

Abstract
The active form of vitamin D, 1,25-dihydroxyvitamin D (1,25(OH)2D), is a potent inducer of the antimicrobial protein cathelicidin, CAMP (LL37). In macrophages this response is dependent on intracrine synthesis of 1,25(OH)2D from precursor 25-hydroxyvitamin D (25OHD), catalyzed by the enzyme 25-hydroxyvitamin D-1alpha-hydroxylase (CYP27B1). In view of the fact that trophoblastic cells also express abundant CYP27B1, we postulated a similar intracrine pathway for induction of CAMP in the placenta. Analysis of placenta explants, primary cultures of human trophoblast, and the 3A trophoblastic cell line treated with 1,25(OH)2D (1–100 nM) revealed dose-dependent induction of CAMP similar to that observed with primary cultures of human macrophages. Also consistent with macrophages, induction of trophoblastic CAMP was enhanced via intracrine conversion of 25OHD to 1,25(OH)2D. However, in contrast to macrophages, induction of CAMP by vitamin D in trophoblasts was not enhanced by costimulation with Toll-like receptor ligands, such as lipopolysaccharide. Despite this, exposure to vitamin D metabolites significantly enhanced antibacterial responses in trophoblastic cells: 3A cells infected with Escherichia coli showed decreased numbers of bacterial colony-forming units compared with vehicle-treated controls when treated with 25OHD (49.6% ± 10.9%) or 1,25(OH)2D (45.4% ± 9.2%), both P < 0.001. Treatment with 25OHD (1–100 nM) or 1,25(OH)2D (0.1–10 nM) also protected 3A cells against cell death following infection with E. coli (13.6%–26.9% and 22.3%–40.2% protection, respectively). These observations indicate that 1,25(OH)2D can function as an intracrine regulator of CAMP in trophoblasts, and may thus provide a novel mechanism for activation of innate immune responses in the placenta.
Keywords: immunology, placenta, steroid hormones, trophoblast, vitamin D
Synthesis of the active form of vitamin D by trophoblastic cells acts to enhance innate immune responses in the placenta by increasing bacterial killing and decreasing trophoblast cell death.
INTRODUCTION
The active form of vitamin D, 1,25-dihydroxyvitamin D (1,25(OH)2D), is a potent modulator of human immune responses [1–3]. In particular, recent studies have shown that 1,25(OH)2D promotes innate immunity by stimulating synthesis of the antimicrobial protein, cathelicidin (CAMP) [1–5]. This response is due to direct transcriptional regulation of the gene for CAMP [6, 7], and 1,25(OH)2D-induced CAMP expression has been demonstrated in a variety of cell types expressing the intracellular vitamin D receptor (VDR) [7–10]. Although this mechanism appears to play a central role in the combating infection by Mycobacterium tuberculosis [5, 8, 11], CAMP is known to be an effective inhibitor of other pathogens such as Staphylococcus aureus [12–14], as well as exhibiting antiviral properties [15–17]. In a similar fashion, although vitamin D-induced CAMP-mediated bacterial killing is a pivotal feature of macrophages [8, 11], an equivalent mechanism has also been described for keratinocytes following epidermal injury [9]. Thus, CAMP expression has been linked not only to tuberculosis, but other diseases, such as sepsis [18], inflammatory bowel disease [19], and inflammatory lung disease [20]. Collectively, these observations support the hypothesis that a key nonclassical function of 1,25(OH)2D is to enhance innate immunity through localized production of CAMP.
As indicated above, the ability of vitamin D to promote antibacterial activity is dependent on the expression of VDR by cells such as macrophages. The availability of ligand for the VDR may be due to endocrine production of 1,25(OH)2D in the kidneys, catalyzed by the enzyme 25-hydroxyvitamin D-1α-hydroxylase (CYP27B1), which is expressed in the proximal tubules [21, 22]. However, current data suggest that innate immune responses to vitamin D are more likely to involve an intracrine mechanism, with cells such as macrophages acting as an extrarenal site for expression of CYP27B1 and synthesis of 1,25(OH)2D [23, 24]. Consistent with this, several studies have demonstrated induction of CAMP following treatment with the inactive precursor metabolite, 25-hydroxyvitamin D (25OHD), as well as active 1,25(OH)2D [8, 9, 25].
Another key extrarenal site for expression of CYP27B1 is the placenta, with both fetal trophoblast and maternal decidua demonstrating synthesis of 1,25(OH)2D [26, 27]. Expression of placental CYP27B1 is induced early in pregnancy [28, 29], and we have postulated that a key function of the enzyme in this tissue is to support both innate and adaptive immunity in utero [29]. In previous studies, we have described intracrine induction of CAMP by 25OHD in maternal decidual cells from first-trimester human placentas [25]. Using the data presented here, we have explored this function of vitamin D in trophoblasts, a cell type closely linked to innate immune responses within the placenta [30]. Consistent with previous studies of decidual cells, trophoblasts potently expressed CAMP in response to treatment with either 25OHD or 1,25(OH)2D, confirming an intracrine mode of action. This innate immune response was similar to that observed with macrophages. In common with macrophages, the induction of CAMP by 1,25(OH)2D enhanced antibacterial responses in trophoblasts, and also abrogated the cell death that occurs following infection of these cells. However, in contrast to macrophages in which the vitamin D system is activated via Toll-like receptors (TLR), trophoblast responses to vitamin D metabolites appears to be due to constitutive expression of VDR and CYP27B1. Collectively, these data suggest that intracrine induction of placental innate immunity may be an important function for vitamin D during pregnancy.
MATERIALS AND METHODS
Cell Lines and Reagents
Human trophoblastic cell lines, 3A and JEG3, were obtained from American Type Tissue Culture Collection. Cells were cultured as adherent monolayers in modified Eagle medium (MEM; Invitrogen Life Technologies, Carlsbad, CA) supplemented with 10% fetal calf serum (FCS), 10 mM Hepes, 1 mM sodium pyruvate, and 100 nM penicillin/streptomycin (Invitrogen Life Technologies). Macrophages, 3A trophoblastic cells, JEG-3 cells, placenta explants, and primary trophoblasts were treated with 1,25(OH)2D (1–100 nM), 25OHD (1–100 nM), or vehicle (0.1% ethanol) for 24–48 h. The 3A trophoblastic cells were treated with reagents from a TLR ligand kit (Invivogen, San Diego, CA). These reagents included: Pam3CSK4, TLR1/2 agonist; HKLM, TLR2 agonist; Poly(I:C), TLR3 agonist; lipopolysaccharide (LPS) (Escherichia coli K12), TLR4 agonist; flagellin Salmonella typhimurium, TLR5 agonist; FSL1, TLR6/2 agonist; imiquimod, TLR7 agonist; ssRNA40/LyoVec, TLR8 agonist; ODN2006, TLR9 agonist. All TLR agonists were added to culture medium at the manufacturer's recommended dose (Invivogen).
