

Abstract
Imidazoline (I1)-evoked hypotension is linked to enhanced phosphorylated extracellular signal-regulated kinase (pERK)1/2 production in the rostral ventrolateral medulla (RVLM). Recent cell culture findings suggest that nischarin is a candidate for the I1 receptor. In the present study, nischarin antisense oligode-oxynucleotide (ODN) (AS1 or AS2), designed according to nischarin cDNA sequence, was administered intracisternally (i.c., 2 nmol/rat for 2 days) to knockdown central nischarin expression; control rats received the corresponding mismatched ODN (MM1 or MM2) or artificial cerebrospinal fluid (aCSF). We investigated the effects of AS1 or AS2 on nischarin expression in the RVLM, and on the hypotension and RVLM pERK1/2 production elicited by the I1-selective agonist rilmenidine (25 μ g/rat i.c.). Compared with aCSF, the mismatched ODN (MM1 or MM2) had no significant effect on RVLM nischarin expression or the cardiovascular and cellular (RVLM pERK1/2) responses elicited by rilmenidine. However, either antisense ODN substantially (>80%) reduced nischarin expression in the RVLM (AS1/MM1, 3 ± 1 versus 32 ± 2 positive cells; AS2/MM2, 4 ± 1 versus 31 ± 2 positive cells) and abrogated rilmenidine (I1)-evoked hypotension (AS1/MM1, −4.1 ± 0.9 versus −10.8 ± 1.9 mm Hg; AS2/MM2, −2.1 ± 1.1 versus −15.3 ± 2.5 mm Hg) and ERK1/2 activation in the RVLM (AS1/MM1, 10 ± 1 versus 15 ± 2 positive cells; AS2/MM2, 9 ± 1 versus 18 ± 2 positive cells). Finally, pERK1/2 generated by central I1 receptor activation is colocalized with nischarin in the RVLM neurons. This is the first evidence in vivo that nischarin plays a critical role in I1 receptor-mediated pERK1/2 production in the RVLM and the subsequent hypotension.
Evidence in support of the I1 imidazoline receptor (I1R) as a functional entity has been hampered by the lack of pure I1R agonists; the most commonly used selective I1 agonists, rilmenidine and moxonidine, exhibit weak α2-adrenergic receptor (α2AR) agonist activity (Szabo, 2002). Therefore, it is important to use molecular approaches to delineate the signaling and function of the I1R. The discovery that the mouse nischarin is a homolog of the human imidazoline receptor antisera-selective (IRAS) has made it possible to delineate the signaling and function of these proteins in vitro (Lim and Hong, 2004; Reddig et al., 2005).
Our recent findings in rat pheochromocytoma (PC12) cells (Zhang and Abdel-Rahman, 2006), supported by a subsequent study in the same cell line (Sun et al., 2007), suggest that nischarin serves as, or at least shares, a common signaling pathway with the I1R. Nischarin was first identified as a mouse homolog of human IRAS (Alahari et al., 2000; Alahari, 2003). It is noteworthy that human IRAS has been considered the I1R since the transfection of IRAS cDNA into the Chinese hamster ovary cells led to the expression of high-affinity I1 binding sites for moxonidine and rilmenidine (Piletz et al., 2000). In addition, the Chinese hamster ovary cells stably expressing IRAS exhibit enhanced phosphatidyl-choline-specific phospholipase C and extracellular signal-regulated kinase (ERK) signaling (Li et al., 2006); the latter signaling pathway is triggered by rilmenidine or moxonidine in PC12 cells (Zhang et al., 2001), which lack the α2AR (Ernsberger et al., 1995; Separovic et al., 1996). It is noteworthy that pERK1/2 generation in the RVLM has been implicated in the hypotension caused by I1R activation (Zhang and Abdel-Rahman, 2005), and nischarin homology exists among different species (Piletz et al., 2003; Sun et al., 2007). It is imperative also to note that despite differences in the methodologies used for the development of polyclonal nischarin antibodies and anti-nischarin antisense oligode-oxynucleotides (ODNs), these molecular probes facilitated the identification of nischarin expression and the impact of its knockdown on I1-mediated signaling in PC12 cells (Zhang and Abdel-Rahman, 2006; Sun et al., 2007). However, these valuable molecular probes have not been exploited to help delineate the biological function of the nischarin/I1R in vivo.
In the present study, we investigated the effect of knocking down nischarin expression within the central nervous system on the hypotensive response and enhanced neuronal pERK1/2 production in the RVLM elicited by central I1R activation. To achieve this goal, a multidisciplinary approach was adopted whereby the cardiovascular and cellular (RVLM pERK1/2) responses elicited by intracisternal (i.c.) rilmenidine were investigated in rats pre-treated with one of two nischarin antisense ODNs. The latter was administered i.c. for 2 days before rilmenidine administration. Control rats received the corresponding mismatched ODN or artificial cerebrospinal fluid (aCSF). We verified the ability of the intracisternally administered antisense ODNs to effectively knockdown nischarin expression in the RVLM, the major site for the hypotensive effect of rilmenidine (Ernsberger and Haxhiu, 1997; Zhang and Abdel-Rahman, 2005). The studies were extended to investigate the effect of knocking down nischarin expression on I1R (rilmenidine)-mediated enhancement of pERK1/2 production in the RVLM and the associated hypotensive response. Finally, given the key role of pERK1/2 generation in the RVLM neurons in I1-mediated hypotension (Zhang and Abdel-Rahman, 2005), we investigated whether pERK1/2, generated by the I1R activation, is co-localized with nischarin in the RVLM neurons.
