

Abstract
Experimental autoimmune encephalomyelitis (EAE) models, in animals, many characteristics of multiple sclerosis, for which there is no adequate therapy. We investigated whether lithium, an inhibitor of glycogen synthase kinase-3 (GSK3), can ameliorate EAE in mice. Pretreatment with lithium markedly suppressed the clinical symptoms of EAE induced in mice by myelin oligodendrocyte glycoprotein peptide (MOG35–55) immunization and greatly reduced demyelination, microglia activation, and leukocyte infiltration in the spinal cord. Lithium administered postimmunization, after disease onset, reduced disease severity and facilitated partial recovery. Conversely, in knock-in mice expressing constitutively active GSK3, EAE developed more rapidly and was more severe. In vivo lithium therapy suppressed MOG35–55-reactive effector T cell differentiation, greatly reducing in vitro MOG35–55-stimulated proliferation of mononuclear cells from draining lymph nodes and spleens, and MOG35–55-induced IFN-γ, IL-6, and IL-17 production by splenocytes isolated from MOG35–55-immunized mice. In relapsing/remitting EAE induced with proteolipid protein peptide139–151, lithium administered after the first clinical episode maintained long-term (90 days after immunization) protection, and after lithium withdrawal the disease rapidly relapsed. These results demonstrate that lithium suppresses EAE and identify GSK3 as a new target for inhibition that may be useful for therapeutic intervention of multiple sclerosis and other autoimmune and inflammatory diseases afflicting the CNS.
Multiple sclerosis is the most common autoimmune inflammatory disease of the CNS. Multiple sclerosis is characterized by immune-mediated demyelination and neurodegeneration of the CNS with lesions predominantly occurring in the white matter (1–4). Although multiple sclerosis afflicts more than two million people, its etiology remains unresolved and currently there are no adequate therapeutic interventions.
Experimental autoimmune encephalomyelitis (EAE)4 is widely studied in animals to model many of the clinical, immunological, and neuropathological features of multiple sclerosis (5). EAE is induced in susceptible mice by eliciting an immune response to injected myelin Ags, such as myelin oligodendrocyte glycoprotein (MOG) peptide or proteolipid protein peptide (PLP) (6). In EAE, the integrity of the blood-brain barrier is impaired, allowing perivascular infiltrates in the CNS, leading to demyelination and loss of neuronal function, and culminating in paralysis. CD4+ T cells infiltrating the CNS are the initiator and the early effector cells in the development of EAE, but infiltrating macrophages, dendritic cells, and resident glia are the ultimate effector cells that amplify neuroinflammation and cause demyelination and axonal damage (3, 7).
Lithium has been used for >50 years for the therapeutic treatment of psychiatric diseases in humans (8). The basis of this therapeutic action of lithium remains unresolved, but accumulating evidence indicates that it stems largely from its inhibition of glycogen synthase kinase-3 (GSK3) (9), a serine-threonine protein kinase with regulatory actions affecting many cellular functions (10). The activity of GSK3 is regulated mainly by the inhibitory phosphorylation of N-terminal serines of the two isoforms of GSK3, Ser21 in GSK3α and Ser9 in GSK3β (10). Mutation of the N-terminal serine to alanine provides a form of GSK3 that cannot be inactivated by this mechanism. Recently, knock-in mice were produced in which the regulatory serines of both GSK3 isoforms were mutated to alanines, S9A-GSK3β and S21A-GSK3α, with both GSK3 isoforms expressed at normal levels (11). Thus, in these GSK3 knock-in mice, GSK3α and GSK3β are present at physiological levels, but they cannot be inhibited by serine-phosphorylation, so GSK3 is maximally active.
Growing evidence in the literature indicates that GSK3 is a major regulator of inflammation. By inhibiting GSK3, lithium greatly reduced the production of major proinflammatory cytokines following stimulation of several types of Toll-like receptors in human monocytes and mouse peripheral blood monocytes (12). In rodents, lithium and other inhibitors of GSK3 also increased survival from lethal sepsis (12) and lethal lupus (13), attenuated organ injury associated with sepsis (14), provided significant protection from a wide variety of apoptotic insults in the CNS (15), and ameliorated several inflammatory and immune conditions, such as arthritis, peritonitis, and colitis (14, 16–18). Notably, administration of GSK3 inhibitors reduced the development of inflammation and tissue injury associated with spinal cord trauma, significantly blocking the development of hind limb motor impairments (19). Most importantly for the present study, in 1991, before lithium was known to inhibit GSK3, intraperitoneal injections of lithium in rats was reported to inhibit the development of EAE (20). Unfortunately, high toxic doses of lithium were used and it was concluded that “the immunosuppression was a toxic effect” (20), which appears to have discouraged further studies.
Considering the greater understanding of the effects of GSK3 and the long history of safe usage of lithium in humans, we considered the possibility that administration of low, therapeutically relevant, levels of lithium may provide protection from inflammatory autoimmune diseases affecting the CNS. Lithium at therapeutic levels is nontoxic and is commonly administered to mice in the diet to achieve serum levels equivalent to those attained therapeutically in human patients (21). The results reported in this study show that pretreatment with therapeutically relevant levels of lithium almost completely blocked the onset of EAE, lithium promoted recovery when administered after the development of EAE, and, remarkably, chronic treatment blocked relapse episodes of EAE, which rapidly returned after lithium was withdrawn.