Isolation and Culture of Placental Tissue and Primary Trophoblasts
Tissue was isolated from third-trimester placentas from healthy, full-term pregnant women undergoing elective Caesarian section. All samples were obtained in accordance with Cedars-Sinai Medical Center institutional review board protocols. Unpurified placental tissue was separated from membranes, minced using forceps and scalpel, and then washed in Hank Balanced Salt Solution (HBSS; Invitrogen Life Technologies) and centrifuged three times to remove excess blood and cell debris. The resulting placental explants were then resuspended in Dulbecco MEM (DMEM; Invitrogen Life Technologies) supplemented with 10% normal human serum (Gemini Bio-Products, West Sacramento, CA). Trophoblastic cells were isolated as described previously [31]. Briefly, placental tissue specimens were washed with cold HBSS to remove excess blood. Cells were scraped from the membranes, transferred to trypsin-EDTA (Invitrogen Life Technologies) digestion buffer and incubated at 37°C for 10 min with shaking. An equal volume of DMEM containing 10% FCS was added to inactivate the trypsin. This mixture was vortexed for 20 sec and allowed to sediment, and the supernatant was collected. This was repeated twice, and the collected supernatant was centrifuged at 1500 rpm for 10 min. Contaminating red blood cells were removed by resuspending the cellular pellet with HBSS, layering this over the same volume of Lymphocyte Separation Media (ICN Biomedicals, Irvine, CA), and centrifuging at 2000 rpm for 25 min. The cellular interface containing the trophoblast cells was collected and resuspended in DMEM supplemented with 10% normal human serum and cultured at 37°C/5% CO2 for three passages. Purity of the trophoblast cells was >98%, as determined by immunostaining for cytokeratin-7 (Sigma-Aldrich, St. Louis, MO) [31].
Isolation and Culture of Human Macrophages
Peripheral blood mononuclear cells were isolated from buffy coat preparations of human blood obtained commercially from the Virology Core Unit at the University of California, Los Angeles. Monocytic cells were adhered to 24-well culture plates in serum-free RPMI 1640 (RPMI) culture medium for 2 h, after which nonadherent cells were removed and the remaining adherent monocytes washed with further serum-free RPMI. The monocytes were then cultured for 5 days in RPMI supplemented with 10% FCS and granulocyte-macrophage colony-stimulating factor (Immunex, Seattle, WA). The resulting cells showed increased expression of macrophage markers, such as CD14 [23].
Extraction of RNA and Reverse Transcription
RNA was extracted from mouse tissues using the RNeasy total RNA extraction kit, as detailed by the manufacturer (Qiagen, Valencia, CA). RNA was eluted in RNase-free elution solution, and 1.5 μg aliquots were reverse transcribed using Powerscript MMLV reverse transcriptase (ABI, Foster City, CA), as described by the manufacturer.
Quantitative Real-Time RT-PCR Amplification of cDNAs
Expression of mRNAs for VDR, CYP27B1, 24-hydroxylase (CYP24), CAMP, β-defensin-4 (DEFB4) and other specified genes was quantified using an ABI 7700 sequence detection system (ABI), as described previously [32]. Approximately 50 ng of cDNA was used per reaction. All reactions were multiplexed with the housekeeping 18S rRNA gene (Assays-on-Demand [ABI] primer and probe mix Hs99999901_s1), enabling data to be expressed in relation to an internal reference to allow for differences in sampling. Data were obtained as Ct values (the cycle number at which logarithmic PCR plots cross a calculated threshold line), and used to determine ΔCt values (Ct of target gene − Ct of housekeeping 18S rRNA gene). PCR amplification of target gene cDNA was carried out using the following Taqman human gene expression assays: CYP27B1, forward primer 5′-TTGGCAAGCGCAGCTGTAT-3′, reverse primer 5′-TGTGTTAGGATCTGGGCCAAA-3′, TaqMan probe 5′-TTGCAATTCAAGCTCTGCCAGGCG-3′; VDR, forward primer 5′-CTTCAGGCGAAGCATGAAGC-3′, reverse primer 5′-CCTTCATCATGCCGATGTCC-3′, Taqman probe 5′-AAGGCACTATTCACCTGCCCCTTCAA-3′; CYP24A1, forward primer, 5′-CAAACCGTGGAAGGCCTATC-3′, reverse primer 5′-AGTCTTCCCCTTCCAGGATCA-3′, TaqMan probe 5′-ACTACCGCAAAGAAGGCTACGGGCTG-3′; CD14, ABI primer and probe mix Hs00169122_g1; TLR2, Hs00610101_m1; TLR4, Hs00152939_m1; CAMP, Hs00189038_m1; inducible nitric oxide synthase (NOS2) Hs00167257_m1. All cDNAs were amplified under the following conditions: 50°C for 2 min; 95°C for 10 min followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. All reactions were performed in triplicate and initially expressed as mean (±SD) ΔCt values, which were employed in statistical comparisons. Visual representation of data was carried out by converting ΔCt values to fold change data relative to ΔCt values for control (vehicle-treated) cells using the equation 2−ΔΔCt.
Measurement of 1,25(OH)2D Synthesis by Macrophages and Trophoblasts
Activity of CYP27B1 in 3A trophoblasts and macrophages was assessed by quantifying the conversion of 3[H]-25OHD (180 Ci/mmol; Amersham Biosciences, Piscataway, NJ) to 3[H]-1,25(OH)2D. For each assay, 10 nM 3[H]-25OHD suspended in 0.2 ml serum-free RPMI was added to 3A cells or macrophages and incubated at 37°C for 5 h, with the reaction being terminated by freezing at −20°C. Protein from these samples was initially precipitated with added acetonitrile (1:1). Vitamin D metabolites were then extracted from the reaction mixtures by elution on C18-OH columns according to manufacturer's instructions (Diasorin, Stillwater, MN). The resulting eluent was resuspended in 25 μl of elution solvent hexane:methanol:isopropanol (90:5:5), vortexed for 15 sec, and individual metabolites separated by HPLC using a Beckman Gold system with an Agilent Technologies Zobax Sil normal phase column (Agilent Technologies, Palo Alta, CA) eluted at a rate of 1.5 ml/min for 20 min. Elution profiles for standard vitamin D metabolites (25OHD, 24,25(OH)2D, 1,25(OH)2D) were determined by ultraviolet absorbance at 264 nm. Elution of metabolites of 3[H]-25OHD was assessed using a β-Ram Model 4 in-flow detector (IN/US, Tampa, FL) in conjunction with Ultima-Flo M scintillation fluid (Perkin-Elmer, Boston, MA) at a 2:1 ratio with a 5-sec dwell time to designate the increments for data collection. Lauralite 3 software (LabLogic, Sheffield, UK) was used to quantitate peaks of radioactivity corresponding to 25OHD, 24,25(OH)2D, or 1,25(OH)2D. Data were reported as mean fmoles metabolite synthesized/h/106 cells ± SD following three separate experiments.