Materials and Methods
In total, 33 Sprague-Dawley male rats, weighing 310 to 380 g (Harlan, Indianapolis, IN), were used. All rats were housed in a room with controlled environment at a constant temperature of 23 ± 1°C, humidity of 50 ± 10%, and a 12/12-h light/dark cycle. All experiments were approved by the institutional animal care and use committee and carried out in accordance with the Declaration of Helsinki and with the Institute of Laboratory Animal Resources (1996) as adopted and promulgated by the National Institutes of Health.
Intracisternal Cannulation and Intravascular Catheterization
Under pentobarbital sodium (50 mg/kg i.p.) anesthesia, intracisternal cannulation, and intravascular catheterization were conducted as in our previous studies (Zhang and Abdel-Rahman, 2005). For intracisternal cannulation, a stainless steel guide cannula (23 gauge; Small Parts, Miami. FL) was passed between the occipital bone and the cerebellum so that its tip protruded into the cisterna magna. The cannula was secured by dental acrylic cement (Duralon; Thomas Dental Supply, Raleigh, NC). Each rat received a subcutaneous injection of the analgesic buprenorphine hydrochloride (Buprenex; 0.3 μg/rat) and an intramuscular injection of 60,000 U of penicillin G benzathine and penicillin G procaine in aqueous suspension (Durapen). After i.c. cannulation, the rats were housed individually, and femoral vascular catheterization was performed 3 days later under pentobarbital (50 mg/kg i.p.) anesthesia. A catheter (polyethylene 50) was placed in the abdominal aorta via the femoral artery for measurement of blood pressure (BP) and heart rate (HR). The catheter was inserted ~5 cm into the femoral artery, secured in place with sutures, and flushed with heparinized saline (200 U/ml). The arterial catheter was connected to a Gould-Statham (Oxnard, CA) pressure transducer, and BP was displayed on a Grass polygraph (model 7D; Grass Instruments, Quincy, MA). HR was computed from BP waveforms by a Grass tachograph, and it was displayed on another channel of the polygraph.
Immunohistochemistry
The procedure for immunohistochemistry was modified from Current Protocols in Neuroscience (Gerfen, 1997; Ince and Levey, 1997). In brief, brain fixation was achieved by transcardiac perfusion of the fixative solution (4% paraformaldehyde) after a lethal dose of pentobarbital (i.p.). The brain was carefully removed and placed in a vial containing the fixation solution for postfixation overnight in the refrigerator. Then, the brain was transferred into 30% sucrose in Tris-buffered saline (TBS) for infiltration until the brain sank. Coronal brain sections (16 μm in thickness) were cut serially with a microtome cryostat (HM 505E; Microm International GmbH, Walldorf, Germany). Six to eight sections were collected in each well of a cell culture plate (12 wells; BD Biosciences, San Jose, CA) containing ice-cold TBS. Before immunostaining, a section from each well was stained with thionin (Gerfen, 1997) for the segment mapping according to the atlas of Paxinos and Watson (1982). The sections from the segments (−12.8 to −11.8 mm) were used for immunostaining. The avidin-biotin-peroxidase complex-diaminobenzidine method was used in accordance with the manufacturer’s instruction (Vectastain ABC kit; Vector Laboratories, Burlingame, CA). For detection of the expression of nischarin protein, we used anti-nischarin antibody (1:5000), which was developed in our laboratory using the peptide technique (Zhang and Abdel-Rahman, 2006). The pERK1/2 was detected with phospho-MAPK (p42/44) monoclonal antibody (Cell Signaling Technology Inc., Beverly, MA; 1:400). The brain sections were incubated with the primary antibody for 48 h with shaking in a cold room (4°C). Following three rinses, the sections were incubated with the secondary antibody (biotinylated anti-rabbit or anti-mouse antibody) and the avidin-biotin-peroxidase complex. The positive cells were visualized with diaminobenzidine/H2O2 solution and monitored under a microscope (Diaphot 300; Nikon, Tokyo, Japan). After dehydrating and clearing the tissue, the sections were sealed with Permount (Fisher Scientific Co., Pitts-burgh, PA). For either protein, nischarin, or pERK1/2, the brain sections from treatment and control groups were processed under the same conditions to circumvent potential confounding factors that might influence stain development and hence quantification of the positive cells. The cells were counted on each side of the RVLM from six to eight sections of similar segments of the RVLM from treatment and control rats. The data were presented as number of cells expressing nischarin or pERK1/2 in the RVLM. Images were captured with a true-color digital camera (Micropublisher; Q Imaging, Burnaby, BC, Canada) as in our previous study (Zhang and Abdel-Rahman, 2005).