Materials and Methods
Animals
Male C57BL/6 and female SJL mice were purchased from Frederick Cancer Research. To test whether constitutively active GSK3 exacerbates EAE, GSK3 knock-in mice (11) and matched controls were used. These mice contain serine-to-alanine mutations in the regulatory serines of both GSK3 isoforms, S21A-GSK3α and S9A-GSK3β, in place of endogenous GSK3α/β to disable the inhibitory serine phosphorylation of GSK3, so GSK3α/β retain maximal activities. All mice were housed and treated in accordance with National Institutes of Health and the University of Alabama Animal Care and Use Committee guidelines. For lithium pretreatment, lithium was administered in pelleted food containing 0.2% lithium carbonate (Harlan-Teklad) for 1 wk before immunization and maintained after immunization. This lithium administration to mice in the diet is nontoxic and is commonly used to achieve serum levels equivalent to those attained therapeutically in human patients (21). For lithium treatment after immunization, mice were administered the lithium-containing food and were given two injections of LiCl (100 mg/kg) on the first and second days of lithium treatment to increase lithium levels more rapidly than can be attained by dietary administration alone. The concentration of lithium in the serum was measured by inductively coupled plasma/mass spectrometry performed by Medtox Laboratories.
Induction of active EAE
Male C57BL/6 mice (8–12 wk old, from Frederick Cancer Research) or GSK3 knock-in mice (11) and matched controls were immunized with a s.c. injection of 150 μg of MOG35–55 peptide (Biosynth International) emulsified in CFA on day 0, and an intraperitoneal injection of 500 ng pertussis toxin (List Biological Laboratories) on days 0 and 2. Female SJL mice (8–12 wk old, from Frederick Cancer Research) were immunized with a s.c. injection of 150 μg of PLP139–151 peptide (Biosynth International) emulsified in CFA on day 0. Onset and clinical progression of EAE symptoms were monitored daily using a standard scale of 0 to 6: 0, no clinical signs; 1, loss of tail tone; 2, flaccid tail; 3, incomplete paralysis of one or two hind legs; 4, complete hind limb paralysis; 5, moribund (animals were humanly euthanized); 6, death. To compare the time course of disease development in different groups of mice, the daily average of the clinical scores was calculated for each group. A cumulative disease index (CDI) score for each treatment group was calculated as the average of the sum of the daily clinical scores for each mouse. Statistically significant differences were calculated using a Student’s t test and values of p < 0.05 were considered to be statistically significant.
T cell proliferation and cytokine production
Single cell suspensions from draining lymph nodes and spleen were obtained at 10 days after MOG35–55-immunization, a time at which very efficient MOG35–55-specific responses can be detected (22). Cells were cultured in 96-well plates (2 × 105 cells/well) and stimulated with 0, 1, or 10 μg/ml MOG35–55 peptide, or 1 μg/ml anti-CD3 (145–2C11) in triplicate. After 72 h, cells were labeled with [3H]-thymidine (1 μCi/well) for 18 h, and incorporation of [3H]-thymidine was measured. Single cell suspensions of splenocytes were stimulated with 0, 1, or 10 μg/ml MOG35–55 peptide, or 1 μg/ml anti-CD3, and after 48 h, cytokines in the culture supernatants were measured by ELISA (eBioscience).
Flow cytometry
Mice were anesthetized, spleen and draining lymph nodes removed, and single cell suspension was prepared. Mice were then perfused, and spinal cords were removed and incubated with 2 mg/ml collagenase D (Roche) and 5 U/ml DNase (Sigma-Aldrich) for 1 h at 37°C. Mononuclear cells from the spinal cord were purified by two-step Percoll gradient centrifugation, as described previously (23). Mononuclear cell preparations were incubated with anti-CD16/32 (2.4G2, FcR block); stained with PE-anti-CD8 (53–6.7), PerCP-anti-CD4 (L3T4), FITC-anti-NK 1.1 (DX5), or PE-anti-CD25 (PC61.5); and conjugated to appropriate fluorochromes, as indicated. For intracellular staining, surface stained cells were permeabilized and stained with Alexa647-anti-FoxP3. All Abs were obtained from eBio-sciences. Stained cells were analyzed using a FACSCalibur (BD Biosciences).
NK cell cytotoxicity assay
YAC-1 cells expressing firefly luciferase (YAC-1—Luc) were used as target cells to measure NK activity in spleen cells as described (24). In brief, spleen mononuclear cells from untreated and day 10 lithium-treated mice were incubated with YAC-1—Luc cells at a ratio of 5:1 for 4 h at 37°C in a tissue culture incubator. Separate wells contained only YAC-1 cells. The level of luciferase activity was determined at the end of the incubation by a chemiluminescence assay according to the manufacturer’s instructions (Promega). For each target, three replicates of the internal references for the 0% viability background (MIN) and the 100% viability maximal signal (MAX) were run. The 0% viability reference point was determined by plating target cells in media with a final concentration of 1% SDS (MINSDS). The 100% viability reference point (MAXmedia) was determined by plating target cells in media without effector cells. Percent viability was calculated as the mean luminescence of the experimental sample minus background (MINSDS) divided by the mean luminescence of the input number of target cells used in the assay (MAXmedia) minus background (MINSDS). Percent-specific lysis is equal to (1 — percent viability) × 100 and is calculated as follows: % specific lysis = [1 — counts per 5 s (experimental — MINSDS)/(MAXmedia — MINSDS] × 100.
T regulatory (Treg) cell activity assay
Treg cell activity was determined by a proliferation-suppression assay (25). CD4+CD25+ T cells (Treg cells) from spleens and lymph nodes of untreated and day 10 lithium-treated mice and CD4+CD25- (responder) T cells from C57BL/6 mice were fractionated using magnetic bead chromatography (Stem Cell Technologies). Cultures of Treg cells and responder cells at a ratio of 2:1 were stimulated with 1 μg/ml anti-CD3 (145–2C11) in the presence of gamma-irradiated T-depleted spleen cells that served as APCs for 48 h. Proliferation was assessed by incubation with 1 μCi of [3H]-thymidine for an additional 18 h of culture. Separate wells contained anti-CD3 and cultures of responder T cells plus APC only, Treg cells plus APC, and APC only. The purity of responder cells or Treg cells prepared as above are routinely greater than 95%.
