

Abstract
Melanocortin 4 receptor (MC4R) plays an important role in the regulation of food intake and body weight. To determine the molecular basis of human MC4R (hMC4R) responsible for α-melanocortin-stimulating hormone (α-MSH) binding, in this study, we utilized both receptor domain exchange and site-directed mutagenesis studies to investigate the molecular determinants of hMC4R responsible for α-MSH binding and signaling. α-MSH is a potent agonist at hMC4R but not at hMC2R. Cassette substitutions of the second, third, fourth, fifth, and sixth transmembrane regions (TM) of the hMC4R with the homologous regions of hMC2R were performed and α-MSH binding and signaling were examined. Our results indicate that each chimeric receptor was expressed at the cell surface and the expression levels remain similar to that of the wild-type receptor. The cassette substitutions of the second, fourth, fifth, and sixth TMs of the hMC4R with homologous regions of the hMC2R did not significantly alter α-MSH binding affinity and potency except substitution of the TM3 of the hMC4R, suggesting that the conserved residues in TMs of the hMC4R are crucial for α-MSH binding and signaling. Further mutagenesis studies indicate that conserved residues Glu100 in TM2, Asp122, Asp126 in TM3 and Trp258, Phe261, His264 in TM6 are involved in α-MSH binding and signaling. In conclusion, our results suggest that the conserved residues in the TM2, TM3, and TM6 of the hMC4R are responsible for α-MSH binding and signaling.
Obesity is a common and rapidly growing health problem in the United States and worldwide (1-8). The melanocortin-4 receptor (MC4R)2 plays a key role in the regulation of food intake and body weight. Mutations of human MC4R (hMC4R) are the most common monogenic cause of human obesity described to date, accounting for up to 6% of all cases of severe early onset obesity (9-11). MC4R is a seven-transmembrane G protein-coupled receptor expressed mainly in the hypothalamus (12-16). The endogenous agonist, α-melanocyte-stimulating hormone (α-MSH), activates MC4R and inhibits food intake, whereas agouti-related protein, the endogenous antagonist, inhibits MC4R action and induces food intake (13, 17, 18). MC4R is a member of the family of G protein-coupled receptors and consists of a single polypeptide featuring seven α-helical transmembrane domains (TMs), an extracellular N terminus, three extracellular loops, three intracellular loops, and an intracellular C terminus. Pharmacologic studies have identified that the tripeptide Phe7-Arg8-Trp9 in α-MSH is the minimal active fragment retaining biological activity at MC4R (85). The ligand receptor interaction between α-MSH and MC4R has been extensively studied based on the model generated by computer (19-24). However, detailed information about ligand receptor interaction between α-MSH and MC4R remains unclear.
In this study, we utilize both chimeric receptor and site-directed mutagenesis study to determine the molecular basis of hMC4R responsible for α-MSH binding and signaling. Both hMC4R and hMC2R are subtypes of the melanocortin receptor family and share a number of structural, functional, and pharmacological similarities. In the putative TM regions, both hMC4R and hMC2R show overall identities of 58% and similarities of 91%. They have high binding affinities for agonist ACTH and antagonist agouti signaling protein. However, hMC4R and hMC2R exhibit a number of pharmacologic differences (25-27). Endogenous agonists, α-MSH, β-MSH, and γ-MSH, are potent agonists at MC4R but not at hMC2R. Agouti-related protein is a potent antagonist for hMC4R but not at MC2R. Therefore, MC2R is a good candidate to determine the molecular determinants of hMC4R responsible for α-MSH binding and signaling. Our results indicate that six conserved amino acid residues in TM2, TM3, and TM6 of the hMC4R are crucial for MSH binding and signaling.
EXPERIMENTAL PROCEDURES
[Nle4-D-Phe7]-MSH (NDP-α-MSH), an analogue of α-MSH, was purchased from Peninsula Laboratories (Belmont, CA). 3-Isobutylmethylxanthine was from Sigma and [125I]NDP MSH was from Amersham Biosciences. Dulbecco's modified Eagle's medium and Lipofectamine were from Invitrogen.