Double-Labeling Immunofluorescence
Brain sections from rats treated with rilmenidine were subjected to the procedures used in our previous study (Wang and Abdel-Rahman, 2005). The brain sections containing the RVLM were blocked with 2% bovine serum albumin in 0.1 mM TBS containing 0.1% Triton X-100 for 1 h at room temperature. The sections were incubated with both primary antibodies, anti-nischarin polyclonal antibody (rabbit; 1:500) and phospho-MAPK (p42/44) monoclonal antibody (mouse; 1:250) overnight in a cold room. After washing three times with ice-cold TBS for 15 min, the sections were incubated with the secondary antibodies anti-rabbit IgG-conjugated with fluorescein isothiocyanate (1:50) and anti-mouse IgG conjugated with Texas Red (1:50) for 1 h at room temperature. After three washings, 15 min each, the sections were put on slides, and they were sealed with VectaShield mounting medium (Vector Laboratories). The fluorescence was examined by confocal microscopy (LSM 510; Carl Zeiss, Jena, Germany). A green and a red color reflected the expression of nischarin and pERK1/2, respectively.
Design and Synthesis of Nischarin Antisense and Mismatched ODNs
As detailed in our previous study (Zhang and Abdel-Rahman, 2006), two pairs of end-capped phosphorothioate modified ODNs were designed according to the cDNA sequence of the murine nischarin (Alahari et al., 2000). Antisense 1 (AS1) targeted the initial area (ATG) of the nischarin cDNA, 5′-C*GATGAGTG-TTAGATTC*T-3′ (* represents phosphorothioated), whereas mismatched ODN1 (MM1) served as its control, 5′-C*GTTGTGTCT-TAGAATC*A-3′. Antisense 2 (AS2) was designed to target the cDNA area that encodes the C-terminal of nischarin, 5′T*CCACTATCCAT-TGCCT*G-3′, and mismatched 2 (MM2) was its control, 5′-T*CGAC-AATCCAAAGCCT*C-3′. The antisense and mismatched ODNs were synthesized by Invitrogen (Carlsbad, CA).
Experimental Protocols
Five days after intracisternal cannulation and 2 days after intravascular catheterization, the rats were randomly divided into five groups (Table 1). The rats in each group received aCSF (123 mM NaCl, 0.86 mM CaCl2, 3 mM KCl, 0.89 mM MgCl2, 25 mM NaHCO3, 0.5 mM NaH2PO4, and 0.25 mM Na2HPO4, pH 7.4), one of the nischarin antisense ODNs (AS1 or AS2), or the corresponding mismatched ODNs (MM1 or MM2). The treatment groups received two intracisternal injections (2 nmol in 2 μl of aCSF) of nischarin antisense ODN (AS1 or AS2) or the corresponding mismatched ODN (MM1 or MM2), with 24 h elapsing between treatments. The control (vehicle) group received the same volume of aCSF. Twenty-four hours after the second ODN or aCSF injection, the rats were anesthetized with pentobarbital, and the arterial catheter was connected to a pressure transducer for measurement of BP and HR, as mentioned above. After a stabilization period of at least 30 min, the selective I1 receptor agonist rilmenidine (rilmenidine dihydrogen phosphate; gift from Technologies Server, Cedex, France) was administrated intracisternally (25 μg/rat in 5 μl of aCSF). BP and HR were recorded for 15 min, and the rats were euthanized; the 15-min period was chosen because it coincides with maximal hypotension caused by rilmenidine (Zhang and Abdel-Rahman, 2005). The brains were processed for immunohistochemistry or double-labeling immunofluorescence studies as detailed above.
TABLE 1.
MAP and HR values obtained 24 h after 2-day intracisternal injections of nischarin antisense ODN (AS1 or AS2), its mismatched ODN (MM1 or MM2), or aCSF
| Group | MAP | HR | n |
|---|---|---|---|
| mm Hg | beats/min | ||
| aCSF | 80.3 ± 3.2 | 295 ± 11 | 5 |
| MM1 | 77.8 ± 6.1 | 274 ±13 | 6 |
| AS1 | 71.3 ±4.2 | 246 ±17 | 9 |
| MM2 | 82.2 ±4.8 | 315 ±16 | 6 |
| AS2 | 77.8 ±5.1 | 272 ± 14 | 7 |
The MAP and HR values represent the baseline values before rilmenidine administration. The values are presented as mean ± S.E.; n is the number of rats in each group
Statistical Analysis
Values are presented as mean ± S.E. Mean arterial pressure (MAP) was calculated as diastolic + [(systolic − diastolic)/3]. Statistical comparison was made by analysis of variance followed by post hoc multiple comparisons of the mean with Student-Newman-Keuls test. The probability level less than 0.05 was considered significant.
Results
Nischarin Antisense ODNs Had No Effect on the Baseline Blood Pressure and Heart Rate
MAP and HR measured 24 h after the second daily intracisternal injections of the nischarin antisense ODN (AS1 or AS2), the mismatched ODN (MM1 or MM2), or aCSF are shown in Table 1. There were no differences between the MAP and HR values of the treatment and control groups (Table 1), which indicates that the intracisternal administration of nischarin antisense ODN or its mismatched ODN for 2 days had no effect on the baseline blood pressure or heart rate.