Histopathology and immunohistochemistry
Cross-sections made through the whole length of the spinal cords were immersion-fixed in Bouin’s fixative and paraffin-embedded, and six sections (5 μm) from a minimum of three animals per group were deparaffinized and stained with Luxol fast blue for evaluation of demyelination, or with biotin-conjugated Griffonia simplicifolia lectin (GS-I-B4) for staining microglia. For immunohistochemistry, sections were deparaffinized, followed by Ag retrieval and inhibition of endogenous peroxidase activity, and blocked for 30 min (1% BSA, 0.2% skim milk, 0.3% Triton X-100 in PBS for rabbit Abs, or 5% horse serum, 0.3% Triton X-100 in PBS for the goat Abs). Sections were incubated overnight at 4°C with rabbit anti-myeloperoxidase (Lab Vision) or with goat anti-mouse CD4 (R&D Systems) for detection of neutrophils and CD4+ T cells, respectively, followed by PBS washes and application of HRP-conjugated anti-rabbit or anti-goat secondary Abs (Jackson ImmunoResearch Laboratories) for 1 h at room temperature. After three washes in PBS, cyanine 3-conjugated tyramide was deposited according to the manufacturer’s protocol (TSA Plus, PerkinElmer Life Science Products). Sections were washed and counter-stained with Hoechst 33258 (Sigma-Aldrich), coverslipped with PBS: glycerol (1:1), and viewed with a Zeiss-Axioscope microscope (Carl Zeiss) equipped with epifluorescence. Digital images were captured with a Zeiss Axiocam and Zeiss Axiovision software. All sections used for analysis were processed in parallel for detection in the same staining group, using the same reagents, concentrations, and incubation times. Images were collected at the same time using identical settings with respect to image exposure time and image compensation settings.
Statistics
Statistical significance between untreated and lithium-treated mice was calculated using Student’s t test and values of p < 0.05 were considered to be statistically significant.
Results
Lithium administration ameliorates clinical symptoms of EAE
To assess whether lithium is protective and anti-inflammatory in EAE, C57BL/6 mice with or without lithium pretreatment were immunized with MOG35–55 peptide to induce EAE. Lithium-free mice developed clinical EAE after 19.5 ± 0.7 days with an incidence of 100% and a CDI of 48.5 ± 3.1 (Fig. 1A). Lithium pretreatment completely prevented EAE in 81% (13/16) of mice and the afflicted 19% of mice had a delayed onset (28 ± 2.1 days) and greatly reduced severity, with a CDI of only 3.4 ± 2.0 (p < 0.05). The lithium concentration in the serum of mice on a lithium diet for 5 wk after EAE induction was 0.53 ± 0.03 mEq/l (n = 4).
FIGURE 1.

Lithium treatment improves clinical symptoms of EAE. A, Mice were pretreated with dietary lithium for 1 wk before immunization with MOG35–55 peptide and were maintained on lithium for the duration of the experiment (n = 15–16). B, Mice were immunized with MOG35–55 peptide and lithium was administered after mice reached a clinical score of 2. Because mice reached this score on different days after immunization, data are presented beginning on the day each mouse achieved criteria (n = 8–9). C, Mice were immunized with MOG35–55 peptide and lithium was administered after 20 days (n = 10). Values shown are means ± SEM. In all three experimental paradigms, lithium treatment significantly (p < 0.05) reduced the CDI.
To test whether lithium administration was capable of ameliorating ongoing EAE, MOG35–55-immunized mice were treated with lithium after onset of clinical symptoms. In one protocol, lithium treatment was initiated when mice attained a clinical score of 2, which was achieved on day 16.8 ± 1.3 postimmunization, and mice were monitored to day 53. Whereas lithium-free mice with EAE continued to deteriorate with increased clinical scores, mice with EAE given lithium upon reaching a score of 2 stabilized at that level of disease and did not worsen (Fig. 1B). The overall severity of EAE as measured by the CDI was significantly lower at 77.8 ± 4.4 for lithium-treated EAE mice compared with 107.6 ± 5.1 for lithium-free mice (p < 0.05).
A more challenging protocol was also tested, in which lithium treatment was initiated 20 days postimmunization, at the peak of the acute phase (Fig. 1C). The CDI from day 0 to 19 (before lithium treatment) was 23.5 ± 3.4 for the mice that were not going to be treated with lithium, and 24.4 ± 3.2 for the mice that were subsequently administered lithium. The CDI from days 20 to 72 was 97.0 ± 16.7 for the lithium-free mice and 69.3 ± 11.0 for the lithium-treated mice (p < 0.05). Therefore, lithium treatment enabled a significant recovery in the clinical course of EAE. Thus, lithium treatment before immunization with MOG35–55 peptide rendered mice resistant to the development of EAE and lithium treatment after establishment of EAE-lowered disease severity and/or facilitated partial recovery.
Lithium administration ameliorates neuropathology associated with EAE
Spinal cords examined 33 days after MOG35–55 peptide immunization contained activated microglia that colocalized with extensive demyelination, which were absent in the spinal cords of lithium-pretreated mice (Fig. 2, A and B, respectively). The spinal cords from mice with EAE that were treated with lithium after reaching a clinical score of 2 displayed lower microglial activation and less demyelination than lithium-free mice with EAE (Fig. 2, A and B, respectively). Thus, pre- or posttreatment with lithium attenuated clinical progression, demyelination, and microglia activation in mice with EAE.
FIGURE 2.

