

Abstract
Murine oligodendrocytes express the gap junction (GJ) proteins connexin32 (Cx32), Cx47, and Cx29. CNS phenotypes in patients with X-linked Charcot-Marie-Tooth disease may be caused by dominant effects of Cx32 mutations on other connexins. Here we examined the expression of Cx31.3 (the human ortholog of murine Cx29) in human brain and its relation to the other oligodendrocyte GJ proteins Cx32 and Cx47. Furthermore, we investigated in vitro whether Cx32 mutants with CNS manifestations affect the expression and function of Cx31.3. Cx31.3 was localized mostly in the gray matter along small myelinated fibers similar to Cx29 in rodent brain and was coexpressed with Cx32 in a subset of human oligodendrocytes. In HeLa cells Cx31.3 was localized at the cell membrane and appeared to form hemichannels but no GJs. Cx32 mutants with CNS manifestations were retained intracellularly, but did not alter the cellular localization or function of co-expressed Cx31.3. Thus, Cx31.3 shares many characteristics with its ortholog Cx29. Cx32 mutants with CNS phenotypes do not affect the trafficking or function of Cx31.3, and may have other toxic effects in oligodendrocytes.
Keywords: CMT1X, connexin29, gap junctions, protein trafficking, hemichannels
Introduction
Gap junctions (GJs) are channels that allow the diffusion of ions and small molecules between apposed cell membranes. Vertebrate GJs are formed by a family of molecules called connexins, comprised of about 20 different genes in mammals (Willecke et al. 2002). Six connexin molecules form a hemichannel and two apposed hemichannels form a functional channel. Rodent oligodendrocytes express at least three connexins - connexin32 (Cx32) (Scherer et al. 1995), Cx47 (Kleopa et al. 2004; Menichella et al. 2003; Odermatt et al. 2003) and Cx29 (Altevogt et al. 2002). Each one has a distinct expression pattern at the cellular and subcellular level; this may differ between different types of oligodendrocytes (Altevogt and Paul 2004; Kamasawa et al. 2005; Kleopa et al. 2004; Li et al. 2004). Connexins may have overlapping functions in oligodendrocytes, since mice deficient for either Cx32 or Cx47 have minimal or no pathology in the CNS, whereas mice lacking both Cx32 and Cx47 develop severe CNS demyelination (Menichella et al. 2003; Menichella et al. 2006; Odermatt et al. 2003; Scherer et al. 1998). Cx32 and Cx47 mediate the GJ coupling of astrocytes to oligodendrocytes (A:O coupling), which may serve the spatial buffering of K+ that is elaborated during the propagation of action potentials (Altevogt and Paul 2004; Kamasawa et al. 2005; Nagy et al. 2003b; Rash et al. 2001). Cx29 is coexpressed with Cx32 and Cx47 in oligodendrocytes especially in the gray matter. In contrast to Cx32, Cx29 is mostly localized at the adaxonal membrane and does not appear to form GJs, but may instead form hemichannels (Altevogt et al. 2002).
Mutations in the GJB1/Cx32 gene encoding Cx32 cause the X-linked form of Charcot-Marie-Tooth disease (CMT1X), a demyelinating peripheral neuropathy (Bergoffen et al. 1993) (http://molgen-www.uia.ac.be/ CMTMutations/). A subset of these mutations are also associated with acute or chronic clinical CNS manifestations, including spasticity, hyperactive reflexes, extensor plantar responses, or even acute encephalopathy with reversible white matter abnormalities (Kleopa and Scherer 2006; Kleopa et al. 2002; Paulson et al. 2002; Taylor et al. 2003). Compensatory effects by other oligodendrocytic connexins expressed may account for the lack of such CNS phenotypes in most CMT1X patients. Expression studies of several CMT1X mutations have shown that many of the mutated proteins, including all the ones known to cause CNS phenotypes, are retained intracellularly in the Golgi apparatus or endoplasmic reticulum (ER), with reduced or absent GJ formation (Deschênes et al. 1997; Kleopa et al. 2002; Kleopa et al. 2006; Oh et al. 1997; Omori et al. 1996; VanSlyke et al. 2000; Yum et al. 2002). Patients who have a GJB1/Cx32 deletion do not appear to have a CNS phenotype (Hahn et al. 2000), indicating that loss of Cx32 function alone is not detrimental to oligodendrocytes. Cx32 mutations that cause clinical CNS manifestations are therefore likely to cause abnormal gain of function, for example through trans-dominant negative effects on other human oligodendrocytic connexins. There are precedents for connexin mutants having such gain of function: Cx32 mutations that cause CMT1X caused impairment of GJ function of the coexpressed wild type Cx32 in transfected cells (Omori et al. 1996). Expression of the Golgi-retained R142W Cx32 mutation in myelinating Schwann cells of transgenic mice resulted in decreased levels of wild type mouse Cx32 and caused demyelination (Jeng et al. 2006). Furthermore, dominant Cx26 mutations that cause hearing loss suppressed channel activity of coexpressed wild type Cx26, but only those mutants additionally causing skin disease inhibited intercellular conductance of co-expressed wild type Cx43 (Richard et al. 1998; Rouan et al. 2001). Here we describe the expression of Cx31.3, the human ortholog of Cx29 (Altevogt et al. 2002), and its relation to the other oligodendrocyte GJ proteins Cx32 and Cx47. Furthermore, we investigated in vitro whether Cx32 mutants with CNS manifestations affect the expression and function of Cx31.3.
Materials and Methods
Cx31.3 and Cx32 expression constructs
Human GJB1/Cx32 mutations were generated by site-directed mutagenesis using the QuickChange kit (Stratagene, La Jolla, CA) with mutagenic oligonucleotide primers and PfuTurbo DNA polymerase as previously described (Kleopa et al. 2002; Yum et al. 2002). Mutated inserts were subcloned into the pREP9 or pcDNA vectors with the G418 resistance gene (Invitrogen). For the Cx31.3 construct, we used primers developed from a human Cx31.3 gene sequence (GenBank accession number AY297109) to amplify the entire putative open reading frame of Cx31.3 from human brain RNA (Clontech, Mountain View, CA) using RT-PCR (Superscript II, Invitrogen, Carlsbad, CA), adding AflII and BamHI restriction sites at the 5′ and 3′ ends, respectively: 5′-CAGGATGTGTGGCAGGTTC-3′ and 5′-TCAGGCATCTCTGGGTCCA-3′. The PCR product was cloned into the pIRESpuro3 vector (Clontech, Mountain View, CA) and sequenced.
Generation of the Cx31.3 antisera
Peptides from the intracellular loop domain (sequence: EEETLIQGREGNTDVPGAGSL, corresponding to amino acids 111-131) and the carboxy-terminus (sequence: STRRHKKATDSLPVVETKEQF, corresponding to amino acids 237-257) of Cx31.3 were synthesized. BLAST database searches showed that these sequences were unique to Cx31.3. Peptides were conjugated to the carrier protein keyhole limpet hemocyanin (KLH) (Synpep, Dublin, CA) via a cysteine residue added at the C-terminus. Polyclonal antisera from two rabbits per peptide were developed using incomplete Freund's adjuvant at Covance Research Products (Denver, PA). Each antiserum was screened by immunostaining HeLa cells expressing Cx31.3. Only the antisera against the carboxy-terminus of Cx31.3 provided specific binding and were used for all the experiments described in this report.