Pretreatment with Nischarin Antisense ODN Attenuated Rilmenidine-Evoked Hypotension
In this experiment, we investigated the effect of nischarin antisense ODNs on the hypotensive response elicited by the activation of central I1 receptor. Rilmenidine (25 μg/rat) was microinjected into the cisterna magna of rats pretreated with the nischarin antisense ODN (AS1 or AS2; 2 nmol/day for 2 days), the corresponding mismatched ODN (MM1 or MM2; 2 nmol/day for 2 days), or equal volume of aCSF. As shown in Table 1, the blood pressure and heart rate values of the treatment and control groups were similar before rilmenidine administration. Two-day pretreatment with the nischarin antisense ODNs significantly (p < 0.05) attenuated the hypotensive response elicited by central I1 receptor activation; the maximal reductions in MAP caused by i.c. rilmenidine were −4.1 ± 0.9 mm Hg (AS1), −2.1 ± 1.1 mm Hg (AS2), −10.8 ± 1.9 mm Hg (MM1), −15.3 ± 2.5 mm Hg (MM2), and −13.6 ± 1.2 mm Hg (aCSF). The virtual abolition, by AS1 or AS2 pretreatment, of rilmenidine-evoked hypotension was accompanied with a significant attenuation of the associated bradycardic response (Fig. 1).
Fig. 1.

Effect of intracisternal administration of one of the nischarin anti-sense ODNs (AS1 or AS2) or the mismatched ODNs (MM1 or MM2), compared with equal volume of aCSF, on rilmenidine (25 μg/rat i.c.)-evoked hypotension and bradycardia. Values are mean ± S.E., and the number in parentheses indicates the number of rats in each group. * or #, p < 0.05, compared with the corresponding mismatched ODN and aCSF-treated group, respectively.
Suppression of Nischarin Expression in the RVLM of Nischarin Antisense-Pretreated Rats
This experiment verified the ability of intracisternally administered nischarin antisense ODNs to knockdown nischarin expression in the RVLM of rats that exhibited attenuated I1-evoked hypotension. Intracisternal treatment with the nischarin antisense ODNs AS1 or AS2 resulted in substantial (>80%) reduction in the nischarin expression in the RVLM. As shown in Fig. 2, the number of RVLM neurons that exhibited nischarin immunoreactivity was significantly (p < 0.05) reduced by AS1 (3 ± 1 cells) or AS2 (4 ± 1 cells) compared with the corresponding values of the rats pretreated with the mismatched ODNs, MM1 (32 ± 2 cells) or MM2 (31 ± 1 cells), or with aCSF (30 ± 2 cells).
Fig. 2.

Effect of intracisternal administration of one of the nischarin antisense ODNs (AS1 or AS2) or the mismatched ODNs (MM1 or MM2), compared with equal volume of aCSF on the expression of nischarin in the RVLM. The photomicrograph shows the immunohistochemical images of nischarin-immunoreactive neurons in the RVLM. The low-power image demonstrates the location of the RVLM (marked by square) from which the immunoreactive neurons were counted. High-power images display the effects of the different treatments. Scale bar, 100 μm. Bar graphs show the number of nischarin-immunoreactive neurons in the RVLM. Values are expressed as mean ± S.E., and the number in parentheses indicates the number of rats in each group. *, p < 0.05, compared with the aCSF control group. # or @, p < 0.05, compared with the corresponding mismatched ODN group.
I1 (Rilmenidine)-Evoked Activation of ERK1/2 in the RVLM Is Suppressed in Nischarin Antisense ODN-Pre-treated Rats
Under basal conditions, pERK1/2 expression is scarce in the RVLM, but it increases in response to central I1 receptor activation (Zhang and Abdel-Rahman, 2005). This experiment determined whether pretreatment with nischarin antisense ODN (AS1 or AS2), which resulted in nischarin knockdown in the RVLM (Fig. 2), abrogates the I1 receptor-mediated activation of ERK1/2 in the RVLM. The activation of central I1 receptor, by i.c. rilmenidine, resulted in significant and similar increases in pERK1/2 expression in the RVLM of rats pretreated with aCSF or mismatched nischarin antisense ODN (MM1 or MM2); the number of pERK1/2-immunoreactive neurons in these groups (Fig. 3) is significantly greater than basal expression level (7 ± 1 positive cells) reported in the same preparation in our previous study (Zhang and Abdel-Rahman, 2005). By contrast, in rats pre-treated with either nischarin antisense ODN (AS1 or AS2), rilmenidine-evoked increase in pERK1/2 expression in the RVLM was abrogated (Fig. 3).
Fig. 3.