Construction of Melanocoritn Receptor Chimeric Receptors
The amino acid sequences of the hMC2R and hMC4R were examined by hydrophobicity plot (Genetics Computer Group, Inc., Madison, WI) and manually by comparing their sequences to a previously published alignment of seven transmembrane G protein-coupled receptor α-helices (28). A sequence comparison of the transmembrane domains of the hMC2R and hMC4R is shown in Fig. 1. The chimeras utilized in these studies are schematically diagramed in Fig. 2. The chimeric receptors were constructed by PCR using Pfu polymerase (Stratagene, La Jolla, CA) (29). The human MC4R served as templates. During an initial round of PCR, partial-length receptor fragments were generated. The sequence of one of the PCR primer oligonucleotides consisted of the transmembrane domain of interest, which was then coupled to a portion of the extracellular domain required to form the chimeric receptor. The second oligonucleotide primer consisted of the 5′ or 3′ end of the MC4R. Receptor fragments were separated by agarose gel electrophoresis and used in a second round ofPCR in which full-length chimeric receptor constructs were assembled by cycling the appropriate fragments together for 10 cycles prior to adding both 5′ and 3′ receptor primers. The chimeric receptors were subcloned into the eukaryotic expression vector pcDNA 3.1 (Invitrogen).
FIGURE 1. Receptor sequence comparison between MC4R and MC2R.

The conserved amino acid residues in the upper regions of the hMC4R TMs are highlighted with black. The abbreviation e represents extracellular loop and i represents intracellular loop of the receptor.
FIGURE 2. Schematic representation of the chimeric hMCRs utilized in these studies.

A schematic depicts the seven transmembrane structures of the “wild-type” (WT) MC4R (drawn with heavy lines) and MC2R (drawn with thin lines). B depicts the structure of the chimeric MC4R.
Site-directed Mutagenesis of hMC4R
Single mutations was constructed using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The entire coding region of the mutated receptors was sequenced to confirm that the desired mutation sequences were present and that no sequence errors had been introduced by University of Alabama, Birmingham Sequence Core. The mutated receptors are shown in Fig. 5. The mutant receptors were then subcloned into the eukaryotic expression vector pCDNA 3.1 (Invitrogen).
FIGURE 5. Two-dimensional representation of the seven TM structure of the hMC4R.

The conserved TM residues mutated in these experiments are denoted by gray highlighting. Those TM residues whose mutation significantly affected NDP-MSH binding as determined are highlighted by black.
Cell Culture and Transfection
The OS3 cell line was used for hMC4R, hMC2R, and chimeric receptor transfection. The cells transfected with receptor were cultured in Dulbecco's modified Eagle's medium containing 10% bovine fetal serum and HEPES. Cells at 80% confluence were washed twice, and the receptor constructs were transfected into cells using Lipofectamine (Invitrogen) (30).
Binding Assays
Binding experiments were performed using the conditions previously described (19). Briefly, after removal of the media, cells were incubated with non-radioligand NDP-MSH from 10-10 to 10-6 M in 0.5 ml of minimal essential medium containing 0.2% bovine serum albumin and 2 × 105 cpm of 125I-NDP-MSH for 1 h. The binding reactions were terminated by removing the media and washing the cells twice with minimal essential medium containing 0.2% bovine serum albumin. The cells were then lysed with 0.2 N NaOH, and the radioactivity in the lysate was quantified in an analytical γ-counter (PerkinElmer). Non-specific binding was determined by measuring the amount of 125I label bound on the cells in the presence of 10-6 M excess unlabeled ligand. Specific binding was calculated by subtracting nonspecifically bound radioactivity from total bound radioactivity. Binding data are reported as Bmax and IC50.
cAMP Assay
Cellular cAMP generation was measured using a competitive binding assay kit (TRK 432, Amersham Biosciences). Briefly, cell culture media was removed, and cells were incubated with 0.5 ml of Earle's balanced salt solution, containing the melanocortin agonist NDP-MSH (10-10-10-6 M), for 1 h at 37 °C in the presence of 10-3 M isobutylmethylxanthine. The reaction was stopped by adding ice-cold 100% ethanol (500 μl/well). cAMP content was measured as previously described, according to instructions accompanying the assay kit (25).
Receptor Expression Using FACs (31)
hMC4R-transfected cells were harvested using 0.2% EDTA and washed twice with phosphate-buffered saline. Aliquots of 3 × 106 cells were centrifuged and fixed with 3% paraformaldehyde in phosphate-buffered saline (pH 7.4). The cells were incubated with 50 μl of 10 μg/ml murine anti-FLAG M1 monoclonal antibody (Sigma, catalog number 316) in incubation buffer for 45 min. Under this condition the primary antibody binds only to receptors located at the cell surface. The cells were collected by centrifugation. The cell pellets were suspended in 100 μl of incubation buffer containing CY3™-conjugated Affinity Pure Donkey Anti-mouse IgG (ImmunoResearch Lab, Inc., West Grove, PA). Flow cytometry was performed on a fluorescence-activated cell sorter (BD Biosciences FACStar plus six parameter cytometer/sorter with a dual argon ion laser). The results were analyzed using the software CellQuest (BD Biosciences Immunocytometry Systems, San Jose, CA).