Cell transfections
Communication-incompetent HeLa cells (originally obtained from Prof. Klaus Willecke, University of Bonn) were grown in low-glucose Dulbecco's modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 μg/ml penicillin/streptomycin) in a humidified atmosphere containing 5% CO2 at 37°C to achieve 80% confluency the day of transfection. For permanent transfection, FuGENE 6 transfection Reagent (Roche) and DNA containing either Cx31.3, wild type human Cx32, or the relevant Cx32 mutations were incubated in Optimem (Gibco-BRL) (ratio 6:1). Cells transfected with Cx32 were selected with 1 mg/ml G418 (Gibco-BRL) and cells transfected with Cx31.3 with puromycin (0.5 μg/ml). After 3 weeks resistant colonies with stable growth were obtained and clonal cells were bulk selected. Untransfected HeLa cells treated with G418 or puromycin did not survive. Cx31.3 were further transfected with mutant or wild type Cx32 constructs and underwent double selection for expression of both Cx32 and Cx31.3 using a combination of G418 (1 mg/ml) and puromycin (0.5 μg/ml). Transient transfections of Neuro2a cells were performed using Lipofectamine 2000 or Lipofectamine LTX (Gibco-BRL) using 0.5 μg DNA per 18 mm well as per manufacturer's instructions. Cells were transfected with either human Cx26 (amplified from genomic DNA by PCR and sequenced) or Cx31.3, both cloned into pIRES2-EGFP, or with vector alone as control.
Immunocytochemistry
HeLa cells were plated in 4-chamber slides (Nalge Nunc International), washed in phosphate buffered saline (PBS), fixed in acetone at -20°C for 10 min, blocked with 5% bovine serum albumin (BSA) in PBS containing 0.1% Triton for 1 hr at room temperature (RT) and incubated with primary antibodies overnight at 4°C. For Cx32 staining, a rabbit antiserum against the intracellular loop (Chemicon; diluted 1:500), and a mouse monoclonal antibody (7C6.C7) against the carboxy-terminus (diluted 1:2) (Li et al. 1997) were used. The rabbit antisera against the carboxy-terminus of Cx31.3 (1:800) were used to label cells expressing Cx31.3. A rabbit antiserum against COPII (Abcam, 1:500) was used to label vesicles that shuttle between the ER and Golgi; a mouse antibody against calnexin (Abcam, 1:100) was used as an ER marker; a mouse antibody against pan-cadherins (Abcam, 1:500) was used as a cell membrane marker; and a mouse antibody against the 58 kDa protein (Abcam, 1:50) was used as Golgi marker. After washing in PBS, cells were incubated with fluorescein- and rhodamine-conjugated donkey cross-affinity purified secondary antibodies (1:100; Jackson ImmunoResearch, West Grove, PA) for 1 hr at RT. Cell nuclei were visualized with 4′,6′-diamidino-2-phenylindole (DAPI; Sigma-Aldrich). Slides were covered with mounting medium (Dako) and images were photographed under a Zeiss fluorescence microscope with a digital camera using the Zeiss Axiovision software (Carl Zeiss MicroImaging, Germany). To confirm co-localization, some images were taken with a Leica DMR confocal microscope (Leica DMR, Microsystems, Heidelberg).
Scrape loading experiments
Confluent HeLa cells were used for scrape loading. Lucifer Yellow (4%) dissolved in PBS (Sigma, MW: 443, 2-) was added in the dark after washing with PBS and a grid was cut into the dish with a scalpel blade. Cells were incubated for 5 min, then washed in Hank's Balanced Salt Solution (HBSS) containing calcium to stop GJ communication and pictures were taken under a Nikon Eclipse TE2000-U microscope using a Nikon digital Camera DXM 1200F. For neurobiotin scrape loading, cells were rinsed in HBSS without calcium or magnesium, 1% neurobiotin (Vector Laboratories, MW: 287, 1+; diluted in HBSS) was added, cells were scraped and incubated for 5 min, washed in HBSS, fixed in 4% paraformaldehyde (PFA) for 10 min, washed in PBS, blocked in 5% BSA with 0.1% Triton X-100 for 30 min at RT, incubated in streptavidin-rhodamine (Vector Laboratories, diluted 1:300) at RT for 1 hour, counterstained with DAPI and imaged as above. For quantification of gap junctional connectivity, we counted the number of fluorescent cells outside the scrape line in a 100×150μ rectangle. For each tracer, we calculated the mean ± standard error (SE) for 3 independent experiments and compared the results with ANOVA and Bonferroni's post test.
Neurobiotin uptake assay
HeLa cells permanently expressing Cx31.3 were grown in 24-well dishes and transiently transfected with selected Cx32 constructs (including wild type Cx32, T55I and R75W mutants) cloned in the pIRES-EGFP vector, to ensure coexpression of Cx31.3, Cx32 and GFP in a subset of cells. Two days after transfection cells were washed in PBS lacking divalent ions and incubated with 2% neurobiotin for 30 min, then washed and fixed for 10 minutes in 4% PFA at 4°C. After blocking in 5% BSA with 0.1% Triton X-100 for 30 min at RT, cells were incubated in streptavidin-Texas red (Vector Laboratories, 1:1000) at RT for 1 hour, counterstained with DAPI and imaged as above. To determine whether the neurobiotin uptake was specifically through hemichannels, we performed the same experiments in the presence of 2 mM octanol (Sigma-Aldrich), a hemichannel blocker, diluted in Optimem for 10 min before and during incubation with neurobiotin. For quantitative analysis of neurobiotin uptake with or without addition of octanol in untransfected or Cx31.3 expressing cells the mean red channel pixel intensity was measured in images captured with constant time exposures using the ImageJ software (NIH image) and at least 50 cells in each condition were compared. In addition, we compared mean red channel pixel intensity in at least 50 GFP+ and GFP- cells in Cx31.3 clonal cells transfected with each of the Cx32 constructs in order to determine whether coexpression of Cx32 mutants affects neurobiotin uptake. Statistical significance was examined using ANOVA with Bonferroni's post test.
Calcein Blue efflux
Transfected Neuro2a cells were plated on 10mm diameter cover slips. Twenty four hours after plating they were loaded with Calcein Blue AM as follows. The cover slip was removed from serum-containing medium, rinsed once in normal Tyrode solution and then incubated for 5 minutes in 0.5 ml of a freshly-prepared Tyrode solution containing 4 μg of Calcein Blue AM per ml. The cover slip was then immediately transferred to the stage bath on a Nikon E600FN upright microscope equipped with epifluorescence illumination. Tyrode solution in the bath had no added Ca++ and 2 mM Mg++. For hemichannel blockade octanol was added at a concentration of 16μl per 50 ml of Tyrode solution. Filters used for visualizing Calcein Blue consisted of a 360nm excitation and 460nm emission. A neutral density filter was used to minimize exposure of cells to light. Time lapse imaging was performed to sample fluorescence intensity: 300 msec exposure every 5 minutes for a 20 minute period. Images were acquired and analyzed using a Retiga 2000R digital camera (QImaging) and IP Lab software. Data was analyzed to obtain a mean change in fluorescence intensity for the aggregate of all cells in the field of view and expressed as a %ΔF/Finitial. Statistical significance was examined with ANOVA with Bonferroni's post test.