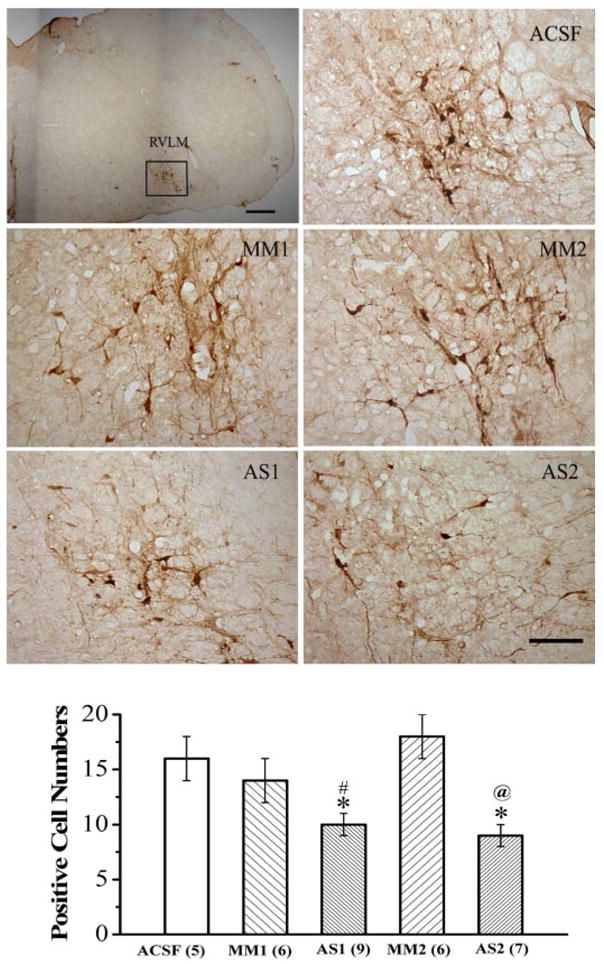
Effect of intracisternal administration of one of the nischarin antisense ODNs (AS1 or AS2) or the mismatched ODNs (MM1 or MM2), compared with equal volume of aCSF, on the phosphorylation of MAPK (pERK1/2) in the RVLM elicited by rilmenidine (25 μg/rat i.c.). The photomicrograph shows the immunohistochemical images of pERK1/2-immunoreactive neurons. The low-power image demonstrates the location of the RVLM (marked by square) from which the pERK1/2-immuno-reactive neurons were counted. Scale bar, 200 μm. High-power images display the effects of the different treatments. Scale bar, 100 μm. Bar graphs show the number of pERK1/2-immunoreactive neurons in the RVLM. Values are expressed as mean ± S.E. *, p < 0.05 compared with the aCSF control group. # or @, p < 0.05 compared with the corresponding mismatched ODN group.
To determine whether the I1 receptor (rilmenidine)-mediated activation of ERK1/2 occurs in nischarin-expressing neurons in the RVLM, double-labeling immunofluorescence studies were conducted. As indicated above, under basal conditions, pERK1/2 expression is scarce, but it increases following rilmenidine administration (Zhang and Abdel-Rahman, 2005). Therefore, the brain sections used to generate the confocal immunofluorescence images were obtained from rilmenidine-treated rats at the maximal hypotensive response. Figure 4 shows nischarin (left) and pERK1/2 (middle) expression in the RVLM. Merging of the two images (right) revealed that nischarin and pERK1/2 are colocalized in the RVLM neurons.
Fig. 4.
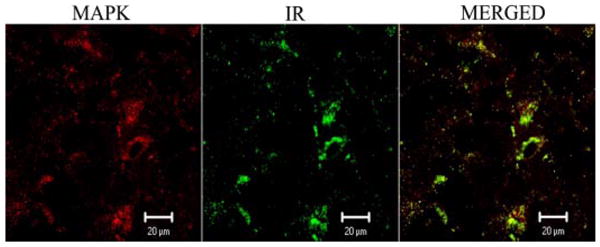
Colocalization of nischarin protein and pERK1/2 detected by double-labeling immunofluorescence technique. pERK1/2 was detected by phospho-p42/44 MAPK monoclonal antibody with anti-mouse IgG secondary antibody conjugated with Texas Red fluorescent label (red). Nischarin in protein (imidazoline receptor, IR) was identified by nischarin polyclonal antibody with anti-rabbit IgG secondary antibody conjugated with fluorescein isothiocyanate fluorescent label (green). The signal of colocalization (yellow) is visualized by merging the two images.
Discussion
Nischarin, a homolog of human IRAS (Piletz et al., 2000), plays an important role in cell signaling and function (Lim and Hong, 2004; Reddig et al., 2005). However, no study has established the neurobiological relevance of nischarin signaling in vivo. Most, if not all, reported findings on human I1R (IRAS) and/or its homolog mouse nischarin were generated in cell culture. Recent evidence suggests that the generation of pERK1/2, a well established signaling product of I1 receptor activation, is dependent on nischarin in PC12 cells (Zhang and Abdel-Rahman, 2006; Sun et al., 2007). The current study presents evidence that nischarin is essential for the initiation of neuronal signaling triggered by I1 receptor activation in the RVLM, which underlies the subsequent hypotensive response. This evidence is supported by the findings that knockdown of nischarin expression in the RVLM virtually abolishes the I1R (rilmenidine)-mediated enhancement of pERK1/2 production in the RVLM and the associated hypotension. Evidence is also presented that pERK1/2, generated in response to central I1 receptor activation, is colocalized with nischarin in the RVLM neurons. Together, these findings support the hypothesis that nischarin serves as, or at least shares a common signaling pathway with the I1-receptor in the brainstem.
We used antisense ODN strategy to investigate the effect of nischarin knockdown on neurochemical and cardiovascular responses elicited by central I1 receptor activation. The nischarin antisense and mismatched ODNs used in the present study were designed according to cDNA sequence from the GenBank. The ability of these antisense ODNs to effectively knockdown nischarin expression was verified in PC12 cells in our previous study (Zhang and Abdel-Rahman, 2006). Furthermore, the antisense ODNs used in the present study were phosphorothioated to prevent their degradation by endogenous enzymes (Putney et al., 1981). The dose regimen of the antisense ODNs was based on reported studies (Satoh et al., 2002; Nikaido et al., 2004). We report a substantial (>80%) reduction in nischarin level in the RVLM of rats treated with nischarin antisense ODN. Given that different proteins exhibit different turnover rates, it was important to choose a treatment time that causes optimal inhibition of nischarin expression. The 2-day treatment regimen was based on our previous findings that demonstrated, using the same ODN probes, optimal inhibition in nischarin expression in PC12 cells (Zhang and Abdel-Rahman, 2006). Interestingly, in a recent study (Sun et al., 2007), despite the use of a different strategy to design the nischarin antisense ODN, a 2-day exposure to nischarin antisense ODN also substantially inhibited nischarin expression in the same cell line. Together, in addition to agreeing with previous in vitro findings in PC12 cells, which exhibit a neuronal phenotype (Zhang and Abdel-Rahman, 2006; Sun et al., 2007), we present the first evidence that intracisternal administration of nischarin antisense ODN effectively and substantially reduced the expression of nischarin in the brainstem (RVLM neurons).