Statistical Analysis
Each experiment was performed in duplicate three separate times. The mean value of the dose-response data of binding and cAMP production was fit to a sigmoid curve with a variable slope factor using non-linear squares regression analysis (GraphPad Prism, GraphPad Software, San Diego, CA). Data were expressed as mean ± S.E. Significant difference was assessed by one-way analysis of variance with p < 0.05 considered to be statistically significant.
RESULTS
Substitutions of the TMs of the hMC4R with the Corresponding Regions of the hMC2R on NDP-α-MSH Binding
The binding affinity of NDP-MSH at wild-type hMC4 and hMC2 receptors was first examined. As shown in Fig. 3A, NDP-α-MSH possessed high affinity binding at wild-type hMC4R but not at hMC2R. To examine the ability of NDP-α-MSH to activate hMC4R or MC2R, cAMP production was determined. NDP-α-MSH dose-dependently increased cAMP generation at hMC4R but not at hMC2R as expected (Fig. 3B).
FIGURE 3. Binding affinity and potency of NDP-MSH at the wild-type hMC4R and hMC2R.

Cells transfected with hMC4R or hMC2R were incubated with 125I-NDP-MSH at 37 °C for 1 h in the presence of the indicated amounts of unlabeled ligands. The total 125I-NDP-MSH binding was then determined on duplicate wells as described under “Experimental Procedures.” Data points represent the mean ± S.E. of at least three independent experiments. A depicts the binding affinity of NDP-MSH at hMC4R-WT and hMC2R-WT. B demonstrates the ability of NDP-MSH to stimulate the production of intracellular cAMP at hMC4R-WT and hMC2R-WT.
To investigate which regions of the hMC4R are responsible for NDP-MSH binding and specific activity, a domain-exchange strategy was used to localize regions of the hMC4R responsible for NDP-MSH binding. Cassette substitutions of the TM2, TM3, TM4, TM5, and TM6 of the hMC4R with homologous regions of the hMC2R were performed (Fig. 2). The first and seventh TMs were not chosen for investigation because our previous data suggested that they were not important in NDP-MSH binding (21, 30, 32).
To determine whether chimeric receptor proteins are expressed at the cell surface and to quantify the receptor expression level we have utilized the antigenic epitope FLAG to examine the receptor expression. The FLAG sequence was inserted into the N terminus of the hMC4R using polymerase chain reaction. The FLAG protein is an eight amino acid peptide (Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys) and has been widely used to determine protein expression by flow cytometry (33). Our results indicate that FLAG signal fluorescence emitted by cells expressing the hMC4R-WT and chimeric receptors can be detected by FACs. The protein expression levels of the chimeric receptors showed no significant variation compared with that of the wild-type receptor (Table 1).
TABLE 1.
Receptor domain exchanges of hMC4R on 125I-NDP-MSH binding and siganaling
| Receptor expression | NDP-MSH Ki | NDP-MSH EC5O | |
|---|---|---|---|
| %WT | nM | ||
| hMC4R-WT | 100 | 3.6 ± 0.1 | 0.8 ± 0.2 |
| hMC4R/TM2 hMC2R | 95 ± 11.5 | 7.2 ± 0.2 | 1.1 ± 0.3 |
| hMC4R/TM3 hMC2R | 97 ± 6.4 | 45.6 ± 11.2a | 32 ± 7.2a |
| hMC4R/TM4 hMC2R | 92 ± 7.7 | 6.9 ± 0.3 | 1.7 ± 0.1 |
| hMC4R/TM5 hMC2R | 91 ± 5.5 | 5.1 ± 0.6 | 1.4 ± 0.3 |
| hMC4R/TM6 hMC2R | 90 ± 8.5 | 6.1 ± 0.4 | 1.3 ± 0.1 |
| hMC2R-WT | 100 | No | NRb |
p < 0.05 compared with WT receptor.
NR, no response.
To assess the binding affinity of ligand at these chimeric receptors, an experiment of unlabeled NDP-MSH to displace labeled 125I-NDP-MSH was performed (Fig. 4A). Our results indicate that these chimeric receptors did not significantly alter NDP-MSH binding affinity except for the chimeric receptor, hMC4R/TM3hMC2R, their Ki values are summarized in Table 1. To examine the ability of NDP-MSH to activate hMC4/ hMC2 chimeric receptors, cAMP production was determined. Our results indicate that NDP-MSH increased cAMP generation in a dose-dependent manner at these hMC4/hMC2 chimeric receptors (Fig. 4B) and their EC50 values are shown in Table 1. Substitution of the hMC4R TM3 with the corresponding region of the hMC2R (hMC4R/TM3hMC2R) significantly decreased NDP-MSH binding affinity and potency. Their EC50 values are shown in Table 1.