Lithium administration ameliorates neuropathology associated with EAE. Spinal cord sections from unimmunized mice, mice immunized with MOG35–55 peptide without lithium treatment, after 1 wk of lithium treatment, or treated with lithium after reaching a clinical score of 2, as shown in Fig. 1, were analyzed at the end of scoring clinical symptoms. A—D, Spinal cord sections were stained to evaluate activated microglia (A), demyelination (B), CD4+ T cells (C), or neutrophils (D). E, Mononuclear cells from spinal cords of mice treated with lithium after reaching a score 2 were surface stained with anti-CD4 and anti-CD8 and analyzed by flow cytometry to determine the proportion of CD4+ and CD8+ T cells in spinal cords. Dot plots represent CD4 and CD8 staining of cells within the mononuclear cell gate from untreated and lithium treated mice. Each section shown is a representative of six serial sections of the spinal cord from at least three mice per group.
Amelioration of EAE by lithium treatment was further confirmed by examinations of leukocyte infiltration into the CNS. Spinal cords from MOG35–55-immunized mice examined after the lithium pretreatment and lithium post-treatment paradigms described in Fig. 1, A and B contained much lower evidence of infiltrated CD4+ T cells (Fig. 2C) and neutrophils (Fig. 2D) than matched spinal cords from MOG35–55-immunized mice not given lithium. Surface staining of mononuclear cells from spinal cords of MOG35–55-immunized mice treated with lithium after reaching score 2 confirmed that there was a much lower proportion of CD4+ T cells in spinal cords of lithium-treated than in spinal cords of lithium-free mice (Fig. 2E).
Lithium administration reduces effector T cells
The resistance to EAE provided by lithium treatment could be due to attenuated generation of MOG35–55-specific T cells. Therefore, we measured the in vitro-stimulated proliferation of T cells isolated from draining lymph nodes and spleens 10 days after MOG35–55-immunization, with or without in vivo lithium pretreatment. The MOG35–55-stimulated proliferation of T cells from primed mice was greatly reduced in cells of both tissues prepared from lithium-pretreated mice (Fig. 3A). The low response of T cells from lithium-treated MOG35–55-immunized mice to Ag restimulation could be due to compromised effector cell generation and/or intrinsic loss of T cell ability to be activated. To determine whether lithium treatment compromises the ability of T cells to be activated, we examined the proliferative response to anti-CD3, and this was similar between T cells from lithium-treated and untreated mice (Fig. 3B). These results indicate that in vivo lithium pretreatment selectively inhibits the generation of MOG35–55-specific effector T cells. This conclusion was further supported by measurements of MOG35–55-stimulated production of cytokines by splenocytes isolated from MOG35–55-immunized mice 10 days postimmunization. In cells from mice pretreated in vivo with lithium, the MOG35–55-induced productions of IFN-γ, IL-6, and IL-17 were much less than the amounts produced by splenocytes isolated from MOG35–55-immunized mice not treated with lithium, whereas anti-CD3-induced IFN-γ, IL-6, and IL-17 production was unaffected by lithium treatment (Fig. 4). IL-10 is a regulatory cytokine in inflammatory autoimmune diseases and its elevated expression is associated with amelioration of, or protection from, EAE (26–28). We therefore determined whether lithium treatment induced generation of IL-10-producing effector T cells. Restimulation of splenocytes isolated from lithium-treated or untreated MOG35–55-immunized mice with MOG peptide did not result in the production of detectable levels of IL-10 (data not shown). However, stimulation of splenocytes from MOG-immunized lithium-treated mice but not untreated mice with anti-CD3 resulted in IL-10 production (67.9 pg/ml). This result suggests that one mechanism of beneficial action of lithium in EAE is by the generation of IL-10-producing T cells, but this is limited to non-Ag (MOG35–55) specific T cells.
FIGURE 3.
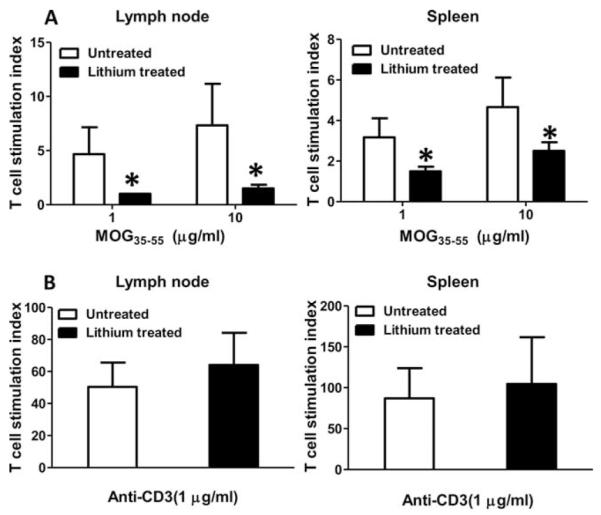
Lithium pretreatment reduces Ag-specific T cell proliferation. Mice were immunized with MOG35–55 peptide with or without lithium pretreatment and analyzed after 10 days. Mononuclear cells isolated from draining lymph nodes or spleens were stimulated with 0, 1, or 10 μg/ml MOG35–55 peptide (A), or 0 or 1 μg/ml anti-CD3 (B), and proliferation was measured. For each experiment, cells from two mice were pooled. Means ± SEM (n = 6); *, p < 0.05 compared with mice not pretreated with lithium.
FIGURE 4.

Lithium pretreatment reduces Ag-specific T cell production of cytokines. Mice were immunized with MOG35–55 peptide with or without lithium pretreatment and analyzed after 10 days. Isolated splenocytes were stimulated with 0, 1, or 10 μg/ml MOG35–55 peptide (A), or 0 or 1 μg/ml anti-CD3 (B), and the production of IFN-γ, IL-6, and IL-17 were measured. For each experiment, cells from two mice were pooled. Means ± SEM (n = 3); *, p < 0.05 compared with mice not pretreated with lithium.