Recording from transfected mammalian cell lines
Patch clamping in the dual whole cell, single whole-cell, on-cell, and outside-out configurations were performed as previously described (Abrams et al. 2003). Recording solutions: pipette solution, 145 mM CsCl, 5 mM EGTA, 0.5 mM CaCl2, 10.0 mM HEPES pH 7.2; standard bath solution: 150 mM NaCl, 4 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM Dextrose, 2 mM Pyruvate, 10 mM HEPES; pH 7.4. All baths for metabolic inhibition (MI) contained 10 μM carbonyl cyanide p-(trifluoro-methoxy)phenyl-hydrazone (FCCP, Sigma) and 1mM Iodoacetic acid (IAA, Sigma). Both solutions for preincubation to achieve MI contained minimal or no added Ca2+: either 150 mM NaCl, 4 mM KCl, 2 mM MgCl2, 10 mM HEPES; or 150 mM NaCl, 4 mM KCl, 0.1 mM CaCl2 2mM EGTA,1mM EDTA, 7mM Sucrose 10 mM HEPES; pH 7.4 were used. No difference was seen between these two bath solutions, so the results were combined for analysis. The latter solution was always used for perfusion to induce MI. Unless otherwise noted input resistance (Rinput) was determined by measuring instantaneous Im responses to Vm pulses from -55 to -90mV and applying Ohm's law. Values are presented as mean ± SEM, and were compared using Kruskal-Wallis test with Dunn's post-test for multiple comparisons. Activation curves were determined using a holding potential of -60 mV with one 12.5 second pulse from -80 to +80 applied each 30 second cycle.
Immunohistochemistry of postmortem human brain
Postmortem human brain tissue samples, either snap-frozen or fixed up to 4 hours in 4% PFA were provided by the UK MS Brain Tissue Bank. All tissues have been collected following fully informed consent by the donors via a prospective donor scheme according to Ethics Committee approval. Sections were permeabilized in cold methanol for 10 min, washed 3 times in PBS, blocked in normal serum for 45 min and incubated overnight at 4°C with rabbit antisera against Cx31.3 (1:500), Cx32 (Chemicon, 1:500), or aspartoacylase (ASPA; gift of Dr. Jim Garbern, Wayne State University, 1:1,000), an oligodendrocyte marker (Madhavarao et al. 2005). After washing, sections were incubated with biotinylated secondary anti-rabbit antibody (Vector Laboratories, 1:200) for 1 hr at RT, followed by streptavidin-Texas Red (1:800). All sections for double immunofluorescence were subsequently blocked with biotin/streptavidin (Vector Laboratories) for 15 min and then incubated overnight at 4°C with mouse antibodies against Cx47 (Invitrogen, diluted 1:500), neurofilament light (NFL)/SMI31 (Abcam, 1:5,000), Cx32 (7C6.C7, 1:2) myelin basic protein (MBP; Abcam, 1:500), Caspr (gift of Dr. Elior Peles, Weizmann Institute of Science; 1:50), myelin associated glycoprotein (MAG; clone 513; Boehringer Mannheim; 1:100), or myelin oligodendrocyte glycoprotein (MOG, 1:200; clone Z12, S. Piddlesden, University of Cardiff). After washing, sections were incubated with biotinylated secondary anti-mouse antibody (1:200) for 1 hr followed by streptavidin-FITC (1:800) and counterstained with DAPI. Finally, they were treated with an autofluorescence eliminator reagent (Chemicon) for 10 min and imaged as above. To prove the specificity of Cx31.3 antiserum labeling, sections were stained in parallel after preincubation of the antiserum with the immunogenic peptide.
Immunoblots
HeLa or Neuro2a cells harvested in cold PBS or frozen postmortem human brain tissue samples were lysed in ice-cold NP40 buffer (0.5% NP-40, 10 mM Tris-HCl, pH 7.4, 2 mM EDTA, 100 mM NaCl and 0.017 mg/ml phenylmethylsulfonyl fluoride (Sigma), followed by a brief sonication on ice with a dismembrator (Fisher Scientific). Proteins (100 μg) from cells and tissue lysates were fractionated by 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a Hybond-C extra membrane (Amersham). Membranes were blocked with 5% non-fat milk in Tris-buffered saline containing 0.1% Tween 20 (TBS-T) for 1 hr at RT and incubated with the rabbit anti-Cx31.3 antiserum (diluted 1:10,000) in 5% milk-TBS-T at 4°C overnight. After washing, the blots were incubated with an anti-rabbit-horseradish peroxidase conjugated secondary antibody (Jackson ImmunoResearch, diluted 1:10,000) in 5% milk-TBS-T for 1 hour. The bound antibody was detected with enhanced chemiluminescence (ECL; Amersham). To test the specificity of the rabbit Cx31.3 antisera, blots were processed in parallel after preincubation of the corresponding antiserum with the cognate peptide that was used to immunize animals at RT for 1 hr.
Results
The cellular localization of Cx31.3 in transfected cells
We generated two antisera raised against the C-terminus of Cx31.3. Immunoblot analysis of HeLa cells stably expressing Cx31.3 using the C-terminus antisera showed a ∼30 kDa band that was not present in parental HeLa cells. Furthermore, this band was strongly attenuated by preincubating these antisera with the peptide used as the immunogen (Fig. 1A). Both antisera labeled cells stably expressing Cx31.3, but not parental HeLa cells or cells stably expressing Cx32 (Suppl. Fig. 1). Cx31.3-immunoreactivity in the transiently transfected HeLa cells appeared to be localized to the cell membrane, especially apposed cell membranes, but GJ plaques were not seen. Cx31.3 was similarly localized in HeLa cells that stably expressed Cx31.3 as further demonstrated by its similarity to cadherin staining at the cell membrane, and its distinction from calnexin, which was localized intracellularly (Fig. 2).
Fig. 1.
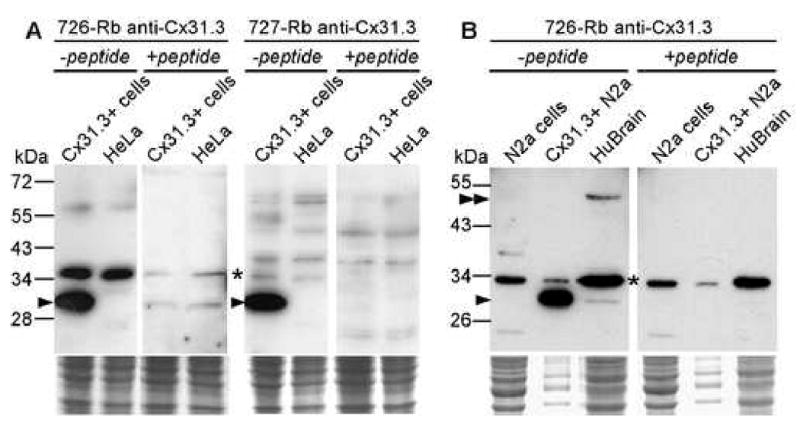
Immunoblot analysis of Cx31.3. A. Fractionated lysates from parental or cloned HeLa cells that stably express Cx31.3 were hybridized with one of two rabbit antisera against a peptide derived from the carboxy-terminus of Cx31.3 as indicated. Both antisera detect a specific band around 30 kDa (arrowhead) in Cx31.3-expressing, but not in parental HeLa cells, as well as a prominent non-specific band (asterisk) found in both transfected and untransfected cells. The specific band is absent when each antiserum is preincubated with the peptide from the Cx31.3 carboxy-terminus that was used as immunogen, confirming the specificity of Cx31.3 labeling. B. Similar findings using lysates of Cx31.3-expressing and parental Neuro2a cells, as well as human brain protein. The same size band found in transfected Neuro2a cells is also weakly present in human brain (arrowhead), along with a stronger band (double arrowhead) at around 50 kDa, possibly representing the dimer. The monomer and the potential dimer bands are absent after preincubation with the immunogenic peptide, in contrast to non-specific bands (asterisk). The lower panels are images of the Coomassie-stained gels following transfer, and document that each lane contained a similar amount of protein.
