioinform. 2007;8:333–346. doi: 10.1093/bib/bbm031. [DOI] [PubMed] [Google Scholar]
- 22.Kohler S, Bauer S, Horn D, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 2008;82:949–958. doi: 10.1016/j.ajhg.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu X, Jiang R, Zhang MQ, Li S. Network-based global inference of human disease genes. Mol. Syst. Biol. 2008;4:189. doi: 10.1038/msb.2008.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu J, Li Y. Discovering disease-genes by topological features in human protein-protein interaction network. Bioinformatics. 2006;22:2800–2805. doi: 10.1093/bioinformatics/btl467. [DOI] [PubMed] [Google Scholar]
- 25.Chen JY, Shen C, Sivachenko AY. Mining Alzheimer disease relevant proteins from integrated protein interactome data. Pac. Symp. Biocomput. 2006:367–378. [PubMed] [Google Scholar]
- 26.Ortutay C, Vihinen M. Identification of candidate disease genes by integrating Gene Ontologies and protein-interaction networks: case study of primary immunodeficiencies. Nucleic Acids Res. 2009;37:622–628. doi: 10.1093/nar/gkn982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen J, Aronow BJ, Jegga AG. Disease candidate gene identification and prioritization using protein interaction networks. BMC Bioinformatics. 2009;10:73. doi: 10.1186/1471-2105-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Junker BH, Koschutzki D, Schreiber F. Exploration of biological network centralities with CentiBiN. BMC Bioinformatics. 2006;7:219. doi: 10.1186/1471-2105-7-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berger SI, Posner JM, Ma'ayan A. Genes2Networks: connecting lists of gene symbols using mammalian protein interactions databases. BMC Bioinformatics. 2007;8:372. doi: 10.1186/1471-2105-8-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Popescu M, Keller JM, Mitchell JA. Fuzzy measures on the gene ontology for gene product similarity. IEEE/ACM Trans. Comput. Biol. Bioinformatics. 2006;3:263–274. doi: 10.1109/TCBB.2006.37. [DOI] [PubMed] [Google Scholar]
- 31.White S, Smyth P. In KDD '03: Proc 9th ACM SIGKDD Int. Conf. Knowledge Discov. Data Mining. ACM, New York: 2003. Algorithms for estimating relative importance in networks; pp. 266–275. [Google Scholar]
- 32.Bader GD, Betel D, Hogue CW. BIND: the Biomolecular Interaction Network Database. Nucleic Acids Res. 2003;31:248–250. doi: 10.1093/nar/gkg056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Breitkreutz BJ, Stark C, Reguly T, Boucher L, Breitkreutz A, Livstone M, Oughtred R, Lackner DH, Bahler J, Wood V, et al. The BioGRID Interaction Database: 2008 update. Nucleic Acids Res. 2008;36:D637–D640. doi: 10.1093/nar/gkm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peri S, Navarro JD, Kristiansen TZ, Amanchy R, Surendranath V, Muthusamy B, Gandhi TK, Chandrika KN, Deshpande N, Suresh S, et al. Human protein reference database as a discovery resource for proteomics. Nucleic Acids Res. 2004;32:D497–D501. doi: 10.1093/nar/gkh070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le-Niculescu H, Patel SD, Bhat M, Kuczenski R, Faraone SV, Tsuang MT, McMahon FJ, Schork NJ, Nurnberger J.I., Jr., Niculescu A.B., 3rd. Convergent functional genomics of genome-wide association data for bipolar disorder: comprehensive identification of candidate genes, pathways and mechanisms. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2009;150B:155–181. doi: 10.1002/ajmg.b.30887. [DOI] [PubMed] [Google Scholar]
- 36.Dhandapany PS, Sadayappan S, Xue Y, Powell GT, Rani DS, Nallari P, Rai TS, Khullar M, Soares P, Bahl A, et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat. Genet. 2009;41:187–191. doi: 10.1038/ng.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunt KA, Zhernakova A, Turner G, Heap GA, Franke L, Bruinenberg M, Romanos J, Dinesen LC, Ryan AW, Panesar D, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat. Genet. 2008;40:395–402. doi: 10.1038/ng.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fisher SA, Tremelling M, Anderson CA, Gwilliam R, Bumpstead S, Prescott NJ, Nimmo ER, Massey D, Berzuini C, Johnson C, et al. Genetic determinants of ulcerative colitis include the ECM1 locus and five loci implicated in Crohn's disease. Nat. Genet. 2008;40:710–712. doi: 10.1038/ng.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, Baba N, Libioulle C, Belaiche J, Bitton A, et al. Common variants in the NLRP3 region contribute to Crohn's disease susceptibility. Nat. Genet. 2009;41:71–76. doi: 10.1038/ng285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat. Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Franke A, Balschun T, Karlsen TH, Hedderich J, May S, Lu T, Schuldt D, Nikolaus S, Rosenstiel P, Krawczak M, et al. Replication of signals from recent studies of Crohn's disease identifies previously unknown disease loci for ulcerative colitis. Nat. Genet. 2008;40:713–715. doi: 10.1038/ng.148. [DOI] [PubMed] [Google Scholar]
- 42.Renstrom F, Payne F, Nordstrom A, Brito EC, Rolandsson O, Hallmans G, Barroso I, Nordstrom P, Franks PW. Replication and extension of genome-wide association study results for obesity in 4923 adults from Northern Sweden. Hum. Mol. Genet. 2009;18:1489–1496. doi: 10.1093/hmg/ddp041. [DOI] [PMC free article] [PubMed] [Google Scholar]