Analysis of 3A Trophoblastic Cell Infection with E. coli
The 3A cells were seeded at 3 × 104 cells/well in a 24-well plate. Cells were cultured with MEM plus 10% FBS at 37°C, 5% CO2, for 72 h, and then treated with either 10 nM 1,25(OH)2D or 100 nM 25OHD for 48 h. E. coli were cultured in 5 ml LB broth overnight with shaking at 250 rpm and 37°C. From these cultures, a 100-μl aliquot of bacteria was added to 10 ml fresh LB medium, and culture continued at 37°C with shaking at 250 rpm for approximately 2–3 h until the optical density (OD) at 600 nm reached 0.4. The resulting bacteria were then centrifuged at 3000 rpm for 10 min, and the pellet washed twice with PBS and then resuspended in MEM plus 10% FBS. From this suspension, E. coli was added into each well of cultured 3A cells at a dose of 50 bacteria/cell in a total volume of 200 μl/well of 3A cells. The cell culture plates were then centrifuged at 500 rpm for 5 min and then incubated at 37°C with 5% CO2 for 30 min. Extracellular E. coli was then removed by aspiration and the 3A cells washed once with PBS. Any residual extracellular bacteria were then killed by the addition of 5 μg/ml gentamicin in MEM plus 10% FBS. At 24 h after the addition of gentamicin, the antibiotic was removed by aspiration and cells were washed three times with PBS. The 3A cells were then lysed by the addition of 1 ml distilled water to each well. The resulting lysates were then removed from each well, transferred to an Eppendorf tube, and then vortexed to ensure complete disruption of the cells. Serial dilutions of each lysate tube were added to LB plates and incubated at 37°C overnight, after which the resulting colonies were counted.
Analysis of 3A Trophoblastic Cell Death Following Infection with E. coli
Assays for E. coli infection were performed essentially as described previously [33]. Briefly, E. coli (strain DH5α; Invitrogen) were grown in TH Broth to mid-log phase (∼108 colony-forming units [cfu]/ml; OD600 = 0.4), washed in PBS, resuspended in RPMI with 10% FBS, and used to infect the 3A trophoblastic cells. The resulting cell cultures were centrifuged at 700 × g for 5 min to settle the bacteria on the monolayer surface, then incubated at 37°C in 5% CO2 for 1 h. To kill extracellular bacteria, cells were washed three times with sterile PBS and incubated with media containing gentamicin (100 μg/ml) and penicillin (5 μg/ml). The concentration and purity of each E. coli inoculum was confirmed by quantitative culture on THB agar plates. The 3A cell death was assessed by measuring cell culture supernatant lactate dehydrogenase (LDH) levels by colorimetric LDH assay (Roche Diagnostics), according to the manufacturer's instructions. Data were reported as the LDH released upon Triton X-100 treatment of cells relative to control infected cells.
Statistical Analyses
Data were expressed as mean ± SD unless otherwise stated. Statistical analysis of data was carried out using a Student t-test applied to raw data, including ΔCt values from real-time RT-PCR assays. All statistical analyses were carried out using SigmaStat 3.1 software (Systat Inc., San Jose, CA).
RESULTS
1,25(OH)2D Induces Expression of CAMP in Human Trophoblasts
To determine whether vitamin D stimulates innate immune response in placental cells, we assessed the effects of 1,25(OH)2D on expression of CAMP in various in vitro trophoblastic cell models relative to human macrophages. Results in Figure 1 showed a dose-dependent induction of CAMP in placental explant tissue cultures, primary human trophoblasts, and 3A transformed human trophoblasts in response to 24-h treatment with 1,25(OH)2D (1–100 nM). The sensitivity and magnitude of this response was similar to that observed with primary cultures of human macrophages, the principal target cell for 1,25(OH)2D induction of CAMP. Similar studies were also carried using the choriocarcinoma trophoblastic cell line, JEG-3, but this showed no response to treatment with 1,25(OH)2D (data not shown).
FIG. 1.

Induction of CAMP in macrophages, placenta, and trophoblastic cells by 1,25(OH)2D. Varying doses of 1,25(OH)2D (1–100 nM) were used to treat primary cultures of human peripheral blood-derived macrophages (macrophages), placental tissue explants from third-trimester human pregnancy (placenta), primary trophoblasts isolated from third-trimester human placenta (trophoblast), and 3A human trophoblastic cells (3A cells). Each set of tissue/cell cultures was incubated with 1,25(OH)2D for 24 h, after which total RNA was isolated. Subsequent real-time RT-PCR was used to assess the expression of mRNA for the antibacterial protein CAMP. Data are shown as the mean (± SD) fold induction of CAMP mRNA relative to vehicle (0.1% ethanol)-treated controls (C) for n = 3 tissue/cell cultures. *Statistically different from vehicle samples, P < 0.05; ***statistically different from vehicle samples, P < 0.001.
Expression and Activity of CY27B1 in Human Trophoblasts
Further RT-PCR analyses indicated that vitamin D-activating enzyme, CYP2B1, was expressed by human trophoblasts and by the 3A trophoblastic cell line (Fig. 2). Levels of mRNA for CYP27B1 were relatively low in the trophoblastic cultures compared with human macrophages. However, measurement of actual CYP27B1 activity (conversion of 25OHD to 1,25(OH)2D utilizing radiolabeled 25OHD as substrate) revealed comparable levels of 1,25(OH)2D synthesis in 3A trophoblasts (13.2 ± 3.6 fmoles/h/106 cells) relative to macrophages (16.7 ± 5.1 fmoles/h/106 cells). Furthermore, trophoblasts and 3A cells showed levels of mRNA for the VDR that were two- to three-times higher than macrophages, underlining the potential for autocrine/intracrine response to endogenously synthesized 1,25(OH)2D in these cells (Fig. 2). The trophoblastic cells and macrophages showed similar levels of mRNA for the feedback control enzyme, CYP24, which is potently induced in response to exogenous or endogenous 1,25(OH)2D. However, enzyme activity analyses showed enhanced production of the CYP24 metabolite 24,25(OH)2D in 3A trophoblasts (19.0 ± 4.6 fmoles/h/106 cells) compared with macrophages (4.3 ± 2.7 fmoles/h/106 cells).
FIG. 2.

Trophoblastic cells express VDRs and vitamin D metabolic enzymes. Expression of mRNAs for the VDR, 25-hydroxyvitamin D-1α-hydroxylase (CYP27B1), and 24-hydroxylase (CYP24) by human macrophages (MΦ), primary human trophoblasts (tropho.), and 3A trophoblastic cells (3A). Data are shown as the mean (± SD) fold induction of mRNA relative to vehicle (0.1% ethanol)-treated controls for n = 3 tissue/cell cultures.
Intracrine Vitamin D-Induction of Innate Immune Responses in 3A Trophoblasts Is Specific for CAMP
Having demonstrated expression and activity of CYP27B1 in trophoblastic cells, we then sought to determine whether this was sufficient to induce expression of CAMP in an intracrine fashion. Results in Figure 3 show that incubation of 3A cells with 10 or 100 nM 25OHD for 24 h stimulated expression of CAMP to a similar degree as 1 and 10 nM 1,25(OH)2D. Furthermore, this response was specific to CAMP, as neither 25OHD nor 1,25(OH)2D affected expression of other putative antibacterial gene products, such as DEFB4 or NOS2.
FIG. 3.