Fig. 2.
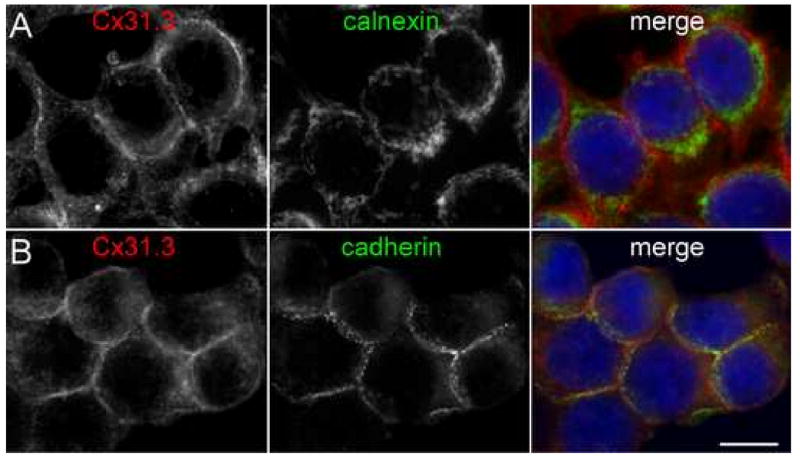
Cx31.3 is localized to cell membranes of transfected cells. These are images of cloned HeLa cells that stably express Cx31.3, immunostained with a rabbit antiserum against Cx31.3 (red) and a mouse monoclonal antibody (green) against either calnexin (A) or “pan-cadherins” (B). Cx31.3-immunoreactivity is localized to the cell membrane, largely external to calnexin-immunoreactivity (marking the ER; A), but similar to cadherins labeling at the cell membrane (B). Scale bar: 10 μm.
Functional properties of Cx31.3
The lack of GJ plaques in Cx31.3-expressing cells suggested that it does not form functional channels. To examine this possibility, we performed scrape-loading of cells that stably expressed Cx31.3, as well as clonal cells that stably expressed wild type Cx32 and parental HeLa cells. In this assay (el-Fouly et al. 1987), cells are wounded with a razor or scalpel blade in the presence of GJ-permeant dye, such as Lucifer Yellow (443 Da) or Neurobiotin (387 Da). Both Lucifer Yellow and neurobiotin diffused to neighboring cells away from the scrape line in cells expressing Cx32, but not in cells expressing Cx31.3 or in parental cells. Identical results were obtained in 3 independent experiments. To quantify these results, we counted the number of fluorescent cell bodies in a rectangle on one side of the scrape line (Suppl. Fig. 2). The number of fluorescent cells was significantly higher in Cx32-expressing cells than in Cx31.3-expressing or untransfected parental cells, whereas Cx31.3-expressing cells did not significantly differ from parental HeLa cells (Suppl. Fig. 2). Because dual whole cell patch clamping is a more sensitive assay for GJ coupling, we examined the coupling between isolated pairs of cells expressing the connexin of interest. Previous work on mCx29, the homologue of Cx31.3, showed that expression of this connexin failed to induce junctional coupling when assayed by dual whole cell patch clamp (Altevogt et al. 2002). Similarly, mammalian cells transfected with Cx31.3 show no junctional coupling above that attributable to the low expression of endogenous connexin. Four of eleven Cx31.3 expressing cell pairs showed low but measurable coupling (average 56±38 pS); this was similar to that seen in cells transfected with empty vector (three of six cell pairs showed low levels of coupling, average 105±87 pS). In addition, all three pairs examined expressing human Cx26 were well coupled. Thus, Cx31.3 does not appear to form functional GJs, at least in cultured cells.
Since Cx31.3 localized to the plasma membrane, but failed to form morphologic or functional GJs, we undertook to examine whether Cx31.3 induces formation of functional hemichannels, such as have been reported for a number of other connexins (González et al. 2006; Trexler et al. 1996; Valiunas 2002; Valiunas and Weingart 2000). As shown in Suppl. Fig. 3A, the input resistances (measured in Ca2+ and Mg2+ containing bath solution as described in the Methods) of Neuro2a cells expressing Cx31.3 are similar to cells transfected with the empty vector as control, suggesting that expression of Cx31.3 did not lead to substantial hemichannel activity at voltages near the resting potential. In addition, extensive examination using whole cell recordings at a variety of holding potentials (n=15) showed no channel activity in the Cx31.3 cells beyond that demonstrable in control cells. On the other hand, cells transfected with a vector containing the gene for human Cx26, a connexin shown to produce functional hemichannel currents (González et al. 2006), showed a significant reduction in input resistance (Suppl. Fig. 3A) compared with the Cx31.3 expressing cells (p<0.05) and control (p<0.01). Unitary conductance changes similar to those previously reported for human Cx26 were seen in 8/8 cells transfected with Cx26 and having input resistances ≥ 0.6 GΩ.
Because low calcium has been reported to activate connexin hemichannel activity (Sáez et al. 2005), we examined the effect of removing calcium and magnesium from the bath solution. 15 cells expressing Cx31.3 and 6 control cells were examined in whole cell configuration with standard bath solution. Perfusion with divalent free bath solution was accompanied by the appearance of 120 pS unitary conductance transitions in 6 of 15 Cx31.3 expressing cells and in 2 of 6 control cells, but no activity unique to Cx31.3 transfected cells was seen. We then examined whether metabolic inhibition (MI) induced opening of Cx31.3 hemichannels. As shown in Suppl. Fig. 3A columns four and five, the input resistances of control and Cx31.3 cells preincubated with FCCP and IAA (as described in the methods) were virtually identical, though much lower than those for cells not undergoing MI. We also examined the effect of MI after establishment of whole cell configuration (Suppl. Fig. 3A columns six and seven) and found no significant difference between conductance induced in control and Cx31.3 cells. Unfortunately, the low input resistances of these cells made it impossible to completely rule out low levels of unique activity in the Cx31.3 transfected cells in the whole cell configuration. For this reason we also examined cell-attached and outside–out patches from control and Cx31.3 transfected cells pre-incubated to induce MI (n=5 for each recording configuration, for control and for Cx31.3 expressing). No channel activity unique to the Cx31.3 expressing cells was seen.