Lithium administration does not affect NK cells, and reduces the number but not the activity of Treg cells
NK cells and Treg cells have a role in modulating disease activity in EAE (29–33). Therefore, lithium could alter the development and/or severity of EAE by altering the numbers and functions of NK or Treg cells. To address these possibilities, we evaluated the number and activity of NK and Treg cells in spleens of mice treated for 10 days with lithium compared with untreated mice. The results show that the number of NK1.1-expressing NK cells was similar in both lithium-treated and untreated mice (Fig. 5A). The NK cell activity within spleen mononuclear cells to YAC-1 targets was also similar in lithium-treated and untreated mice (data not shown). Remarkably, lithium treatment resulted in a significant reduction in the proportion of Treg cells compared with untreated mice (Fig. 5B). However, the ability of Treg cells from lithium-treated mice to suppress the proliferation of responder CD4+ T cells was comparable to that of untreated mice (Fig. 5C). Overall, these results demonstrate that lithium-mediated protection and/or amelioration of EAE is not due to changes in NK cell number or activity or enhanced Treg cell numbers or activity.
FIGURE 5.

Effects of lithium treatment of mice on NK cells and Treg cell. A, Lithium treatment did not alter the number of NK cells. The histogram depicts the percent of NK1.1 positive cells from each of lithium-untreated and treated mice. The scatter plot (lower panel) shows the number of NK1.1 cells per million spleen cells ± SD from lithium-untreated and treated mice (n = 5). B, Treg cells are reduced in spleens of lithium-treated mice. The dot plot shows the percentage of CD25+FoxP3+ T cells within a CD4+ gated population of cells from spleens of lithium-untreated and treated mice. The scatter plot shows the number of CD4-gated CD25+FoxP3+ T cells per million spleen cells from lithium-untreated and treated mice ± SD (n = 5). C, Lithium treatment did not alter Treg cell activity. Treg cells from lithium-treated and untreated mice were evaluated for their ability to suppress the anti-CD3-induced proliferation of CD4+CD25- responder T cells from C57BL/6 mice in a [3H]-thymidine incorporation assay. Data represent mean ± SEM (n = 3).
Increased severity of EAE in constitutively active GSK3 knock-in mice
GSK3α/β knock-in mice containing serine-to-alanine mutations in the regulatory serines of both GSK3 isoforms, S21A-GSK3α and S9A-GSK3β, and matched wild-type mice were immunized with MOG35–55 peptide to test whether constitutively active GSK3 promoted EAE. Wild-type mice developed symptoms of EAE similar to C57BL/6 mice (Fig. 6). The development of the acute phase of the disease was accelerated in the constitutively active GSK3 knock-in mice compared with wild-type mice. Furthermore, during the chronic phase, constitutively active GSK3 knock-in mice exhibited more severe disease compared with wild-type mice. Overall, severity of EAE was significantly different between the two groups of mice, as the CDI was 54.5 ± 4.6 for wild-type mice and 80.3 ± 28.9 for constitutively active GSK3 knock-in mice (p < 0.05). Incidence of disease was 6/6 in the wild-type and 5/6 in the GSK3 mutant knock-in mice. Thus, mice expressing constitutively active GSK3 exhibited more severe EAE than wild-type mice.
FIGURE 6.
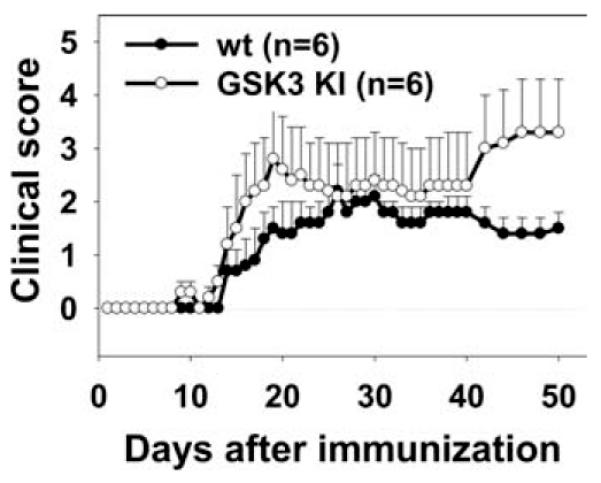
EAE is more severe in GSK3 knock-in mice. EAE was induced in constitutively active GSK3 knock-in mice and in matched wild-type controls with MOG35–55 peptide, and clinical symptoms were scored. Means ± SEM (n = 6). Disease severity was significantly (p < 0.05) increased in the GSK3 knock-in mice.
Lithium administration controls relapsing EAE
A major form of clinical multiple sclerosis is a relapsing/remitting disease, which is modeled in female SJL mice immunized with PLP (34). PLP139–151-immunized mice developed an acute episode of clinical EAE, followed by remission (Fig. 7). During the first remission, 20 days after immunization, half the mice were administered lithium. All mice displayed a secondary relapse episode, but in the lithium-treated mice the severity was approximately half that displayed by the lithium-free mice, which reached clinical scores equivalent to the first episode. Subsequently, the lithium-free mice displayed a third episode of clinical EAE, which stabilized in a chronic progressive phase with an average clinical score near 2. In contrast, the lithium-treated mice stabilized with mild symptoms, which remained below an average clinical score of 1, to 90 days postimmunization. To determine whether continuous lithium treatment was blocking an active disease process, lithium treatment was withdrawn on day 90. Remarkably, after a washout period of a few days, the mice that had been treated with lithium relapsed to reach a clinical score equivalent to the mice that had never received lithium. Restoration of lithium treatment on day 109 promoted recovery. Thus, chronic lithium treatment suppressed an ongoing disease process, which was reactivated upon withdrawal of lithium, demonstrating that lithium treatment is therapeutic in relapsing/remitting EAE.
FIGURE 7.