FIGURE 4. Binding affinity and potency of NDP-MSH at the chimeric receptors.

OS3 cells transfected with hMC4R chimera were incubated with 125I-NDP-MSH at 37 °C for 1 h in the presence of the indicated amounts of unlabeled ligands, and total 125I-NDP-MSH binding was determined. For the cAMP assay, the cells transfected with hMC4R chimera were incubated with the indicated amounts of NDP-MSH for 30 min and total cAMP accumulation was determined on duplicated wells. Data points represent the mean ± S.E. of at least three independent experiments. A depicts the binding affinity of NDP-MSH at the chimeric receptors. B demonstrates the ability of NDP-MSH to stimulate the production of intracellular cAMP at the chimeric receptors.
Mutations of the Conserved Amino Acid Residues in TM2, TM3, TM4, TM5, and TM6 of the hMC4R for NDP-MSH Binding and Signaling
The substitutions of the TM2, TM3, TM4, TM5, and TM6 of the hMC4R with the corresponding regions of the hMC2R did not significantly alter or abolish NDP-MSH specific activity, suggesting that a conserved amino acid residue of the MC4R TMs may be crucial for NDP-MSH binding and activity. Based on the model of MC1R, electrostatic and hydro-phobic forces have been proposed to be involved in MSH binding and receptor activation (32), the conserved ionic and aromatic amino acid residues in TMs of the hMC4R were selected in this study. The conserved neutral amino acids isoleucine, leucine, and pro-line are therefore not included. Moreover, MSH is a 13-amino acid peptide and cannot access the receptor deep transmembrane region, the conserved ionic and aromatic amino acid residues in the upper region of the transmembrane regions of hMC4R are therefore potential candidates for NDP-MSH binding and signaling. A sequence alignment of hMC4R and hMC2R were compared and 15 amino acid residues in TM2, TM3, TM4, TM5, and TM6 are identified conserved between hMC4R and hMC2R in the upper regions of the transmembrane regions of hMC4R (Fig. 5). To determine whether these amino acid residues are involved in NDP-MSH binding and signaling, these residues were individually mutated with alanine and NDP-MSH binding affinity and potency were evaluated. Our results indicate that all of the mutant receptors were expressed at the cell surface and their expressions are shown in Table 2. The expression levels of mutations D90A, E100A, D122A, D126A, F261A, and H264A are lower than that of the hMC4R-WT. The protein expression levels of mutations M91A, S94A, S127A, S131A, T178A, F201A, F254A, and W258A are not significantly different from that of the hMC4R-WT. Receptor function study indicates that NDPMSH dose dependently displaced 125I-NDP-MSH binding at the mutations of the amino acid residues, E100A, D122A, F261A, and H264A but their binding affinities for NDPMSH were significantly reduced compared with that of the hMC4R-WT (Figs. 6A and 7A). Mutation D126A abolished NDP-MSH binding but remains weak signaling. However, mutations of M91A, S94A, S127A, S131A, T178A, F201A, M204A, and W258A remain with similar binding affinity compared with that of the hMC4R-WT. Consistent with the binding results, the mutations of amino acid residues, E100A, D122A, D126A, F261A, and H264A, significantly reduced NDP-MSH-mediated cAMP production but M91A, S94A, S127A, S131A, T178A, F201A, M204A, and W258A maintain high NDPMSH-stimulated cAMP production (Figs. 6B and 7B). Their Ki and EC50 values are shown in Table 2.
TABLE 2.
Effect of the substitutions of the conserved amino acid residues of hMC4R on NDP-MSH binding and cAMP production
| Receptor expression | NDP-MSH Ki | NDP-MSH EC5O | |
|---|---|---|---|
| %WT | nM | ||
| hMC4R-WT | 100 | 3.6 ± 0.1 | 0.8 ± 0.2 |
| D90A | 47 ± 6.3 | 7.4 ± 0.1 | NRa |
| M91A | 97 ± 3.3 | 4.8 ± 0.1 | 1.8 ± 0.7 |
| S94A | 97 ± 6.3 | 4.5 ± 0.1 | 1.4 ± 0.5 |
| E100A | 82 ± 11 | 12.0 ± 2.9b | 5.7 ± 0.5b |
| D122A | 78 ± 11 | 10.9 ± 1.6b | 9.8 ± 3.2b |
| D126N | 35 ± 10 | NDc | 353 ± 43.2b |
| S127A | 91 ± 8.3 | 4.3 ± 0.5 | 1.6 ± 0.8 |
| S131A | 94 ± 4.3 | 4.7 ± 0.9 | 1.7 ± 0.6 |
| T178A | 94 ± 3.6 | 3.1 ± 0.2 | 1.0 ± 0.1 |
| F201A | 95 ± 5.8 | 4.3 ± 0.5 | 1.2 ± 0.2 |
| M204A | 98 ± 5.8 | 3.0 ± 0.9 | 1.1 ± 0.1 |
| F254A | 91 ± 6.3 | 4.0 ± 0.9 | 9.2 ± 0.1 |
| W258A | 89 ± 3.9 | 14.0 ± 1.9b | 9.2 ± 0.3b |
| F261A | 81 ± 8.3 | 28 ± 2.9b | 19 ± 1.8b |
| H264A | 78 ± 4.5 | >103b | 141 ± 13.1b |
NR, no response.