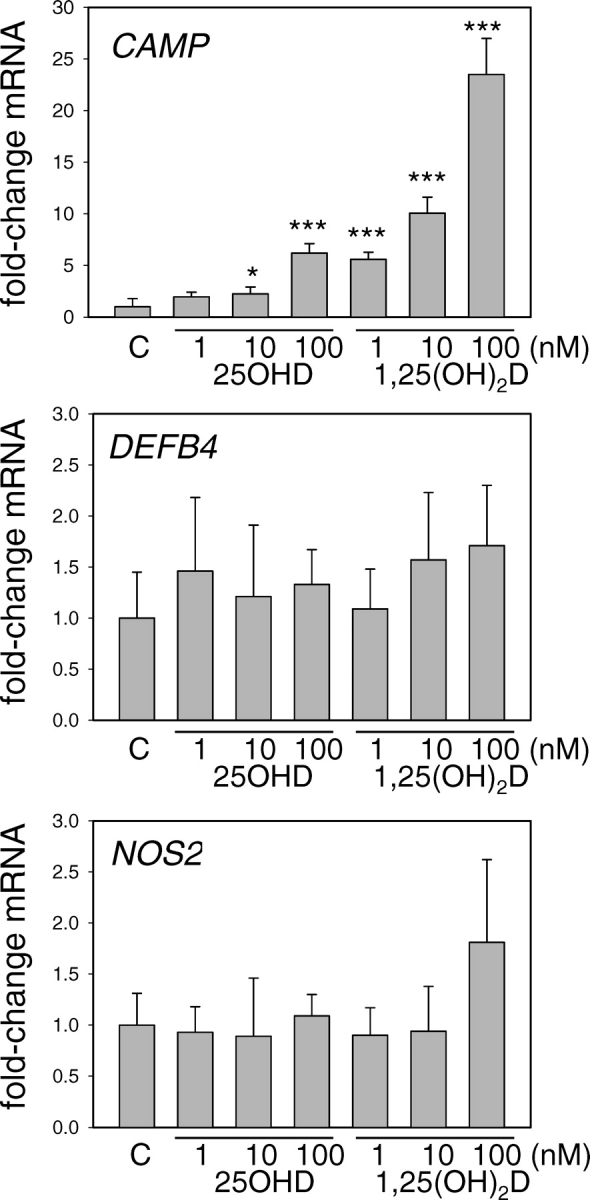
Regulation of antibacterial gene expression by vitamin D metabolites in 3A trophoblastic cells. Expression of mRNAs for CAMP, DEFB4, and NOS2 was analyzed in 3A trophoblastic cells treated for 24 h with either 25OHD or 1,25(OH)2D (1–100 nM for both treatments). Data are shown as the mean (± SD) fold induction of CAMP mRNA relative to vehicle (0.1% ethanol)-treated controls (C) for n = 3 tissue/cell cultures. *Statistically different from vehicle samples, P < 0.05; ***statistically different from vehicle samples, P < 0.001.
Intracrine Induction of Trophoblast CAMP by Vitamin D Is Independent of TLR Responses
Results in Figure 4 show that the TLR4 ligand LPS had no effect on CAMP induction by either 25OHD or 1,25(OH)2D by either cell type. Interestingly, in primary trophoblasts, LPS added alone suppressed expression of CAMP, but this effect was abrogated by coincubation with 25OHD. Results in Figure 5 show that, in macrophages, LPS induced expression of CYP27B1, VDR, TLR2, or TLR4, and this was unaffected by cotreatment with 25OHD. By contrast, in 3A cells, LPS had no effect on expression of CYP27B1, VDR, TLR2, or TLR4, despite the fact that TLR4 was detectable in 3A cells. Further studies using a panel of ligands to TLR1–TLR9 also showed no effect on VDR or CYP27B1 expression in 3A cells (see Supplemental Figure 1, available at www.biolreprod.org). In primary trophoblast cultures, LPS stimulated expression of TLR2 in a similar fashion to that observed with macrophages, but paradoxically suppressed levels of mRNA for TLR4. In both cases, these changes were unaffected by cotreatment with 25OHD. In addition, unlike macrophages, treatment of primary trophoblastic cells with either 25OHD or 1,25(OH)2D acted to suppress expression of CYP27B1.
FIG. 4.

Effect of TLR activation on 25OHD or 1,25(OH)2D-induced regulation of CAMP in 3A trophoblastic cells and primary (1°) cultures of human trophoblasts. Cells were treated for 24 h with TLR4 LPS, with or without 100 nM 25OHD (25) or 10 nM 1,25(OH)2D (1,25). The resulting cells were then used to assess expression of mRNA for CAMP. Data are shown as the mean (± SD) fold induction of CAMP mRNA relative to vehicle (0.1% ethanol)-treated controls (C) for n = 3 tissue/cell cultures. **Statistically different from vehicle samples, P < 0.01; ***Statistically different from vehicle samples, P < 0.001; ###statistically different from LPS-only-treated samples, P < 0.001; nd, not done.
FIG. 5.

Effect of TLR activation on 25OHD or 1,25(OH)2D-induced regulation of CYP27B1, VDR, TLR2, and TLR4 in human macrophages, primary (1°) cultures of human trophoblasts, and 3A trophoblastic cells. Cells were treated for 24 h with TLR4 ligand lipopolysaccharide (L), with or without 100 nM 25OHD (25) or 10 nM 1,25(OH)2D (1,25). The resulting cells were then used to assess expression of mRNA for CYP27B1, VDR, TLR2, and TLR4. Data are shown as the mean (± SD) fold induction of mRNA relative to vehicle (0.1% ethanol)-treated controls (C) for n = 3 cell preparations. *Statistically different from vehicle samples, P < 0.05; ***statistically different from vehicle samples, P < 0.001.
Vitamin D-Induced Expression of CAMP in 3A Trophoblasts Suppresses Bacterial Infection and Counteracts Infection-Associated Cell Death
To assess the functional significance of vitamin D-induced expression of CAMP, further studies were carried out using 3A trophoblastic cells infected with E. coli. Analysis of E. coli cfu in 3A cells showed that pretreatment with 25OHD (100 nM) or 1,25(OH)2D (10 nM) significantly suppressed levels of bacterial infection (49.6% ± 10.9% and 45.4% ± 9.2% relative to vehicle-treated controls, both P < 0.001) (Fig. 6A). We have shown previously that infection of trophoblasts with group B streptococcus (GBS) leads to the induction of cell death [33]. Therefore, we carried out similar studies using 3A cells treated with vehicle, 25OHD (1–100 nM), or 1,25(OH)2D (0.1–10 nM). Results in Figure 6B showed that infection of 3A cells with E. coli stimulated the release of LDH, a marker of cell death [33]. However, 3A cells pretreated with 25OHD or 1,25(OH)2D for 48 h showed significantly lower levels of LDH, indicating that these cells were protected against E. coli-induced cell death. Similar studies were also carried out using 3A cells infected with GBS. In this case, 25OHD (100 nM, 24 h) and 1,25(OH)2D (10 nM, 24 h) suppressed LDH release by 12.5% ± 3.1% and 8.3% ± 3.1%, respectively.
FIG. 6.