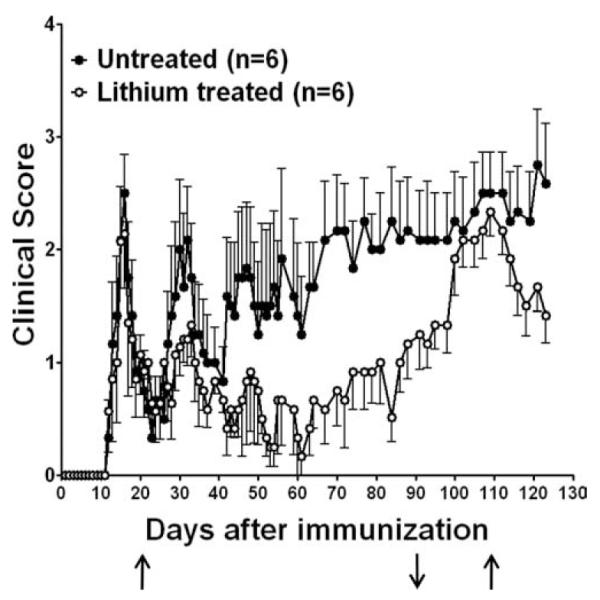
Lithium treatment ameliorates relapsing/remitting EAE. SJL female mice were immunized with PLP139–151 peptide and on day 20, during the first remission, half the mice were administered lithium (↑). On day 90, lithium was withdrawn (↓), and lithium treatment was resumed on day 109 (↑). Disease severity was significantly (p < 0.05) lower in the lithium-treated mice.
Discussion
EAE is a debilitating immune-mediated inflammatory and demyelinating disease of the CNS induced in rodents by the administration of CNS-derived Ags. EAE is widely used to model multiple sclerosis to identify physiological cascades that lead to clinical symptoms and to identify potential therapeutic targets. The results reported in this study show that the symptoms of EAE were significantly relieved in mice using four different lithium treatment protocols: pretreatment, treatment at the onset of EAE, treatment during severe disease, and treatment during remission. Especially notable is the effectiveness of lithium treatment in the relapsing/ remitting EAE paradigm where lithium administered after the first disease episode, during remission, provided long-term suppression of EAE. After nearly 3 mo of protection by lithium, relapse rapidly occurred after lithium was withdrawn, and recovery followed subsequent readministration of lithium. The ability to repress or induce clinical symptoms of EAE at any time after immunization by lithium administration or withdrawal, respectively, provides a unique and valuable model for assessing the disease process long after initial onset.
Attenuation of the clinical symptoms of EAE by lithium treatment was accompanied by reduced leukocyte infiltration into the spinal cord, reduced demyelination, and reduced microglial activation. Remarkably, the extent of demyelination in spinal cords of mice treated with lithium after onset of disease was less than in untreated mice. This could be due to lithium inhibiting demyelination, promoting remyelination, or a combination of both. Lithium could inhibit demyelination by suppressing microglia activation and inflammatory cytokine production. This is consistent with several studies highlighting microglia as a major mediator of neuronal damage in EAE and multiple sclerosis (35–39). Our data also suggest the intriguing possibility that lithium promotes remyelination. However, this is a speculative observation and needs to be examined further.
The broad effectiveness of lithium on characteristic signs of EAE indicated that it affects an early stage in the immunological cascade leading to EAE, and this was confirmed by the finding that lithium attenuated the generation of MOG35–55 peptide-responsive T cells. This block likely stems, in part, from the recent finding that GSK3, which is inhibited by lithium (9), is crucial for the differentiation and activation of proinflammatory dendritic cells (40). The Th17 lineage of CD4+T cells has recently been identified as the major effector T cell for EAE development (41–43), and IL-6 has a crucial role in inducing IL-21, which in cooperation with TGFβ is necessary for the generation of Th17 cells (44–46). Notably, in vivo lithium treatment significantly reduced the in vitro MOG35–55-stimulated production of both IL-6 and IL-17 by splenocytes isolated from MOG35–55-immunized mice, indicating that lithium reduced the development of MOG35–55-responsive Th17 cells, which would retard the development of EAE. However, lithium did not selectively only block the development of Th17 cells, as indicated by the finding that lithium treatment also greatly reduced the in vitro MOG35–55-induced production of IL-6 and of IFN-γ, which are produced by Th1 cells and a population of Th cells that coexpress both IL-17 and IFN-γ, cells that correlate closely with disease severity in EAE (23, 47, 48). Thus, it appears that by inhibiting GSK3, lithium suppresses the development and differentiation of Ag-responsive T cells, possibly at the level of dendritic cell activation. Consistent with this possibility, the ability of T cells obtained from lithium-treated mice to be activated and produce inflammatory cytokines following direct engagement of the Ag receptor with anti-CD3 was unaffected. We also found that lithium-mediated inhibition of GSK3 did not lead to increases in numbers or activity of Treg or NK cells. These findings extend to a therapeutically relevant lithium administration paradigm the previous report that high doses of lithium were capable of blocking EAE in rats (20), which preceded the discovery that lithium is a selective inhibitor of GSK3 (9). The importance of GSK3 is also highlighted by the finding that EAE was moderately but significantly promoted in GSK3 knock-in mice in which the GSK3 is constitutively active and unable to be inhibited by the phosphorylation of regulatory serines (11).
Lithium has also been shown to reduce inflammatory cytokine production by inhibiting GSK3-dependent activation of NF-κB transcriptional activity (12, 17, 49). Inhibitors of GSK3, in some cases including lithium, previously were reported to reduce LPS-induced production of inflammatory cytokines, such as IL-6, in monocytes and other cells (12), and to reduce disease severity in animal models of sepsis, arthritis, peritonitis, and colitis (12, 14, 16–19). This study demonstrates that lithium, likely by inhibiting GSK3, has profound actions in both the innate and adaptive immune systems and in signaling mechanisms controlling the production of inflammatory molecules.