p < 0.05 compared with WT receptor.
ND, no binding.
FIGURE 6. Binding affinity and potency of NDP-MSH at the mutations of the conserved amino acid residues of hMC4Rs.

A shows the binding affinity of unlabeled NDP-MSH to displace 125I-NDP-MSH. B shows that NDP-MSH stimulates cAMP production. Data points represent the mean ± S.E. of at least three independent experiments.
FIGURE 7. Binding affinity and potency of NDP-MSH at the mutations of the conserved amino acid residues of hMC4Rs.

A shows the binding affinity of unlabeled NDP-MSH to displace 125I-NDP-MSH. B shows that NDP-MSH stimulates cAMP production. Data points represent the mean ± S.E. of at least three independent experiments.
Mutations of Non-conserved Amino Acid Residues in TM3 of the hMC4R for NDP-MSH Binding and Signaling
Because substitution of TM3 of the hMC4R with the corresponding TM3 region of hMC2R significantly alters NDPMSH binding affinity and potency, we speculate that non-conserved amino acid residues of TM3 of the hMC4R may also be involved in NDP-MSH specific binding. The sequence of amino acids in TM3 between hMC4R and hMC2R was compared, and depicted in Fig. 1. A sequence alignment of hMC2R and hMC4R revealed that five amino acid residues are non-conserved in TM3. To determine whether these amino acid residues are involved in NDP-MSH specific binding and activity, these residues were individually mutated with alanine and evaluated. Our results indicate that these nonconserved mutant receptors were expressed at the cell surface and the expression levels showed no significant variation compared with that of the wild-type receptor (Table 3). NDP-MSH dose dependently displaced 125I-NDP-MSH binding at mutations N123A, I129A, S131A, C138A, and S142A, and their binding affinities were not significantly altered compared with that of hMC4R-WT. However, mutation N123D significantly reduced NDP-MSH binding affinity compared with that of hMC4R-WT (Fig. 8A). Consistent with the binding results, mutations N123A, I129A, S131A, C138A, and S142A did not significantly alter NDP-MSH-mediated cAMP production, but N123D mutation did greatly reduce NDP-MSH-stimulated cAMP production (Fig. 8B). Their Ki and EC50 values are shown in Table 3.
TABLE 3.
Mutations of the non-conserved residues in TM3 of the hMC4R on 125I-NDP-MSH binding and cAMP production
| Receptor expression | NDP-MSH Ki | NDP-MSH EC5O | |
|---|---|---|---|
| %WT | nM | ||
| hMC4R-WT | 100 | 3.6 ± 0.1 | 0.8 ± 0.2 |
| N123A | 96 ± 2.2 | 4.3 ± 0.2 | 1.6 ± 0.5 |
| N123D | 76 ± 12.2 | 69.3 ± 0.2a | 45.6 ± 7.1a |
| I129A | 87 ± 3.5 | 4.21 ± 0.1 | 1.3 ± 0.3 |
| C130A | 92 ± 7.7 | 2.9 ± 0.3 | 1.8 ± 0.7 |
| S131A | 90 ± 8.1 | 4.3 ± 0.5 | 2.1 ± 0.1 |
| C138A | 91 ± 5.5 | 4.9 ± 0.6 | 0.8 ± 0.1 |
| S142A | 93 ± 3.5 | 5.6 ± 0.8 | 1.9 ± 0.9 |
p < 0.05 compared with WT receptor.
FIGURE 8. Binding affinity and potency of NDP-MSH at the mutations of non-conserved amino acid residues of hMC4Rs.

A shows the binding affinity of unlabeled NDP-MSH to displace 125I-NDP-MSH. B shows that NDP-MSH stimulates cAMP production. Data points represent the mean ± S.E. of at least three independent experiments.