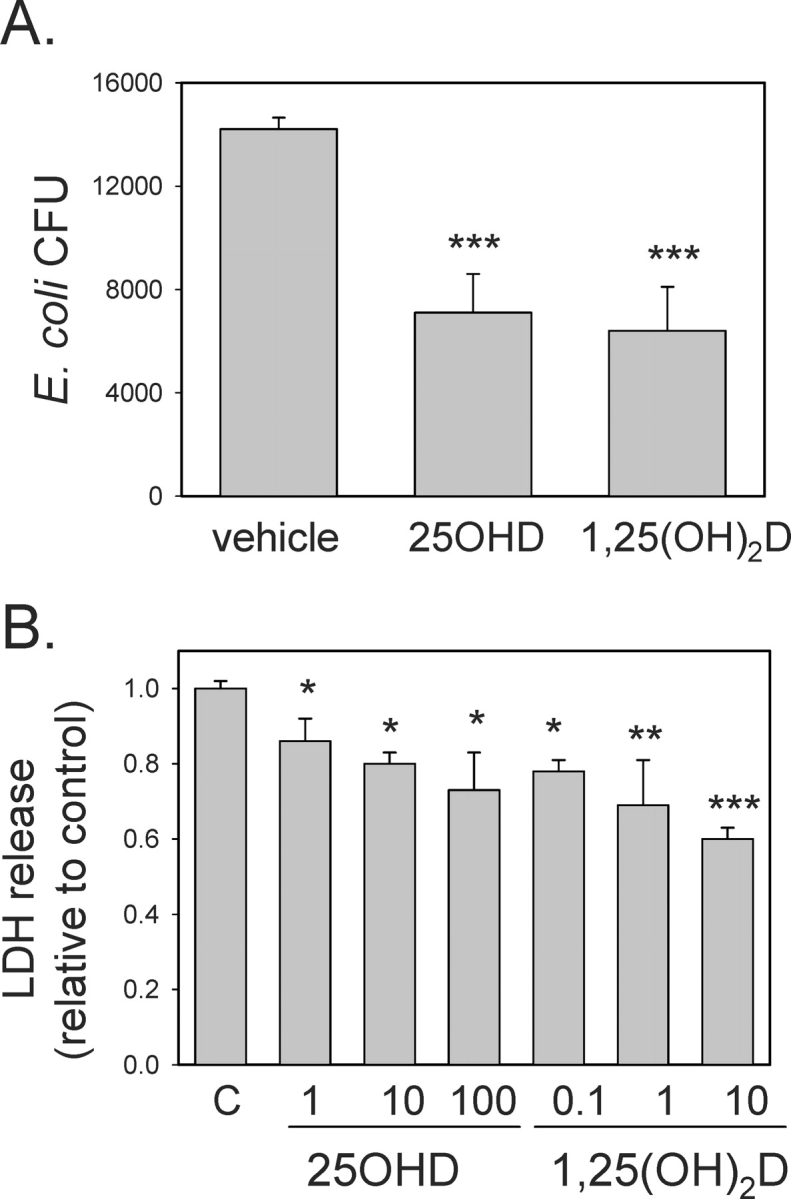
Effect of 25OHD and 1,25(OH)2D on bacterial infection and infection-associated cell death in trophoblastic cells. A) The 3A cells were pretreated with 25OHD (100 nM), 1,25(OH)2D (10 nM), or vehicle (0.1% ethanol) for 48 h and then infected with E. coli for 24 h. After removal of exogenous bacteria, monolayers of 3A cells were lysed and assessed for surviving intracellular E. coli (cfu). B) The 3A cells were pretreated with 25OHD (1–100 nM), 1,25(OH)2D (0.1–10 nM), or vehicle (0.1% ethanol) for 48 h and then infected with E. coli for 24 h. After removal of exogenous bacteria, cell death was assessed by analysis of the release of LDH into cell culture supernatants. Data are shown as the level of LDH in supernatants from infected 3A cells relative to infected vehicle-treated cells (C). *Statistically different from vehicle samples, P < 0.05; **statistically different from vehicle samples, P < 0.01; ***statistically different from vehicle samples, P < 0.001.
DISCUSSION
The placenta is one of the principal extrarenal sites for synthesis of the active vitamin D metabolite, 1,25(OH)2D [26–28, 34, 35]. In keeping with the classical endocrine function of 1,25(OH)2D, expression of CYP27B1 by the placenta has been linked to the regulation of maternal and fetal calcium homeostasis during pregnancy [36]. However, studies using knockout (−/−) mice for the VDR have cast some doubt on this by showing that VDR−/− or VDR+/− offspring from either VDR−/− or VDR+/− mothers have normal mineral calcium and phosphate levels and skeletal development [37]. In addition, we have shown that the placental CYP27B1 and VDR are induced early in gestation and prior to normal skeletal development [29]. This has raised the question as to whether vitamin D fulfills an alternative function in the placenta. In particular, the accumulating evidence linking vitamin D with the regulation of both innate and adaptive immunity [1–4] has prompted suggestions that the main role of CYP27B1 in the placenta is immunomodulatory [25, 29, 38]. In previous studies using maternal decidual cells, we have shown that intracrine synthesis of 1,25(OH)2D can act to enhance antimicrobial proteins, while suppressing inflammatory cytokine production [25]. The fact that trophoblastic cells also express CYP27B1 [28] suggests that the fetal component of placenta may also be subject to vitamin D-mediated immunoregulation. In the current study, we have investigated the effects of active and inactive metabolites of vitamin D on trophoblast innate antibacterial responses.
Recent studies have highlighted the importance of placental innate immunity as a mechanism for counteracting uterine infection during pregnancy [39, 40]. Although this appears to be mediated in part through pathogen sensing and antibacterial responses in the decidua [39, 41, 42], it is now evident that fetal trophoblasts may also be involved [40, 43]. Human trophoblastic cells have been shown to express various naturally occurring antimicrobial proteins including β-defensins 1–3 [40], secretory leuokocyte protease inhibitor [42] and β-defensin 5 [44]. Moreover, proteomic analysis of bovine conceptus fluid has indicated the presence of antibacterial proteins, such as bovine CAMP-1 [45]. However, to the best of our knowledge, results presented in this study are the first evidence for expression of the CAMP by trophoblastic cells. CAMPs are a family of antimicrobial proteins with a conserved N terminus and homology to the cysteine protease inhibitor, cathelin [46]. In humans, there is a single CAMP, for which the full-length proform protein is known as hCAP-18. However, the N terminus of this protein is cleaved to form an active C-terminal mature peptide of CAMP, LL37, which is incorporated into phagocytic vacuoles [47]. CAMP achieves its antibacterial effects in neutrophils and macrophages by permeabilizing the bacterial cell wall and membrane [47]. In view of the fact that previous studies have highlighted a potential phagocytic function for human trophoblastic cells [43], we hypothesize that concomitant bacterial killing by trophoblasts may be promoted by intracrine activation of vitamin D.