Although these actions of GSK3 may at first seem surprising considering its original identification as a kinase regulating glycogen metabolism, research during the last 10 years has revealed that GSK3 regulates many cellular functions and signaling pathways, such as phosphorylating >20 transcription factors (10). Thus, GSK3 seems to be the most likely target mediating lithium’s therapeutic effects in EAE, but because lithium also has other targets (8), we cannot rule out the possibility that other actions of lithium may contribute to its effects on EAE. Nonetheless, because lithium was found to be highly effective in providing protection from EAE, and it has been used for >50 years in human patients with psychiatric diseases, taken together these findings suggest that lithium treatment and targeting GSK3 may be a rational strategy to diminish the effects of autoimmune diseases as well as of inflammatory diseases affecting the CNS.
Acknowledgments
We thank Dr. Huang-Ge Zhang for assisting us with the NK cell activity assay, Cecelia Latham, Dr. Simer Preet Singh, and Anna Zimjewska for excellent technical assistance, and the University of Alabama Neuroscience Core Facilities (NS47466, NS57098).
Footnotes
Disclosures: The authors have no financial conflict of interest.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This work was supported by grants from the National Multiple Sclerosis Society PP1335 (to P.D.), RG3891A1 (to C.R.), and National Institute of Health Grants MH38752 and NS37768 (to R.S.J.).
- EAE
- experimental autoimmune encephalomyelitis
- MOG
- myelin oligodendrocyte glycoprotein
- PLP
- proteolipid protein peptide
- GSK3
- glycogen synthase kinase-3
- CDI
- cumulative disease index
- Treg
- T regulatory
References
- 1.McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat. Immunol. 2007;8:913–919. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- 2.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 3.Hafler DA. Multiple sclerosis. J. Clin. Invest. 2004;113:788–794. doi: 10.1172/JCI21357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N. Engl. J. Med. 2000;343:938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 5.Steinman L, Zamvil SS. Virtues and pitfalls of EAE for the development of therapies for multiple sclerosis. Trends Immunol. 2005;26:565–571. doi: 10.1016/j.it.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 6.Bernard CC, Carnegie PR. Experimental autoimmune encephalomyelitis in mice: immunologic response to mouse spinal cord and myelin basic proteins. J. Immunol. 1975;114:1537–1540. [PubMed] [Google Scholar]
- 7.Kuchroo VK, Anderson AC, Waldner H, Munder M, Bettelli E, Nicholson LB. T cell response in experimental autoimmune encephalomyelitis (EAE): role of self and cross-reactive antigens in shaping, tuning, and regulating the autopathogenic T cell repertoire. Annu. Rev. Immunol. 2002;20:101–123. doi: 10.1146/annurev.immunol.20.081701.141316. [DOI] [PubMed] [Google Scholar]
- 8.Jope RS. Anti-bipolar therapy: mechanism of action of lithium. Mol. Psychiatry. 1999;4:117–128. doi: 10.1038/sj.mp.4000494. [DOI] [PubMed] [Google Scholar]
- 9.Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 11.McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hart DA, Done SJ, Benediktsson H, Lenz SP. Partial characterization of the enhanced survival of female NZB/W mice treated with lithium chloride. Int. J. Immunopharmacol. 1994;16:825–833. doi: 10.1016/0192-0561(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 14.Dugo L, Collin M, Allen DA, Patel NS, Bauer I, Mervaala EM, Louhelainen M, Foster SJ, Yaqoob MM, Thiemermann C. GSK-3β inhibitors attenuate the organ injury/dysfunction caused by endotoxemia in the rat. Crit. Care Med. 2005;33:1903–1912. doi: 10.1097/01.ccm.0000178350.21839.44. [DOI] [PubMed] [Google Scholar]
- 15.Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 2006;79:173–189. doi: 10.1016/j.pneurobio.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whittle BJ, Varga C, Posa A, Molnar A, Collin M, Thiemermann C. Reduction of experimental colitis in the rat by inhibitors of glycogen synthase kinase-3β. Br. J. Pharmacol. 2006;147:575–582. doi: 10.1038/sj.bjp.0706509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT, Woodgett JR, Ivashkiv LB. IFN-α suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–574. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 18.Cuzzocrea S, Mazzon E, Di Paola R, Muia C, Crisafulli C, Dugo L, Collin M, Britti D, Caputi AP, Thiemermann C. Glycogen synthase kinase-3β inhibition attenuates the degree of arthritis caused by type II collagen in the mouse. Clin. Immunol. 2006;120:57–67. doi: 10.1016/j.clim.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 19.Cuzzocrea S, Genovese T, Mazzon E, Crisafulli C, Di Paola R, Muia C, Collin M, Esposito E, Bramanti P, Thiemermann C. Glycogen synthase kinase-3β inhibition reduces secondary damage in experimental spinal cord trauma. J. Pharmacol. Exp. Ther. 2006;318:79–89. doi: 10.1124/jpet.106.102863. [DOI] [PubMed] [Google Scholar]
- 20.Levine S, Saltzman A. Inhibition of experimental allergic encephalomyelitis by lithium chloride: specific effect or nonspecific stress? Immunopharmacology. 1991;22:207–213. doi: 10.1016/0162-3109(91)90045-z. [DOI] [PubMed] [Google Scholar]
- 21.De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3β phosphorylation by sodium valproate and lithium. Neuro-pharmacology. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- 22.Axtell RC, Webb MS, Barnum SR, Raman C. Cutting edge: critical role for CD5 in experimental autoimmune encephalomyelitis: inhibition of engagement reverses disease in mice. J. Immunol. 2004;173:2928–2932. doi: 10.4049/jimmunol.173.5.2928. [DOI] [PubMed] [Google Scholar]