DISCUSSION
We have identified six conserved amino acid residues in the TMs of the hMC4R that are crucial for NDP-MSH binding and signaling using both chimeric receptor and site-directed mutagenesis studies, Glu100 in TM2, Asp122 and Asp126 in TM3, Trp258, Phe261, and His264 in TM6 of the hMC4R.
Hypothalamic MC4R plays an important role in the regulation of both human and animal food intake. The identification of the essential amino acid residues of the hMC4R responsible for MSH binding should provide important information for the understanding of the signaling events that regulate food intake and provide a rationale basis for the design of drugs for the treatment of obesity. Extensive studies have been performed to examine ligand receptor interaction between NDP-MSH and MC4R based on the receptor model generated by computer (19, 21, 24). However, the computer-generated model for ligand receptor interaction has limitations for prediction of the ligand receptor interaction. The detailed information between NDP-MSH and MC4R remains unclear. Both chimeric receptors and site-directed mutagenesis have been widely utilized for determining the molecular basis of the receptor responsible for ligand binding and signaling. The chimeric receptor approach is useful for identifying domains of the receptors that are important for specifying the unique pharmacology of each receptor subtype, whereas site-directed mutagenesis studies are important for identifying residues that are invariantly important for agonist and/or antagonist binding (20, 29, 34). In this study, we have taken each of these approaches to determine the molecular basis of hMC4R responsible for NDP-MSH binding and signaling. Melanocortin receptor subtypes are believed to have the same basic molecular architecture and we therefore utilized hMC4R and hMC2R to create chimeric receptors and determine the amino acid residue of the hMC4R involved in NDP-MSH binding. The hMC4R and hMC2R share a number of structural similarities. In the putative TM regions, hMC4R and hMC2R show overall identities of 53% identity. However, NDP-MSH is a potent agonist for MC4R but not for MC2R. Because the conserved amino acid residues in TM2, TM3, TM4, and TM6 of hMC2R are crucial for ACTH binding and signaling that is similar to that of hMC4R, the chimeric receptor experiment will provide important information of ligand receptor interaction. We anticipate that if substitution of TM of the hMC4R with the corresponding region of the hMC2R results in the decrease NDP-MSH binding affinity and potency, it will imply that non-conserved residues are involved in NDP-MSH binding and potency. In contrary, if substitution does not alter NDP-MSH binding affinity and potency, it will imply that conserved residues are involved in NDP-MSH binding. Our results support our theory that conserved residues in TMs of the hMC4R are crucial for NDP-MSH binding and activity because substitutions of the hMC4R TM regions with the corresponding regions of the hMC2R did not significantly abolish or decrease NDP-MSH binding affinity and potency. To determine which conserved residue of the hMC4R is involved in NDP-MSH binding, we have compared the sequences of hMC4R and hMC2R and found that only 16 amino acids in the TM2, TM3, TM4, TM5, and TM6 are conserved between hMC4R and hMC2R and these amino acids are therefore potential candidates for NDP-MSH binding. Site-directed mutagenesis studies of these conserved amino acid residues were therefore performed. Six among the 13 conserved amino acid residues have been identified to be crucial for NDP-MSH binding and signaling. These residues are Glu100 in TM2, Asp122 and Asp126 in TM3, and Trp258, Phe261, and His264 in TM6 of the hMC4R.
Electrostatic and hydrophobic forces have been proposed to be involved in MSH binding and receptor activation at MCRs and 14 amino acid residues are proposed in ligand receptor interaction (19, 30, 32). In this study, we have utilized chimeric receptors to identify the conserved residues between MC4R and MC2R involved in NDP-MSH binding and signaling. Further mutagenesis studies narrowed down the amino acid residues crucial for NDP-MSH binding and signaling to six. Similar to hMC1R, our results suggest that an ionic binding pocket also existed in hMC4R. However, unlike MC1R, only TM6 of the hMC4R seems to be involved in a hydrophobic binding pocket. Mutations of the TM6 residues, Trp258, Phe261, and His264, decreased NDP-MSH binding affinity and receptor biological activity, whereas the other aromatic residues in TM4 and TM5 of the hMC4R do not seem to play an important role in NDP-MSH binding and receptor biological activity. Our results suggest that hMC4R shares ionic binding sites with hMC1R but have different binding sites from that of hMC1R. We have incorporated these results into the MC4R model and propose that both electrostatic and hydrophobic forces are involved in MSH binding and receptor activation (Fig. 9). An ionic pocket is formed by amino acid residues, Glu100 in TM2, and Asp122 and Asp126 in TM3, which are proposed to form an ionic interaction with Arg8 of NDP-MSH. A hydrophobic binding pocket is formed by three hydrophobic receptor residues, Trp258, Phe261, and His264 in TM6, which are proposed to form aromatic-aromatic interactions with Trp9 of NDP-α-MSH.