If hemichannels have mean open times much shorter than 1/(filter cutoff frequency), their unitary conductance changes may not be detected. However, they would still contribute to the mean current. The similar input resistances of Cx31.3 transfected and control cells suggested that the contribution of Cx31.3 hemichannels to membrane conductance at large negative voltages was insignificant. We then proceeded to examine whether depolarization could activate macroscopic voltage activated membrane currents. With 1.8 mM Ca2+ in the bath similar fractions of Cx31.3 expressing and control cells show activating currents at voltages greater than +40 mV. In Ca2+ free bath solution, the mean current activated in Cx31.3 expressing cells with pulses smaller than 60 mV is similar to that in control cells (Suppl. Fig. 3B). Mean currents at +80 mV rises 274+/-87 pA for Cx31.3 expressing cells and 121+/-34 for control cells. However, this difference is fully accounted for by large currents in 6 of the 40 Cx31.3 expressing cells examined and is not statistically significant. Thus, there is no clear electophysiologic evidence for functional Cx31.3 hemichannels.
The failure to demonstrate Cx31.3 mediated hemichannel currents could be due to a low efficiency of formation combined with low open probability. Thus, to further test whether Cx31.3 forms functional hemichannels in vitro, we incubated living cells for 30 minutes with neurobiotin in the presence or absence of 2 mM octanol, then fixed the cells and visualized them with streptavidin-Texas red. As shown in Figure 3, clonal cells permanently expressing Cx31.3 showed uptake of neurobiotin, which was at least in part blocked by octanol, a hemichannel inhibitor, and was significantly higher than in parental HeLa cells. To quantify these results, we measured the red channel pixel intensity in at least 50 cells in each condition, and found that Cx31.3+ cells had a higher red fluorescence pixel intensity of 39.74 ± 5.36 (mean ± standard deviation) compared to parental HeLa (20.36 ± 2.73) or Cx31.3+ cells treated with octanol (21.35 ± 2.39). This difference was statistically significant (p<0.001 for both comparisons with ANOVA; Fig. 3E). In addition, Neuro2a cells expressing Cx31.3 or transfected with empty vector were pre-loaded with the AM ester of Calcein Blue. As shown in Figure 4, the rate of dye loss, as revealed by a decrease in Calcein Blue fluorescence intensity, was approximately twice as fast in Cx31.3 transfected cells than in control cells transfected with pIRES-EGFP alone. Furthermore, addition of octanol to the bath decreased dye loss in Cx31.3 cells below the level found in control cells. This may reflect inhibition by octanol of endogenous as well as Cx31.3 related permeability pathways for Calcein Blue. Although not conclusive, these results are consistent with the idea that Cx31.3 forms hemichannels in the cell membrane.
Fig. 3.
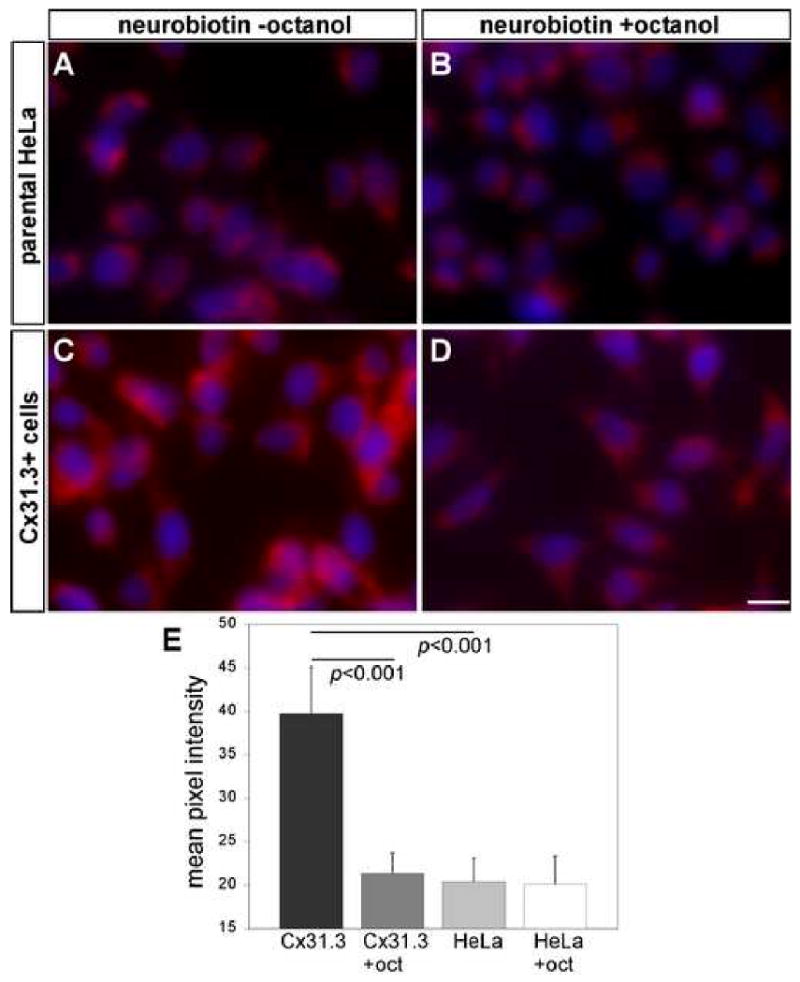
Neurobiotin uptake assay. Clonal HeLa cells that stably express Cx31.3 were incubated in neurobiotin for 30 minutes in the presence or absence of 2 mM octanol, fixed in 4% PFA, and neurobiotin was visualized with streptavidin-Texas red. Compared to parental cells (A), cells stably expressing Cx31.3 showed more Texas red staining (C). Incubating with the hemichannel inhibitor octanol reduced the Texas red staining in cells stably expressing Cx31.3 (D) but not in parental cells (B). To quantify these results, we measured the red channel pixel intensity in at least 50 cells in each condition (E): Cx31.3 expressing cells had a significantly higher pixel intensity (39,74 ± 5,36; mean ± standard deviation) than parental HeLa (20,36 ± 2,73), and octanol significantly reduced the red pixel intensity of Cx31.3-expressing cells (21,35 ± 2,39) compared to untreated cells (p<0.001 for both comparisons).
Fig. 4.
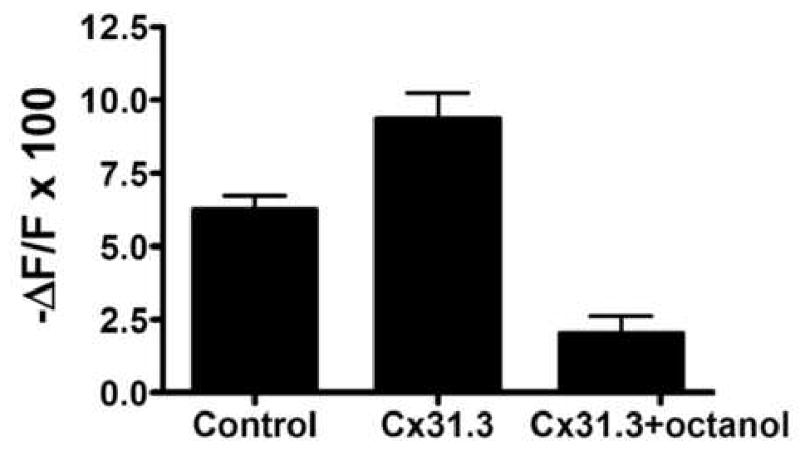
Dye efflux from Neuro2a cells expressing Cx31.3 and controls. Neuro2a cells expressing Cx31.3 or transfected with empty vector were pre-loaded with the AM ester of Calcein Blue. After 20 minutes, the average normalized intensity of cells expressing Cx31.3 dropped 9.4 +/- 0.9% compared with 6.2 +/- 0.5% for control cells (p<0.05). Treatment with octanol decreased the reduction in fluorescence intensity to 2 +/- 0.6%. (p<0.05 compared to Cx31.3 non-octanol treated).