- 23.Axtell RC, Xu L, Barnum SR, Raman C. CD5-CK2 binding/ activation-deficient mice are resistant to experimental autoimmune encephalomyelitis: protection is associated with diminished populations of IL-17-expressing T cells in the central nervous system. J. Immunol. 2006;177:8542–8549. doi: 10.4049/jimmunol.177.12.8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu C, Yu S, Kappes J, Wang J, Grizzle WE, Zinn KR, Zhang HG. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood. 2007;109:4336–4342. doi: 10.1182/blood-2006-09-046201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reddy J, Illes Z, Zhang X, Encinas J, Pyrdol J, Nicholson L, Sobel RA, Wucherpfennig KW, Kuchroo VK. Myelin proteolipid protein-specific CD4+CD25+ regulatory cells mediate genetic resistance to experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA. 2004;101:15434–15439. doi: 10.1073/pnas.0404444101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bettelli E, Das MP, Howard ED, Weiner HL, Sobel RA, Kuchroo VK. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J. Immunol. 1998;161:3299–3306. [PubMed] [Google Scholar]
- 27.Kennedy MK, Torrance DS, Picha KS, Mohler KM. Analysis of cytokine mRNA expression in the central nervous system of mice with experimental autoimmune encephalomyelitis reveals that IL-10 mRNA expression correlates with recovery. J. Immunol. 1992;149:2496–2505. [PubMed] [Google Scholar]
- 28.Segal BM, Dwyer BK, Shevach EM. An interleukin (IL)-10/ IL-12 immunoregulatory circuit controls susceptibility to autoimmune disease. J. Exp. Med. 1998;187:537–546. doi: 10.1084/jem.187.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu W, Fazekas G, Hara H, Tabira T. Mechanism of natural killer (NK) cell regulatory role in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005;163:24–30. doi: 10.1016/j.jneuroim.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 30.Vollmer TL, Liu R, Price M, Rhodes S, La Cava A, Shi FD. Differential effects of IL-21 during initiation and progression of autoimmunity against neuroantigen. J. Immunol. 2005;174:2696–2701. doi: 10.4049/jimmunol.174.5.2696. [DOI] [PubMed] [Google Scholar]
- 31.Reddy J, Waldner H, Zhang X, Illes Z, Wucherpfennig KW, Sobel RA, Kuchroo VK. Cutting edge: CD4+CD25+ regulatory T cells contribute to gender differences in susceptibility to experimental autoimmune encephalomyelitis. J. Immunol. 2005;175:5591–5595. doi: 10.4049/jimmunol.175.9.5591. [DOI] [PubMed] [Google Scholar]
- 32.Jahng AW, Maricic I, Pedersen B, Burdin N, Naidenko O, Kronenberg M, Koezuka Y, Kumar V. Activation of natural killer T cells potentiates or prevents experimental autoimmune encephalomyelitis. J. Exp. Med. 2001;194:1789–1799. doi: 10.1084/jem.194.12.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar V, Stellrecht K, Sercarz E. Inactivation of T cell receptor peptide-specific CD4 regulatory T cells induces chronic experimental autoimmune encephalomyelitis (EAE) J. Exp. Med. 1996;184:1609–1617. doi: 10.1084/jem.184.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tuohy VK, Lu Z, Sobel RA, Laursen RA, Lees MB. Identification of an encephalitogenic determinant of myelin proteolipid protein for SJL mice. J. Immunol. 1989;142:1523–1527. [PubMed] [Google Scholar]
- 35.Ponomarev ED, Shriver LP, Dittel BN. CD40 expression by microglial cells is required for their completion of a two-step activation process during central nervous system autoimmune inflammation. J. Immunol. 2006;176:1402–1410. doi: 10.4049/jimmunol.176.3.1402. [DOI] [PubMed] [Google Scholar]
- 36.Crocker SJ, Whitmire JK, Frausto RF, Chertboonmuang P, Soloway PD, Whitton JL, Campbell IL. Persistent macrophage/microglial activation and myelin disruption after experimental autoimmune encephalomyelitis in tissue inhibitor of metalloproteinase-1-deficient mice. Am. J. Pathol. 2006;169:2104–2116. doi: 10.2353/ajpath.2006.060626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Butovsky O, Landa G, Kunis G, Ziv Y, Avidan H, Greenberg N, Schwartz A, Smirnov I, Pollack A, Jung S, Schwartz M. Induction and blockage of oligodendrogenesis by differently activated microglia in an animal model of multiple sclerosis. J. Clin. Invest. 2006;116:905–915. doi: 10.1172/JCI26836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N, Waisman A, Rulicke T, Prinz M, Priller J, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat. Med. 2005;11:146–152. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- 39.Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J. Mol. Med. 1997;75:165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- 40.Rodionova E, Conzelmann M, Maraskovsky E, Hess M, Kirsch M, Giese T, Ho AD, Zoller M, Dreger P, Luft T. GSK-3 mediates differentiation and activation of proinflammatory dendritic cells. Blood. 2007;109:1584–1592. doi: 10.1182/blood-2006-06-028951. [DOI] [PubMed] [Google Scholar]
- 41.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu. Rev. Immunol. 2007 doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 42.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 44.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 45.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 46.Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suryani S, Sutton I. An interferon-α-producing Th1 subset is the major source of IL-17 in experimental autoimmune encephalitis. J. Neuroimmunol. 2007;183:96–103. doi: 10.1016/j.jneuroim.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 48.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat. Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 49.Steinbrecher KA, Wilson W, III, Cogswell PC, Baldwin AS. Glycogen synthase kinase 3β functions to specify gene-specific, NF-κB-dependent transcription. Mol. Cell. Biol. 2005;25:8444–8455. doi: 10.1128/MCB.25.19.8444-8455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]