In macrophages, the ability of vitamin D to counteract bacterial infection of these cells is dependent on upregulation of both VDR and CYP27B1 to provide an effective intracrine mechanism for induction of CAMP [8]. This appears to be a pivotal facet of macrophage responses to Gram-positive bacteria, such as M. tuberculosis, which enhances vitamin D metabolism and function via TLR2 [5]. Data for trophoblastic cells suggest that basal expression of VDR and CYP27B1 is sufficient to support intracrine synthesis and action of 1,25(OH)2D in the absence of any TLR stimulus. Using a panel of TLR ligands, we were unable to demonstrate any significant effects on VDR or CYP27B1 expression in 3A cells, and the TLR4 ligand LPS did not modulate these genes in primary cultures of trophoblastic cells, despite being sensitive to LPS with respect to expression of TLR2 and TLR4 themselves. The regulation of trophoblast TLR2 expression by LPS was similar to that observed with human macrophages, and was also consistent with previous studies from our group demonstrating LPS induction of TLR2 in human endothelial cells [48]. In both macrophages and endothelial cells, LPS also induced its own receptor TLR4, whereas trophoblastic cells showed LPS autoinhibition of TLR4 (see Fig. 5). The latter effect has been reported previously for mouse macrophages, where LPS can induce transient suppression of TLR4 expression [49]. Thus, it is possible that the LPS suppression of TLR4 mRNA in trophoblastic cells simply represents a temporal variation relative to macrophages.
It is also possible that low TLR expression renders trophoblasts relatively insensitive to TLR ligands, such as LPS, at least with respect to regulation of VDR and CYP27B1 expression. Similar observations have been made for keratinocytes, which have low baseline expression of TLR2 and TLR4, and are therefore unable to induce CYP27B1 or VDR in response to their ligands [9]. In this latter instance, CYP27B1 was induced by transforming growth factor beta, a factor commonly associated with epidermal wounding. The resulting increase in 1,25(OH)2D production stimulated TLR expression, and this in turn increased sensitivity to TLR ligands to enable vitamin D-induced intracrine induction of CAMP similar to that observed with macrophages [9]. We were unable to demonstrate a similar mechanism in trophoblasts, in that treatment with 1,25(OH)2D or 25OHD had no effect on expression of TLR2 or TLR4. Indeed, it was notable that these treatments inhibited expression of CYP27B1, consistent with the feedback control of this enzyme previously described for renal cells [50]. Thus, it would appear that the induction of placental CYP27B1 and VDR that occurs early in gestation is sufficient to support intracrine induction of innate immunity, independent of TLR involvement. However, we cannot exclude the possibility that other factors, such as inflammatory cytokines, may potentiate synthesis of 1,25(OH)2D or expression of VDR. In future studies, it will be important to identify these factors.
Trophoblast and macrophage vitamin D metabolism also differed at the level of catabolism by the enzyme CYP24. Both cell types expressed similar amounts of CYP24 mRNA, but enzyme activity analyses showed enhanced production of the metabolite 24,25(OH)2D in 3A trophoblasts compared with macrophages. One possible explanation for this is that macrophages express a splice variant form of CYP24, encoding a metabolically inactive, truncated protein that is nevertheless still able to bind 25OHD or 1,25(OH)2D [51]. Unlike the wild type CYP24, which functions by catalyzing conversion of 1,25(OH)2D to less active 24-hydroxylated metabolites, the splice variant appears to sequester 25OHD, and may therefore act to attenuate synthesis of 1,25(OH)2D [51].
Induction of CAMP expression is a key feature of innate immune response to infection by a diverse array of pathogens, including organisms such as M. tuberculosis [5, 8, 11] and Gram-positive S. aureus [12–14], and group A streptococcus [52]. CAMP can also target Gram-negative bacteria [53], and, in the current study, we demonstrated that the induction of this CAMP in trophoblasts is associated with killing of E. coli. Gram-positive GBS is an important pathogen during and immediately after pregnancy [54, 55]. However, sensitivity of GBS to antimicrobial peptides is variable, and several strains of this bacterium appear to be resistant to CAMP [56]. In data presented here, we show that induction of CAMP in trophoblasts is not only linked to the antibacterial effects of vitamin D, but may also be involved in protection against infection-associated cell death (see Fig. 6B).
In previous studies, we have shown that infection with GBS induces cell death in trophoblasts and fibroblasts from human placentas [33]. Other groups have shown that, in addition to its antibacterial effects, CAMP is a potent modulator of apoptosis, suppressing programmed cell death in neutrophils, while having the opposite effect in lung epithelial cells [57]. Here, we demonstrate that vitamin D-induced CAMP may also help to protect against trophoblast death following bacterial infection. This novel facet of vitamin D function may help to maximize bacterial killing following induction of CAMP. Recent studies have demonstrated how pathogens such as M. tuberculosis can avoid innate immune surveillance by blocking macrophage apoptosis [58]. Thus, it is possible to speculate that local production of 1,25(OH)2D not only enhances mechanisms for bacterial killing, but also sustains this bacterial killing by promoting host cell viability. Moreover, it is important to recognize that CAMP has also been implicated in diverse biological responses that extend beyond its antibacterial actions [59, 60]. These include effects on angiogenesis [61] and heparin sulfate binding [62], which may also impact on placental function. In future studies, it will be interesting to determine whether these factors are also modulated by vitamin D.
In summary, data presented here show for the first time that induction of trophoblastic CAMP may be a key facet of placental innate immunity. Importantly, we also demonstrate that expression of CAMP by trophoblastic cells is potently enhanced following intracrine activation of vitamin D. Given that circulating levels of the prohormone form of vitamin D (25OHD) vary significantly in pregnant women [63], we hypothesize that placental capacity to synthesize CAMP and mount effective innate immune responses may also be subject to substantial variation. Although the precise function of vitamin D within the placenta remains to be defined, data from this study suggest that local synthesis of active 1,25(OH)2D may play a key role in placental innate immunity. We further postulate that improvement of maternal vitamin D status through dietary supplementation may act to potentiate placental innate immune responses during pregnancy.
Footnotes
1Supported by National Institutes of Health grant RO1AR050626 to M.H.