FIGURE 9. Two-dimensional representation of a proposed three-dimensional model illustrating the synthetic melanocortin NDP-MSH docked inside the hMC4R.
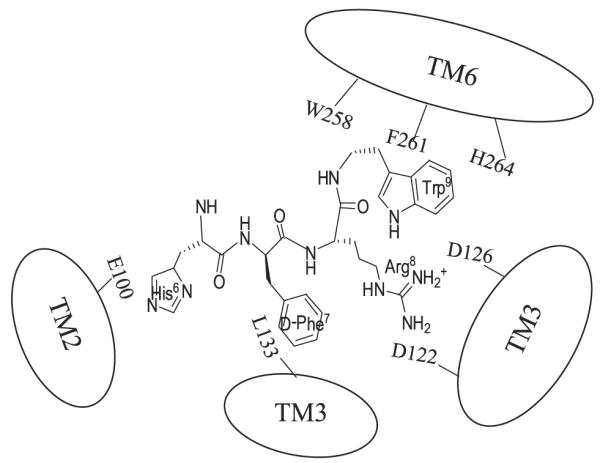
Two receptor binding pockets are hypothesized. The first is a predominantly ionic pocket formed by Glu100, Asp122, and Asp126. The second hydrophobic pocket is formed by aromatic residues Trp258, Phe261, and His264 in TM6.
In conclusion, we have identified that Glu100 in TM2, Asp122 and Asp126 in TM3, and Trp258, Phe261, and His264 in TM6 are important for NDP-MSH binding and signaling. These results provide important information about the molecular determinants of the hMC4R responsible for NDP-MSH binding and signaling.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants R03 HD047312-01A1 (to Y. Y.) and United States Public Health Service, National Institutes of Health Grant DK17420 (to V. H.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
2 The abbreviations used are: hMC4R, human melanocortin-4 receptor; MCR, melanocortin receptor; NDP-MSH, D-[Nle4,Phe7]-α-melanocyte-stimulating hormone; TM, transmembrane domains; FACs, flow cytometry; WT, wild type; ACTH, adrenocorticotropic hormone.
REFERENCES
- 1.Kopelman PG. Nature. 2000;404:635–643. doi: 10.1038/35007508. [DOI] [PubMed] [Google Scholar]
- 2.Flegal KM, Carroll MD, Ogden CL, Johnson CL. J. Am. Med. Assoc. 2002;288:1723–1727. doi: 10.1001/jama.288.14.1723. [DOI] [PubMed] [Google Scholar]
- 3.Allison DB, Fontaine KR, Manson JE, Stevens J, VanItallie TB. J. Am. Med. Assoc. 1999;282:1530–1538. doi: 10.1001/jama.282.16.1530. [DOI] [PubMed] [Google Scholar]
- 4.Zimmermann-Belsing T, Feldt-Rasmussen U. Endocrinology. 2004;145:1501–1502. doi: 10.1210/en.2004-0078. [DOI] [PubMed] [Google Scholar]
- 5.Rugg K. Nurs. Times. 2004;100:28–30. [PubMed] [Google Scholar]
- 6.Kowalski TJ. Appetite. 2004;42:11–14. doi: 10.1016/j.appet.2002.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Damcott CM, Moffett SP, Feingold E, Barmada MM, Marshall JA, Hamman RF, Ferrell RE. Metabolism. 2004;53:303–309. doi: 10.1016/j.metabol.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 8.Geller F, Reichwald K, Dempfle A, Illig T, Vollmert C, Herpertz S, Siffert W, Platzer M, Hess C, Gudermann T, Biebermann H, Wich-mann HE, Schafer H, Hinney A, Hebebrand J. Am. J. Hum. Genet. 2004;74:572–581. doi: 10.1086/382490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lubrano-Berthelier C, Cavazos M, Dubern B, Shapiro A, Stunff CL, Zhang S, Picart F, Govaerts C, Froguel P, Bougneres P, Clement K, Vaisse C. Ann. N. Y. Acad. Sci. 2003;994:49–57. doi: 10.1111/j.1749-6632.2003.tb03161.x. [DOI] [PubMed] [Google Scholar]
- 10.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O'Rahilly S. N. Engl. J. Med. 2003;348:1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 11.Farooqi IS, Yeo GS, Keogh JM, Aminian S, Jebb SA, Butler G, Cheetham T, O'Rahilly S. J. Clin. Investig. 2000;106:271–279. doi: 10.1172/JCI9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 13.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 14.Giraudo SQ, Billington CJ, Levine AS. Brain Res. 1998;809:302–306. doi: 10.1016/s0006-8993(98)00837-3. [DOI] [PubMed] [Google Scholar]