Localization of Cx31.3 in human brain
To determine whether human oligodendrocytes express Cx31.3, we immunostained sections of fixed and unfixed human cortex and subcortical white matter, as well as fixed cervical spinal cord with rabbit antisera against Cx31.3 or Cx32 and mouse monoclonal antibodies against Cx47 or Cx32. In the cerebral cortex (Fig. 5A-I), Cx31.3-immunoreactivity was present along small caliber fibers and in the cytoplasm of oligodendrocytes, whereas Cx47-immunoreactivity was also localized on the cell membrane, sometimes appearing to form GJ plaques (Fig. 5B, Suppl. Fig. 4). Preincubating the Cx31.3 antiserum with the immunogenic peptide abolished the labeling of small myelinated fibers (Suppl. Fig. 5). Cx32-immunoreactivity was sparse and mostly detected along some larger caliber myelinated fibers (Fig.5 E&H). In the subcortical white matter, oligodendrocyte perikarya and proximal processes were Cx47-positive (Fig. 5M), Cx31.3 was not detected (J) and large diameter fibers were Cx32-positive (Fig.5K&N). In the spinal cord (Suppl. Fig. 6) large myelinated fibers in white matter were Cx32-positive and Cx31.3-negative, while small fibers, predominantly in gray matter, were Cx31.3-positive but Cx32-negative. Thus, in spite of the technical challenges in immunostaining human material, the patterns of Cx31.3, Cx32, and Cx47 are similar to what we previously found in rodents (Altevogt et al. 2002; Kleopa et al. 2004): Cx31.3 (Cx29 in rodents) is mainly localized to oligodendrocytes in gray matter and smaller caliber myelinated axons, Cx32 is localized to large myelinated axons, and Cx47 is localized to oligodendrocyte perikarya and proximal processes.
Fig. 5.
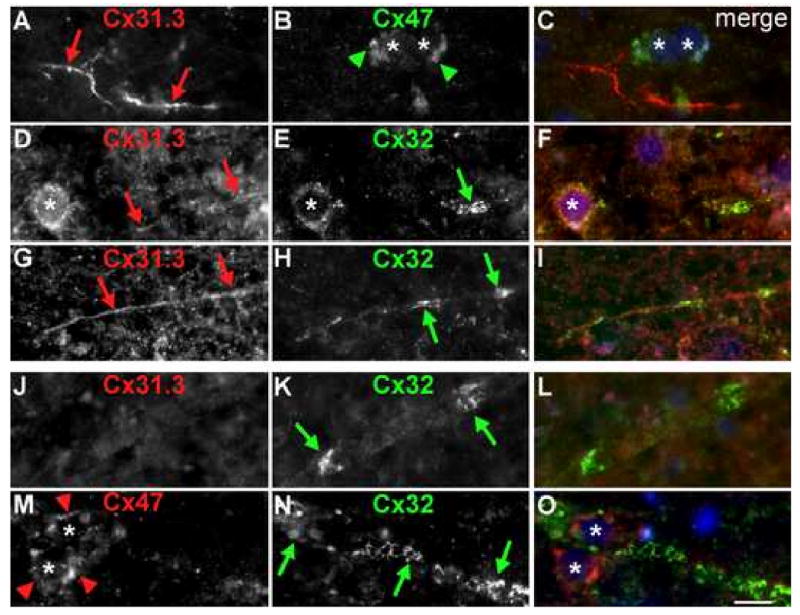
Oligodendrocytic connexins in human brain. These are images of unfixed human brain sections immunostained with combinations of rabbit antisera (red) against Cx31.3 (726) or Cx32, and a mouse monoclonal (green) against Cx32 or Cx47 as indicated and counterstained with DAPI (blue). Merged images are shown in the right column. In the cerebral cortex (A-I), Cx31.3 is expressed along thin fibers (red arrows A, D, G) and diffusely in the cell bodies of oligodendrocytes (asterisk in D). Cx47 is localized to the perikarya and proximal processes of oligodendrocytes possibly forming GJ plaques (green arrowheads in B). Cx32 (E, H) is also expressed by some gray matter oligodendrocytes and possibly forms GJ plaques in myelinated fibers (green arrows). In the subcortical white matter large myelinated fibers express Cx32 (K, N) but not Cx31.3 (J), and Cx47-immunoreactivity is again localized to the perikarya and proximal processes of adjacent oligodendrocytes (red arrowheads in M). Scale bar: 10 μm.
To evaluate further the localization of Cx31.3 in fibers in the gray matter, we double labeled for Cx31.3 and MBP or MAG, which are components of myelin sheaths, or NFL, a subunit of axonal neurofilaments. As shown in Fig. 6, Cx31.3 was co-localized with MBP, MAG, and NFL, demonstrating that Cx31.3 is localized to myelinated fibers. We occasionally saw examples of what might have been Cx31.3 concentrated at juxtaparanodal and/or paranodal areas that were positive for Caspr, a protein localized to paranodes (Fig. 6G-I). To ascertain the identity of oligodendrocytes, we immunostained sections with a rabbit antiserum against ASPA, a marker of oligodendrocytes (Madhavarao et al. 2005). As shown in Suppl. Fig. 4, the cell membranes of ASPA-positive cells were Cx47-positive.
Fig. 6.
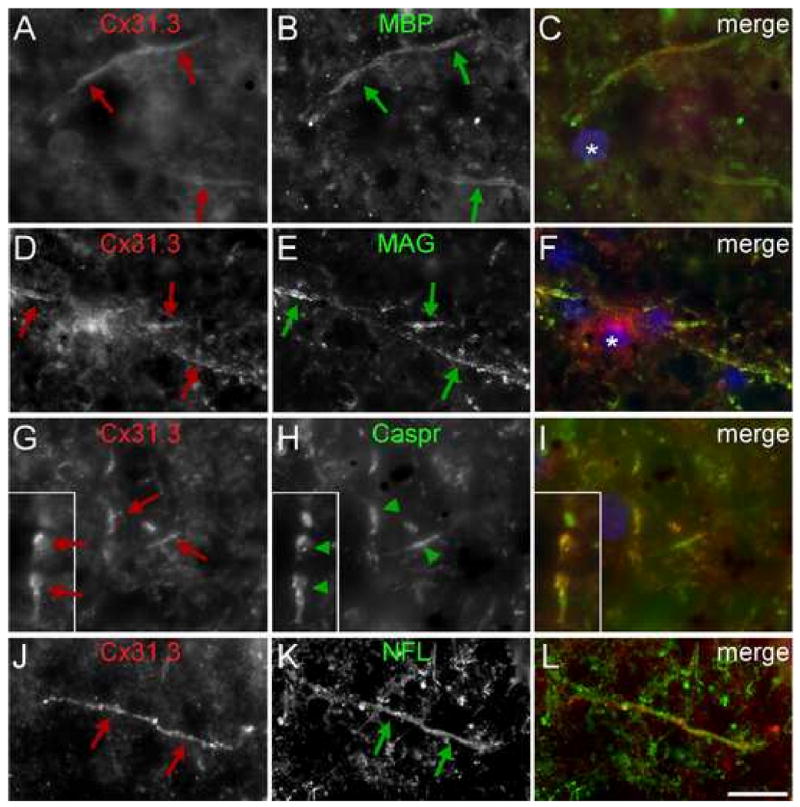
Expression of Cx31.3 in small myelinated fibers. These are images of sections of human cortex, immunostained with a rabbit antiserum against Cx31.3 (726) and a mouse monoclonal antibody against myelin basic protein (MBP), myelin-associated glycoprotein (MAG), Caspr, or neurofilament light (NFL) as indicated. Cx31.3 (red arrows) is co-localizated along thin fibers with MBP and MAG, which are components of the myelin sheath, as well as with NFL, an axonal marker (green arrows). In some myelinated fibers, Cx31.3 appears to be enriched at paranodes flanking the nodes, as it overlaps with Caspr. Scale bar: 10 μm.