REFERENCES
- Adams JS, Hewison M.Unexpected actions of vitamin D: new perspectives on the regulation of innate and adaptive immunity. Nat Clin Pract Endocrinol Metab 2008; 4: 80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JH.Vitamin D signaling, infectious diseases and regulation of innate immunity. Infect Immun 2008; 76: 3837–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavera-Mendoza LE, White JH.Cell defenses and the sunshine vitamin. Sci Am 2007; 297: 62–65, 68–70, 72. [DOI] [PubMed] [Google Scholar]
- Hewison M.Vitamin D and innate immunity. Curr Opin Investig Drugs 2008; 9: 485–490. [PubMed] [Google Scholar]
- Liu PT, Stenger S, Tang DH, Modlin RL.Cutting Edge: Vitamin D-mediated human antimicrobial activity against Mycobacterium tuberculosis is dependent on the induction of cathelicidin. J Immunol 2007; 179: 2060–2063. [DOI] [PubMed] [Google Scholar]
- Wang TT, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, Tavera-Mendoza L, Lin R, Hanrahan JW, Mader S, White JH.Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J Immunol 2004; 173: 2909–2912. [DOI] [PubMed] [Google Scholar]
- Gombart AF, Borregaard N, Koeffler HP.Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J 2005; 19: 1067–1077. [DOI] [PubMed] [Google Scholar]
- Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, Kamen DL, Wagner M, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006; 311: 1770–1773. [DOI] [PubMed] [Google Scholar]
- Schauber J, Dorschner RA, Coda AB, Buchau AS, Liu PT, Kiken D, Helfrich YR, Kang S, Elalieh HZ, Steinmeyer A, Zugel U, Bikle DD, et al. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J Clin Invest 2007; 117: 803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim S, Dhawan P, Ragunath C, Christakos S, Diamond G.Induction of cathelicidin in normal and CF bronchial epithelial cells by 1,25-dihydroxyvitamin D3. J Cyst Fibros 2007; 6: 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau AR, Wilkinson KA, Newton SM, Floto RA, Norman AW, Skolimowska K, Davidson RN, Sorensen OE, Kampmann B, Griffiths CJ, Wilkinson RJ.IFN-gamma- and TNF-independent vitamin D-inducible human suppression of mycobacteria: the role of cathelicidin LL-37. J Immunol 2007; 178: 7190–7198. [DOI] [PubMed] [Google Scholar]
- Ouhara K, Komatsuzawa H, Kawai T, Nishi H, Fujiwara T, Fujiue Y, Kuwabara M, Sayama K, Hashimoto K, Sugai M.Increased resistance to cationic antimicrobial peptide LL-37 in methicillin-resistant strains of Staphylococcus aureus. J Antimicrob Chemother 2008; 61: 1266–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsuzawa H, Ouhara K, Yamada S, Fujiwara T, Sayama K, Hashimoto K, Sugai M.Innate defences against methicillin-resistant Staphylococcus aureus (MRSA) infection. J Pathol 2006; 208: 249–260. [DOI] [PubMed] [Google Scholar]
- Midorikawa K, Ouhara K, Komatsuzawa H, Kawai T, Yamada S, Fujiwara T, Yamazaki K, Sayama K, Taubman MA, Kurihara H, Hashimoto K, Sugai M.Staphylococcus aureus susceptibility to innate antimicrobial peptides, beta-defensins and CAP18, expressed by human keratinocytes. Infect Immun 2003; 71: 3730–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell MD, Jones JF, Kisich KO, Streib JE, Gallo RL, Leung DY.Selective killing of vaccinia virus by LL-37: implications for eczema vaccinatum. J Immunol 2004; 172: 1763–1767. [DOI] [PubMed] [Google Scholar]
- Bergman P, Walter-Jallow L, Broliden K, Agerberth B, Soderlund J.The antimicrobial peptide LL-37 inhibits HIV-1 replication. Curr HIV Res 2007; 5: 410–415. [DOI] [PubMed] [Google Scholar]
- Steinstraesser L, Tippler B, Mertens J, Lamme E, Homann HH, Lehnhardt M, Wildner O, Steinau HU, Uberla K.Inhibition of early steps in the lentiviral replication cycle by cathelicidin host defense peptides. Retrovirology 2005; 2: 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mookherjee N, Rehaume LM, Hancock RE.Cathelicidins and functional analogues as antisepsis molecules. Expert Opin Ther Targets 2007; 11: 993–1004. [DOI] [PubMed] [Google Scholar]
- Wehkamp J, Schmid M, Stange EF.Defensins and other antimicrobial peptides in inflammatory bowel disease. Curr Opin Gastroenterol 2007; 23: 370–378. [DOI] [PubMed] [Google Scholar]
- Hiemstra PS.Defensins and cathelicidins in inflammatory lung disease: beyond antimicrobial activity. Biochem Soc Trans 2006; 34: 276–278. [DOI] [PubMed] [Google Scholar]
- Bland R, Zehnder D, Hewison M.Expression of 25-hydroxyvitamin D3-1alpha-hydroxylase along the nephron: new insights into renal vitamin D metabolism. Curr Opin Nephrol Hypertens 2000; 9: 17–22. [DOI] [PubMed] [Google Scholar]
- Zehnder D, Bland R, Walker EA, Bradwell AR, Howie AJ, Hewison M, Stewart PM.Expression of 25-hydroxyvitamin D3-1alpha-hydroxylase in the human kidney. J Am Soc Nephrol 1999; 10: 2465–2473. [DOI] [PubMed] [Google Scholar]
- Hewison M, Burke F, Evans KN, Lammas DA, Sansom DM, Liu P, Modlin RL, Adams JS.Extra-renal 25-hydroxyvitamin D3-1alpha-hydroxylase in human health and disease. J Steroid Biochem Mol Biol 2007; 103: 316–321. [DOI] [PubMed] [Google Scholar]
- Townsend K, Evans KN, Campbell MJ, Colston KW, Adams JS, Hewison M.Biological actions of extra-renal 25-hydroxyvitamin D-1alpha-hydroxylase and implications for chemoprevention and treatment. J Steroid Biochem Mol Biol 2005; 97: 103–109. [DOI] [PubMed] [Google Scholar]
- Evans KN, Nguyen L, Chan J, Innes BA, Bulmer JN, Kilby MD, Hewison M.Effects of 25-Hydroxyvitamin D3 and 1,25-dihydroxyvitamin D3 on cytokine production by human decidual cells. Biol Reprod 2006; 75: 816–822. [DOI] [PubMed] [Google Scholar]
- Gray TK, Lester GE, Lorenc RS.Evidence for extra-renal 1 alpha-hydroxylation of 25-hydroxyvitamin D3 in pregnancy. Science 1979; 204: 1311–1313. [DOI] [PubMed] [Google Scholar]
- Weisman Y, Harell A, Edelstein S, David M, Spirer Z, Golander A.1 alpha, 25-Dihydroxyvitamin D3 and 24,25-dihydroxyvitamin D3 in vitro synthesis by human decidua and placenta. Nature 1979; 281: 317–319. [DOI] [PubMed] [Google Scholar]
- Zehnder D, Evans KN, Kilby MD, Bulmer JN, Innes BA, Stewart PM, Hewison M.The ontogeny of 25-hydroxyvitamin D(3) 1alpha-hydroxylase expression in human placenta and decidua. Am J Pathol 2002; 161: 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans KN, Bulmer JN, Kilby MD, Hewison M.Vitamin D and placental-decidual function. J Soc Gynecol Investig 2004; 11: 263–271. [DOI] [PubMed] [Google Scholar]
- Guleria I, Pollard JW.The trophoblast is a component of the innate immune system during pregnancy. Nat Med 2000; 6: 589–593. [DOI] [PubMed] [Google Scholar]
- Abrahams VM, Bole-Aldo P, Kim YM, Straszewski-Chavez SL, Chaiworapongsa T, Romero R, Mor G.Divergent trophoblast responses to bacterial products mediated by TLRs. J Immunol 2004; 173: 4286–4296. [DOI] [PubMed] [Google Scholar]