- 15.Cone RD. Trends Endocrinol. Metab. 1999;10:211–216. doi: 10.1016/s1043-2760(99)00153-8. [DOI] [PubMed] [Google Scholar]
- 16.Fisher SL, Yagaloff KA, Burn P. Int. J. Obes. Relat. Metab Disord. 1999;23(Suppl 1):54–58. doi: 10.1038/sj.ijo.0800796. [DOI] [PubMed] [Google Scholar]
- 17.Fong TM, Mao C, MacNeil T, Kalyani R, Smith T, Weinberg D, Tota MR, Van der Ploeg LH. Biochem. Biophys. Res. Commun. 1997;237:629–631. doi: 10.1006/bbrc.1997.7200. [DOI] [PubMed] [Google Scholar]
- 18.Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 19.Yang YK, Fong TM, Dickinson CJ, Mao C, Li JY, Tota MR, Mosley R, Van Der Ploeg LH, Gantz I. Biochemistry. 2000;39:14900–14911. doi: 10.1021/bi001684q. [DOI] [PubMed] [Google Scholar]
- 20.Yang Y, Chen M, Lai Y, Gantz I, Georgeson KE, Harmon CM. J. Biol. Chem. 2002;277:20328–20335. doi: 10.1074/jbc.M201343200. [DOI] [PubMed] [Google Scholar]
- 21.Haskell-Luevano C, Cone RD, Monck EK, Wan YP. Biochemistry. 2001;40:6164–6179. doi: 10.1021/bi010025q. [DOI] [PubMed] [Google Scholar]
- 22.Fleck BA, Chen C, Yang W, Huntley R, Markison S, Nickolls SA, Foster AC, Hoare SR. Biochemistry. 2005;44:14494–14508. doi: 10.1021/bi051316s. [DOI] [PubMed] [Google Scholar]
- 23.Hogan K, Peluso S, Gould S, Parsons I, Ryan D, Wu L, Visiers I. J. Med. Chem. 2006;49:911–922. doi: 10.1021/jm050780s. [DOI] [PubMed] [Google Scholar]
- 24.Pogozheva ID, Chai BX, Lomize AL, Fong TM, Weinberg DH, Nargund RP, Mulholland MW, Gantz I, Mosberg HI. Biochemistry. 2005;44:11329–11341. doi: 10.1021/bi0501840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang YK, Ollmann MM, Wilson BD, Dickinson C, Yamada T, Barsh GS, Gantz I. Mol. Endocrinol. 1997;11:274–280. doi: 10.1210/mend.11.3.9898. [DOI] [PubMed] [Google Scholar]
- 26.Yang YK, Dickinson C, Lai YM, Li JY, Gantz I. Am. J. Physiol. 2001;281:R1877–R1886. doi: 10.1152/ajpregu.2001.281.6.R1877. [DOI] [PubMed] [Google Scholar]
- 27.Yang YK, Thompson DA, Dickinson CJ, Wilken J, Barsh GS, Kent SB, Gantz I. Mol. Endocrinol. 1999;13:148–155. doi: 10.1210/mend.13.1.0223. [DOI] [PubMed] [Google Scholar]
- 28.Baldwin JM. EMBO J. 1993;12:1693–1703. doi: 10.1002/j.1460-2075.1993.tb05814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang YK, Dickinson CJ, Zeng Q, Li JY, Thompson DA, Gantz I. J. Biol. Chem. 1999;274:14100–14106. doi: 10.1074/jbc.274.20.14100. [DOI] [PubMed] [Google Scholar]
- 30.Yang Y, Dickinson C, Haskell-Luevano C, Gantz I. J. Biol. Chem. 1997;272:23000–23010. doi: 10.1074/jbc.272.37.23000. [DOI] [PubMed] [Google Scholar]
- 31.Conrad DM, Hanniman EA, Watson CL, Mader JS, Hoskin DW. J. Cell. Biochem. 2004;92:387–399. doi: 10.1002/jcb.20064. [DOI] [PubMed] [Google Scholar]
- 32.Haskell-Luevano C, Sawyer TK, Trumpp-Kallmeyer S, Bikker JA, Humblet C, Gantz I, Hruby VJ. Drug Des. Discov. 1996;14:197–211. [PubMed] [Google Scholar]
- 33.Mizoue LS, Bazan JF, Johnson EC, Handel TM. Biochemistry. 1999;38:1402–1414. doi: 10.1021/bi9820614. [DOI] [PubMed] [Google Scholar]
- 34.Kristiansen K. Pharmacol. Ther. 2004;103:21–80. doi: 10.1016/j.pharmthera.2004.05.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.