We performed the above immunostainings of specimens from 8 different brains with comparable results. Although both Cx31.3 antisera gave similar results, one antiserum (726) was used for all of the images because it had lower background (diffuse) staining than did the other antiserum. To substantiate these findings, we performed immunoblots on lysates of human brain with the 726 antiserum. As shown in Fig. 1B, this antiserum detected a specific band in Cx31.3-expressing Neuro2a cells that was greatly attenuated by preincubation with the immunogenic peptide. Lysates from a human brain contained a faint band of the same size as well as a stronger band at around 50 kDa, possibly representing dimers of Cx31.3; both bands disappeared after preincubation of the antiserum with the immunogenic peptide. Thus, Cx31.3 appears to be expressed in human brain and is mostly detected in dimer form; these findings are similar to those previously reported for Cx29 in mouse brain (Altevogt et al. 2002).
Co-expression of Cx32 mutants in clonal cells stably expressing Cx31.3
Since Cx32 and Cx31.3 are co-expressed in at least a subset of human oligodendrocytes (Fig. 5), we considered the possibility that Cx32 mutants with CNS manifestations could have a functional effect on co-expressed Cx31.3 in vitro. We first expressed Cx32 mutations associated with clinical CNS manifestations, including two that have not been previously studied, E102del (Hanemann et al. 2003) and C168Y (Paulson et al. 2002). Cloned HeLa cells that stably expressed each mutant were immunostained using a rabbit antiserum against the cytoplasmic loop and a mouse monoclonal antibody against an epitope in the carboxy-terminus of Cx32 as reported previously (Kleopa et al. 2002); both showed the same result. Cells expressing wild type Cx32 had GJ plaques on apposed cell membranes, as well as Cx32-positive cytoplasmic vesicles. Cells expressing Cx32 mutants showed different patterns of abnormal intracellular retention (Table). Like A39V and T55I (Kleopa et al. 2002), C168Y mutants showed diffuse cytoplasmic staining and even some cytoplasmic vesicles, but no formation of GJ plaques, consistent with ER localization. This was confirmed by co-localization with calnexin, an ER marker (Suppl. Fig. 7C and data not shown). Like M93V and R164W (Kleopa et al. 2002), E102del mutants had vesicular staining, often with focal accumulation; the first two also formed rare GJ plaques. These mutants co-localized with the Golgi marker 58 kDa and with COPII, a marker of vesicles that shuttle between the ER and the Golgi (Suppl. Fig. 7A&B and data not shown).
Table. Effects of Cx32 mutants in Cx31.3-expressing HeLa cells.
| Cx32 Mutant | Subcellular localization in Cx31.3 clonal cells* |
|---|---|
| A39V** | ER retention, does not affect Cx31.3 expression |
| A39P | ER retention, does not affect Cx31.3 expression |
| T55I** | ER retention, does not affect Cx31.3 expression |
| M93V** | Golgi retention with cytoplasmic vesicles, does not affect Cx31.3 expression |
| E102del** | ***ER-Golgi retention with cytoplasmic vesicles and rare GJ, does not affect Cx31.3 expression |
| R164W** | Golgi retention with cytoplasmic vesicles, does not affect Cx31.3 expression |
| C168Y** | ***ER retention, does not affect Cx31.3 expression |
| R183S | Golgi retention with cytoplasmic vesicles and rare GJ, does not affect Cx31.3 expression |
| Wild type Cx32 | Forms many GJ-like plaques and some cytoplasmic vesicles, does not affect Cx31.3 expression |
Expression of Cx32 mutants in Cx31.3 clonal cells was identical to their expression in Cx31.3-negative HeLa;
Associated with CNS phenotype;
No previous expression studies.
ER: endoplasmic reticulum; GJ: gap junctions
All cells were labeled with mouse monoclonal antibody to Cx32 and rabbit antiserum to Cx31.3. Cells expressing ER- or Golgi-retained mutants were also labeled with ER-or Golgi markers (see methods), respectively.
We then co-expressed these Cx32 mutants with Cx31.3 by transfecting cells that stably expressed Cx31.3 with each of the Cx32 mutants, followed by double selection with G418 and puromycin to obtain cell lines that stably expressed both connexins. Immunostaining these cells showed that almost all cells expressed Cx31.3, whereas a variable proportion of cells expressed Cx32. In the cells that expressed both Cx31.3 and Cx32, Cx31.3 was localized to the cell membrane, whereas Cx32 was localized identically as described above (Fig. 7 and Table). There was no focal accumulation of Cx31.3 with Cx32 - for the Cx32 mutants that were localized to the ER (A39V, T55I, C168Y) or Golgi (M93V, E102del, R164W) or with wild type Cx32 in GJ plaques. We also examined two mutants that are not known to cause CNS manifestations (Kleopa et al. 2002). Neither A39P (localized to the ER) nor R183S (localized to the Golgi) altered the localization of Cx31.3 (data not shown). We conclude that Cx32 mutants do not appear to alter the trafficking of Cx31.3.
Fig. 7.
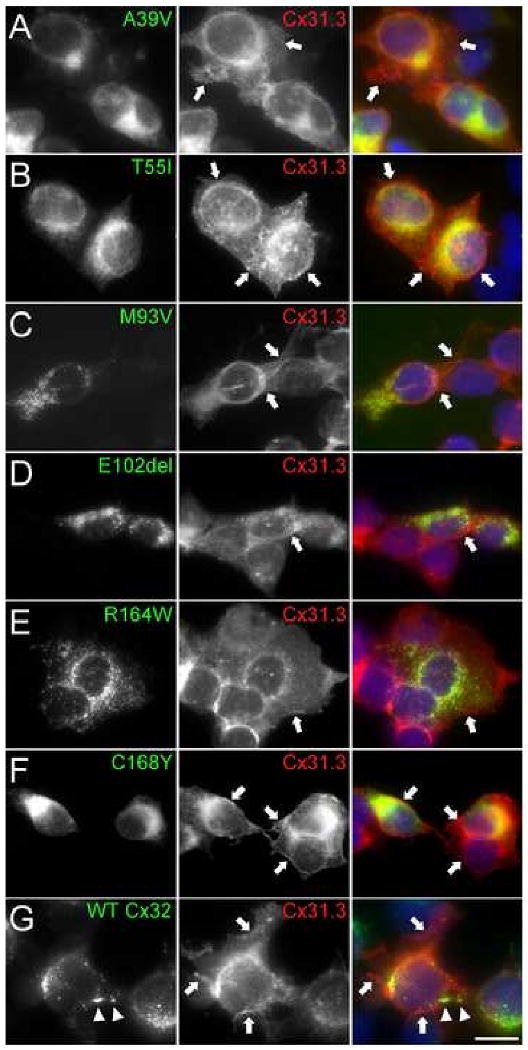
Co-expression of Cx32 mutants with Cx31.3. These are images of HeLa cells that express Cx31.3 and the various Cx32 mutants as indicated. The cells are labeled with a rabbit antiserum against Cx31.3 (726; red) and a mouse monoclonal antibody against Cx32 (green), and counterstained with DAPI; merged images are shown in the right column. Note that all cells appear to express Cx31.3, which is found on the cell membrane (arrows) and diffusely within the cytoplasm, but not in GJ plaques. Not all cells express Cx32. Some Cx32 mutants show diffuse intracellular staining (A, B, F), and others are found in intracellular puncta (C, D, E); only wild type Cx32 (G) forms GJ plaques (arrowheads). Cx31.3-immunoreactivity in double expressing cells is similar to the cells that express only Cx31.3; none of the Cx32 mutants appear to alter the localization of Cx31.3. Scale bar: 10 μm.