Results of the present study show that the hypotensive response elicited by central rilmenidine administration was drastically attenuated in rats that exhibited reduced expression of nischarin in the brainstem. In general, nischarin expression paralleled the reported distribution pattern of the I1 receptor (el-Mas and Abdel-Rahman, 1995; Ernsberger et al., 1995). We focused on the RVLM because reported studies, including ours, have highlighted its role in I1-mediated hypotension (Ernsberger and Haxhiu, 1997). More pertinent to the present study, we previously showed that the hypotensive response elicited by rilmenidine (same dose and route used in the present study) was mediated, at least partly, as a result of pERK1/2 production in the RVLM (Zhang and Abdel-Rahman, 2005).
The present findings show that central I1 receptor activation (i.c. rilmenidine) caused significant increase in pERK1/2 expression in the RVLM, along with the hypotensive response in control (aCSF-pretreated) rats. These neurochemical and blood pressure responses fully agree with our previous findings in the same model (Zhang and Abdel-Rahman, 2005). However, we demonstrate, for the first time, that the neurochemical (RVLM pERK1/2) and blood pressure responses elicited by I1 receptor activation were abrogated by two different nischarin antisense ODNs (AS1 or AS2) and that they were preserved in the presence of the corresponding mismatched antisense ODNs (MM1 or MM2). These findings highlight a pivotal role for nischarin in the RVLM in I1-mediated responses, and they rule out the possibility that neurotoxic effects of the ODNs might have accounted for the observed effects of the nischarin antisense ODNs. In support of this notion was the drastic reduction in nischarin expression in the RVLM caused by nischarin antisense, but not by the mismatched ODN. Together, the findings suggest that the enhanced expression of pERK1/2 in the RVLM and the associated hypotension elicited by I1R activation were tightly related to nischarin expression in the RVLM. These findings confirm and extend similar cell culture findings in PC12 cells, which linked pERK1/2 expression caused by I1R activation to the presence of nischarin (Zhang and Abdel-Rahman, 2006; Sun et al., 2007).
The mechanism by which nischarin modulates I1 receptor signaling remains to be elucidated. Recent findings have delineated the molecular relationship between nischarin (the mouse homolog) and the human IRAS (Lim and Hong, 2004; Reddig et al., 2005). Our present findings are the first to support a functional (in vivo) relationship between nischarin and I1R signaling. However, other signaling mechanisms that might contribute to rilmenidine-evoked hypotension and bradycardia must be considered because 1) although nischarin knockdown virtually abolished the hypotension elicited by rilmenidine, it only reduced the associated bradycardia; and 2) ERK1/2 phosphorylation can be triggered by signaling mechanisms other than the I1 receptor. The α2AR is considered a potential signaling mechanism that might contribute to rilmenidine-evoked reductions in blood pressure and heart rate and the associated increase in RVLM pERK1/2 because 1) rilmenidine exhibits α2AR agonist activity (Szabo, 2002), and 2) α2AR activation enhances pERK1/2 production in cultured cells (Peng et al., 1998). Nonetheless, findings of the present and reported studies argue against a role for the α2AR in rilmenidine-evoked hypotension and the enhancement of RVLM pERK1/2 production in our model system for the following reasons. First, enhanced pERK1/2 production in the RVLM in the present and previous (Zhang and Abdel-Rahman, 2005) studies is elicited by rilmenidine but not by an equal hypotensive dose of the pure α2AR agonist α-methylnorepinephrine. Second, pERK1/2 production is enhanced by rilmenidine or moxonidine in PC12 cells, which lack the α2AR (Ernsberger et al., 1995; Separovic et al., 1996), and the signaling cascade that results in pERK1/2 production is dependent, at least partly, on nischarin (Zhang and Abdel-Rahman, 2006; Sun et al., 2007). Third, in α2AR transfected PC12 cells (Williams et al., 1998), and in adult mouse hippocampus (CA1) cells (Vanhoose et al., 2002), activation of the α2AR did not activate ERK. Conversely, two possibilities might explain the presence of rilmenidine-evoked bradycardia in nischarin antisense ODN-treated rats. First, nischarin expression in the nucleus ambiguous or the dorsal nucleus of the vagus might not be inhibited to the extent achieved in the RVLM. Second, the contribution of brainstem α2AR signaling to rilmenidine evoked bradycardia, which has been reported in a number of studies (see Szabo 2002), cannot be ruled out. The second possibility more likely explains the presence of rilmenidine-evoked bradycardia in AS1 or AS2 ODN-treated rats because a similar bradycardic response is evident following two interventions, I1R blockade or the inhibition ERK1/2 phosphorylation with PD98059, which virtually abolished rilmenidine-evoked hypotension and abrogated the associated increase in RVLM pERK1/2 (Zhang and Abdel-Rahman, 2005). Therefore, given the tight association between nischarin and IRAS (Lim and Hong, 2004; Reddig et al., 2005) and the pivotal role of ERK1/2 phosphorylation in the RVLM in I1R (rilmenidine)-mediated hypotension (Zhang and Abdel-Rahman, 2005), we reasoned that ERK1/2 phosphorylation caused by I1 receptor activation might occur within nischarin-expressing neurons in the RVLM. In support of this notion, we show that pERK1/2, generated in response to central I1 receptor activation, is colocalized with nischarin in RVLM neurons (Fig. 4) of rats that exhibited rilmenidine (I1)-evoked hypotension.