- Evans KN, Taylor H, Zehnder D, Kilby MD, Bulmer JN, Sha F, Adams JS, Hewison M.Increased expression of 25-hydroxyvitamin D-1alpha-hydroxylase in dysgerminomas: a novel form of humoral hypercalcemia of malignancy. Am J Pathol 2004; 165: 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan A, Chung K, Kocak H, Bertolotto C, Uh A, Hobel CJ, Simmons CF, Doran K, Liu GY, Equils O.Group B streptococcus induces trophoblast death. Microb Pathog 2008; 45: 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehnder D, Bland R, Williams MC, McNinch RW, Howie AJ, Stewart PM, Hewison M.Extrarenal expression of 25-hydroxyvitamin D(3)-1 alpha-hydroxylase. J Clin Endocrinol Metab 2001; 86: 888–894. [DOI] [PubMed] [Google Scholar]
- Vanhooke JL, Prahl JM, Kimmel-Jehan C, Mendelsohn M, Danielson EW, Healy KD, Deluca HF.CYP27B1 null mice with LacZreporter gene display no 25-hydroxyvitamin D3–1{alpha}-hydroxylase promoter activity in the skin. Proc Natl Acad Sci U S A 2006; 103: 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs CS, Kronenberg HM.Maternal-fetal calcium and bone metabolism during pregnancy, puerperium, and lactation. Endocr Rev 1997; 18: 832–872. [DOI] [PubMed] [Google Scholar]
- Kovacs CS, Woodland ML, Fudge NJ, Friel JK.The vitamin D receptor is not required for fetal mineral homeostasis or for the regulation of placental calcium transfer in mice. Am J Physiol Endocrinol Metab 2005; 289: E133–E144. [DOI] [PubMed] [Google Scholar]
- Rebut-Bonneton C, Demignon J.Effects of 1,25-dihydroxyvitamin D3 on in vitro lymphocyte reactions: arguments for a role at the maternofetal interface. Gynecol Obstet Invest 1991; 32: 134–138. [DOI] [PubMed] [Google Scholar]
- King AE, Kelly RW, Sallenave JM, Bocking AD, Challis JR.Innate immune defences in the human uterus during pregnancy. Placenta 2007; 28: 1099–1106. [DOI] [PubMed] [Google Scholar]
- King AE, Paltoo A, Kelly RW, Sallenave JM, Bocking AD, Challis JR.Expression of natural antimicrobials by human placenta and fetal membranes. Placenta 2007; 28: 161–169. [DOI] [PubMed] [Google Scholar]
- Koga K, Mor G.Expression and function of Toll-like receptors at the maternal-fetal interface. Reprod Sci 2008; 15: 231–242. [DOI] [PubMed] [Google Scholar]
- King AE, Critchley HO, Kelly RW.Presence of secretory leukocyte protease inhibitor in human endometrium and first trimester decidua suggests an antibacterial protective role. Mol Hum Reprod 2000; 6: 191–196. [DOI] [PubMed] [Google Scholar]
- Amarante-Paffaro A, Queiroz GS, Correa ST, Spira B, Bevilacqua E.Phagocytosis as a potential mechanism for microbial defense of mouse placental trophoblast cells. Reproduction 2004; 128: 207–218. [DOI] [PubMed] [Google Scholar]
- Svinarich DM, Wolf NA, Gomez R, Gonik B, Romero R.Detection of human defensin 5 in reproductive tissues. Am J Obstet Gynecol 1997; 176: 470–475. [DOI] [PubMed] [Google Scholar]
- Riding GA, Jones A, Holland MK, Hill JR, Lehnert SA.Proteomic analysis of bovine conceptus fluids during early pregnancy. Proteomics 2008; 8: 160–177. [DOI] [PubMed] [Google Scholar]
- Tomasinsig L, Zanetti M.The cathelicidins—structure, function and evolution. Curr Protein Pept Sci 2005; 6: 23–34. [DOI] [PubMed] [Google Scholar]
- Risso A.Leukocyte antimicrobial peptides: multifunctional effector molecules of innate immunity. J Leukoc Biol 2000; 68: 785–792. [PubMed] [Google Scholar]
- Faure E, Thomas L, Xu H, Medvedev A, Equils O, Arditi M.Bacterial lipopolysaccharide and IFN-gamma induce Toll-like receptor 2 and Toll-like receptor 4 expression in human endothelial cells: role of NF-kappa B activation. J Immunol 2001; 166: 2018–2024. [DOI] [PubMed] [Google Scholar]
- Nomura F, Akashi S, Sakao Y, Sato S, Kawai T, Matsumoto M, Nakanishi K, Kimoto M, Miyake K, Takeda K, Akira S.Cutting edge: endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J Immunol 2000; 164: 3476–3479. [DOI] [PubMed] [Google Scholar]
- Zehnder D, Hewison M.The renal function of 25-hydroxyvitamin D3-1alpha-hydroxylase. Mol Cell Endocrinol 1999; 151: 213–220. [DOI] [PubMed] [Google Scholar]
- Ren S, Nguyen L, Wu S, Encinas C, Adams JS, Hewison M.Alternative splicing of vitamin D-24-hydroxylase: a novel mechanism for the regulation of extrarenal 1,25-dihydroxyvitamin D synthesis. J Biol Chem 2005; 280: 20604–20611. [DOI] [PubMed] [Google Scholar]
- Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, Pestonjamasp V, Piraino J, Huttner K, Gallo RL.Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 2001; 414: 454–457. [DOI] [PubMed] [Google Scholar]
- Gutsmann T, Larrick JW, Seydel U, Wiese A.Molecular mechanisms of interaction of rabbit CAP18 with outer membranes of Gram-negative bacteria. Biochemistry 1999; 38: 13643–13653. [DOI] [PubMed] [Google Scholar]
- Larsen JW, Sever JL.Group B Streptococcus and pregnancy: a review. Am J Obstet Gynecol 2008; 198: 440–448;.discussion, 448–450. [DOI] [PubMed] [Google Scholar]
- Winn HN.Group B streptococcus infection in pregnancy. Clin Perinatol 2007; 34: 387–392. [DOI] [PubMed] [Google Scholar]
- Jones AL, Mertz RH, Carl DJ, Rubens CE.A streptococcal penicillin-binding protein is critical for resisting innate airway defenses in the neonatal lung. J Immunol 2007; 179: 3196–3202. [DOI] [PubMed] [Google Scholar]
- Barlow PG, Li Y, Wilkinson TS, Bowdish DM, Lau YE, Cosseau C, Haslett C, Simpson AJ, Hancock RE, Davidson DJ.The human cationic host defense peptide LL-37 mediates contrasting effects on apoptotic pathways in different primary cells of the innate immune system. J Leukoc Biol 2006; 80: 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan H, Lee J, Ren F, Chen M, Kornfeld H, Remold HG.Mycobacterium tuberculosis blocks crosslinking of annexin-1 and apoptotic envelope formation on infected macrophages to maintain virulence. Nat Immunol 2008; 9: 1189–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai-Larsen Y, Agerberth B.The role of the multifunctional peptide LL-37 in host defense. Front Biosci 2008; 13: 3760–3767. [DOI] [PubMed] [Google Scholar]
- Zanetti M.Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol 2004; 75: 39–48. [DOI] [PubMed] [Google Scholar]
- Koczulla R, von Degenfeld G, Kupatt C, Krotz F, Zahler S, Gloe T, Issbrucker K, Unterberger P, Zaiou M, Lebherz C, Karl A, Raake P, et al. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J Clin Invest 2003; 111: 1665–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneider NC, Djanani A, Wiedermann CJ.Heparan sulfate proteoglycan-involving immunomodulation by cathelicidin antimicrobial peptides LL-37 and PR-39. ScientificWorldJournal 2007; 7: 1832–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar LM, Simhan HN, Powers RW, Frank MP, Cooperstein E, Roberts JM.High prevalence of vitamin D insufficiency in black and white pregnant women residing in the northern United States and their neonates. J Nutr 2007; 137: 447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]