In summary, we present the first evidence that knocking down nischarin expression in the brainstem by antisense ODN virtually abolished the neurochemical (enhanced pERK1/2 production in the RVLM) and hypotensive responses elicited by central I1 receptor activation. The findings support the hypothesis that brainstem (RVLM) nischarin serves as, or at least shares, a common signaling pathway with the I1 receptor. We have also identified pERK1/2 as an important signaling molecule in the mediation of hypotension that is dependent on the integrity of brainstem nischarin. Further studies are needed to elucidate the mechanisms implicated in nischarin signaling in vivo and its role in central regulation of blood pressure.
Acknowledgments
We thank Kui Sun for excellent technical support.
This work is supported by National Institutes of Health Grant 2R01 AA07839 (to A.A.R.).
ABBREVIATIONS
- I1
imidazoline
- I1R
imidazoline receptor
- α2AR
α2 adrenergic receptor
- IRAS
imidazoline receptor antisera-selective
- ERK
extracellular signal-regulated kinase
- pERK
phosphorylated extracellular signal-regulated kinase
- ODN
oligodeoxynucleotide
- i.c
intracisternally
- aCSF
artificial cerebrospinal fluid
- RVLM
rostral ventrolateral medulla
- BP
blood pressure
- HR
heart rate
- TBS
Tris-buffered saline
- MAPK
mitogen-activated protein kinase
- AS
antisense
- MM
mismatch
- MAP
mean arterial pressure
- PD98059
2′-amino-3′-methoxyflavone
References
- Alahari SK. Nischarin inhibits Rac induced migration and invasion of epithelial cells by affecting signaling cascades involving PAK. Exp Cell Res. 2003;288:415–424. doi: 10.1016/s0014-4827(03)00233-7. [DOI] [PubMed] [Google Scholar]
- Alahari SK, Lee JW, Juliano RL. Nischarin, a novel protein that interacts with the integrin alpha5 subunit and inhibits cell migration. J Cell Biol. 2000;151:1141–1154. doi: 10.1083/jcb.151.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Mas MM, Abdel-Rahman AA. Upregulation of imidazoline receptors in the medulla oblongata accounts for the enhanced hypotensive effect of clonidine in aortic barodenervated rats. Brain Res. 1995;691:195–204. doi: 10.1016/0006-8993(95)00672-d. [DOI] [PubMed] [Google Scholar]
- Ernsberger P, Graves ME, Graff LM, Zakieh N, Nguyen P, Collins LA, Westbrooks KL, Johnson GG. I1-imidazoline receptors. Definition, characterization, distribution, and transmembrane signaling. Ann N Y Acad Sci. 1995;763:22–42. doi: 10.1111/j.1749-6632.1995.tb32388.x. [DOI] [PubMed] [Google Scholar]
- Ernsberger P, Haxhiu MA. The I1-imidazoline-binding site is a functional receptor mediating vasodepression via the ventral medulla. Am J Physiol Regul Integr Comp Physiol. 1997;273:R1572–R1579. doi: 10.1152/ajpregu.1997.273.5.R1572. [DOI] [PubMed] [Google Scholar]
- Gerfen C. Basic neuroanatomical methods. In: Crawley JN, Gerfen CR, Rogawski MA, Sibley DR, Skolnick P, Wray S, editors. Current Protocols in Neuroscence. John Wiley & Sons, Inc.; New York: 1997. pp. 1.1.1–1.1.11. [Google Scholar]
- Ince E, Levey A. Immunohistochemical Localization of Neurochemicals. In: Crawley JN, Gerfen CR, Rogawski MA, Sibley DR, Skolnick P, Wray S, editors. Current Protocols in Neuroscence. John Wiley & Sons, Inc.; New York: 1997. pp. 1.2.1–1.2.12. [Google Scholar]
- Institute of Laboratory Animal Resources. Guide for the Care and Use of Laboratory Animals. 7. Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council; Washington DC: 1996. [Google Scholar]
- Li F, Wu N, Su RB, Zheng JQ, Xu B, Lu XQ, Cong B, Li J. Involvement of phosphatidylcholine-selective phospholipase C in activation of mitogen-activated protein kinase pathways in imidazoline receptor antisera-selected protein. J Cell Biochem. 2006;98:1615–1628. doi: 10.1002/jcb.20806. [DOI] [PubMed] [Google Scholar]
- Lim KP, Hong W. Human Nischarin/imidazoline receptor antisera-selected protein is targeted to the endosomes by a combined action of a PX domain and a coiled-coil region. J Biol Chem. 2004;279:54770–54782. doi: 10.1074/jbc.M411315200. [DOI] [PubMed] [Google Scholar]
- Nikaido H, Tsunoda H, Nishimura Y, Kirino T, Tanaka T. Potential role for heat shock protein 72 in antagonizing cerebral vasospasm after rat subarachnoid hemorrhage. Circulation. 2004;110:1839–1846. doi: 10.1161/01.CIR.0000142615.88444.31. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; Sydney, Australia: 1982. [Google Scholar]
- Peng M, Li Y, Luo Z, Liu C, Laties AM, Wen R. alpha2-Adrenergic agonists selectively activate extracellular signal-regulated kinases in Muller cells in vivo. Invest Ophthalmol Vis Sci. 1998;39:1721–1726. [PubMed] [Google Scholar]
- Piletz JE, Ivanov TR, Sharp JD, Ernsberger P, Chang CH, Pickard RT, Gold G, Roth B, Zhu H, Jones JC, et al. Imidazoline receptor antisera-selected (IRAS) cDNA: cloning and characterization. DNA Cell Biol. 2000;19:319–329. doi: 10.1089/10445490050043290. [DOI] [PubMed] [Google Scholar]
- Piletz JE, Wang G, Zhu H. Cell signaling by imidazoline-1 receptor candidate, IRAS, and the nischarin homologue. Ann N Y Acad Sci. 2003;1009:392–399. doi: 10.1196/annals.1304.053. [DOI] [PubMed] [Google Scholar]
- Putney SD, Benkovic SJ, Schimmel PR. A DNA fragment with an alpha-phosphorothioate nucleotide at one end is asymmetrically blocked from digestion by exonuclease III and can be replicated in vivo. Proc Natl Acad Sci U S A. 1981;78:7350–7354. doi: 10.1073/pnas.78.12.7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddig PJ, Xu D, Juliano RL. Regulation of p21-activated kinase-independent Rac1 signal transduction by nischarin. J Biol Chem. 2005;280:30994–31002. doi: 10.1074/jbc.M502546200. [DOI] [PubMed] [Google Scholar]
- Satoh M, Parent AD, Zhang JH. Inhibitory effect with antisense mitogen-activated protein kinase oligodeoxynucleotide against cerebral vasospasm in rats. Stroke. 2002;33:775–781. doi: 10.1161/hs0302.103734. [DOI] [PubMed] [Google Scholar]
- Separovic D, Kester M, Ernsberger P. Coupling of I1-imidazoline receptors to diacylglyceride accumulation in PC12 rat pheochromocytoma cells. Mol Pharmacol. 1996;49:668–675. [PubMed] [Google Scholar]
- Sun Z, Chang CH, Ernsberger P. Identification of IRAS/Nischarin as an I1-imidazoline receptor in PC12 rat pheochromocytoma cells. J Neurochem. 2007;101:99–108. doi: 10.1111/j.1471-4159.2006.04413.x. [DOI] [PubMed] [Google Scholar]
- Szabo B. Imidazoline antihypertensive drugs: a critical review on their mechanism of action. Pharmacol Ther. 2002;93:1–35. doi: 10.1016/s0163-7258(01)00170-x. [DOI] [PubMed] [Google Scholar]
- Vanhoose AM, Emery M, Jimenez L, Winder DG. ERK activation by G-protein-coupled receptors in mouse brain is receptor identity-specific. J Biol Chem. 2002;277:9049–9053. doi: 10.1074/jbc.M108309200. [DOI] [PubMed] [Google Scholar]
- Wang X, Abdel-Rahman AA. Effect of chronic ethanol administration on hepatic eNOS activity and its association with caveolin-1 and calmodulin in female rats. Am J Physiol Gastrointest Liver Physiol. 2005;289:G579–G585. doi: 10.1152/ajpgi.00282.2004. [DOI] [PubMed] [Google Scholar]
- Williams NG, Zhong H, Minneman KP. Differential coupling of alpha1-, alpha2-, and beta-adrenergic receptors to mitogen-activated protein kinase pathways and differentiation in transfected PC12 cells. J Biol Chem. 1998;273:24624–24632. doi: 10.1074/jbc.273.38.24624. [DOI] [PubMed] [Google Scholar]
- Zhang J, Abdel-Rahman AA. Mitogen-activated protein kinase phosphorylation in the rostral ventrolateral medulla plays a key role in imidazoline (i1)-receptor-mediated hypotension. J Pharmacol Exp Ther. 2005;314:945–952. doi: 10.1124/jpet.105.087510. [DOI] [PubMed] [Google Scholar]
- Zhang J, Abdel-Rahman AA. Nischarin as a functional imidazoline (I1) receptor. FEBS Lett. 2006;580:3070–3074. doi: 10.1016/j.febslet.2006.04.058. [DOI] [PubMed] [Google Scholar]
- Zhang J, El-Mas MM, Abdel-Rahman AA. Imidazoline I(1) receptor-induced activation of phosphatidylcholine-specific phospholipase C elicits mitogen-activated protein kinase phosphorylation in PC12 cells. Eur J Pharmacol. 2001;415:117–125. doi: 10.1016/s0014-2999(01)00834-2. [DOI] [PubMed] [Google Scholar]