Finally, to test whether representative Cx32 mutants had a functional effect on possible hemichannel function of coexpressed Cx31.3, we transfected Cx31.3 clonal cells with Cx32 constructs cloned in pIRES-EGFP. A subset of cells expressed in addition to Cx31.3 either the ER-retained T55I mutant, the Golgi-retained R75W (Yum et al. 2002), or wild type Cx32 as a control, and these cells were identified by green fluorescence. Neurobiotin uptake assay was performed in transfected cells, without or with the presence of the hemichannel inhibitor octanol. As shown in Figure 8, neurobiotin uptake in cells that coexpressed the Cx32 mutants with GFP was not impaired compared to cells expressing Cx31.3 alone. To quantify these results, we counted at least 50 GFP+ and 50 GFP- cells in each condition, and compared their mean red pixel intensity (Fig. 8G). There was no statistically significant difference in red pixel intensity corresponding to neurobiotin uptake in GFP+ Cx31.3 clonal cells co-expressing the T55I (38,25 ± 7.45 without octanol and 18,96 ± 3,79 with octanol; mean ± standard deviation) or R75W (38,54 ± 7,49 without and 18,65 ± 5,16 with octanol) mutants or wild type Cx32 (38,26 ± 8,11 without and 18,83 ± 3,86 with octanol) compared to GFP- cells expressing Cx31.3 alone: for T55I 37,53 ± 6,93 without octanol and 18,21 ± 3,72 with octanol; for R75W 38,26 ± 7,83 without and 17,92 ± 5,36 with octanol; and for wild type Cx32 38,66 ± 6,14 without and 18,56 ± 3,70 with octanol (p>0.05 for all comparisons among octanol treated or untreated cells using ANOVA with Bonferroni's post test). Thus, Cx32 mutants do not affect the function of Cx31.3, at least in this in vitro model.
Fig. 8.
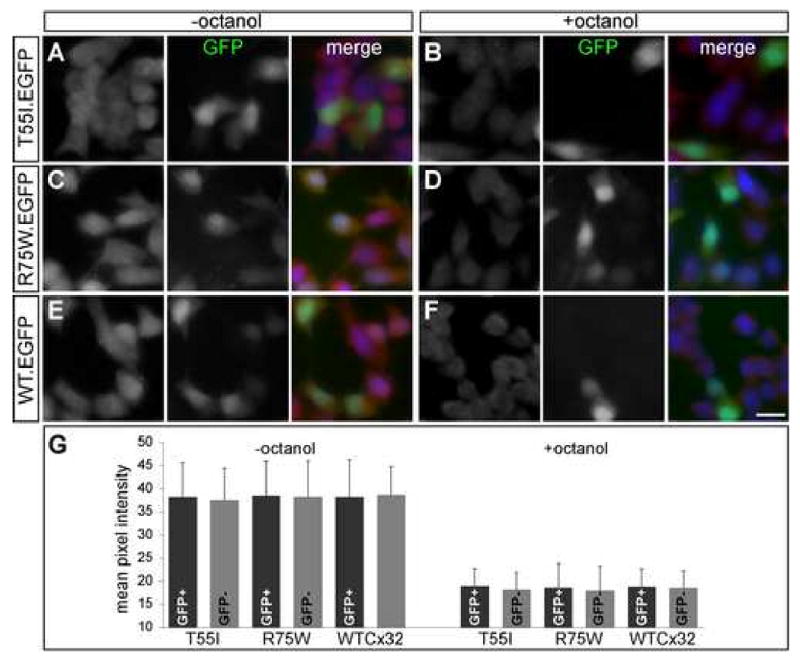
Neurobiotin uptake assay in cells coexpressing Cx31.3 and selected Cx32 mutants. Cloned cells stably expressing Cx31.3 were transiently transfected with Cx32 constructs in pIRES-EGFP vector (to coexpress the Cx32 mutants along with green fluorescent protein; GFP). The cells were incubated in neurobiotin for 30 minutes in the presence (B,D,F) or absence (A,C,E) of 2 mM octanol, fixed in 4% PFA, and neurobiotin was visualized with streptavidin-Texas red (left columns). A subset of cells expresses Cx32 and GFP (middle columns); merged images are shown in the right columns. In the absence of octanol, note the neurobiotin uptake in almost all cells, which does not appear to be impaired in cells coexpressing GFP (A,C,E), and that octanol blocks most of the neurobiotin uptake (B,D,F). Scale bar: 10 μm. G. Quantitative analysis of red channel pixel intensity in at least 50 GFP+ and GFP- cells for each of the Cx32 constructs without and with addition of octanol shows no significant difference among different constructs in each condition (p>0.05 for all conditions, ANOVA), indicating that Cx32 mutants have no effect on Cx31.3 function.
Discussion
Here we demonstrate for the first time that human oligodendrocytes express Cx31.3. As previously shown by CLUSTAL analysis (Altevogt et al. 2002), Cx31.3 and its rodent ortholog, Cx29, are among the most divergent members of the connexin family. Subsequent phylogenetic analysis using connexins from humans, mouse, and zebrafish showed a 100% bootstrap value for the orthologous relationship between Cx29 and Cx31.3 (Cruciani and Mikalsen 2006; Eastman et al. 2006). Cx31.3 and Cx29 differ mainly in the C-terminal domain; this also accounts for the difference in their molecular masses. The additional/alternative C-terminal sequence of Cx31.3 mRNA arises from an exon that is 5.2 kb downstream of the original ORF; this has not been identified in the murine gene (Altevogt et al. 2002). This C-terminal sequence of the Cx31.3 gene has been validated by RT-PCR of human brain mRNA; additional transcripts corresponding to another possible version of the human gene (Cx30.2), originally proposed as the human ortholog of Cx29 (Sohl et al. 2001), were not detected (Altevogt et al. 2002).
This is the first report to localize Cx31.3, Cx32, and Cx47 in human brain. We had to examine many specimens to find ones with preservation adequate to allow even an approximation of the cellular localization that one routinely obtains in rodent tissue. Cx31.3 is localized in the perikarya of oligodendrocytes and along small diameter fibers, especially in the superficial layers of the cortex and spinal cord gray matter, and is rarely expressed in small fibers of the subcortical and spinal cord white matter. In contrast, Cx32 is rarely found in medium size fibers in the gray matter, where it is coexpressed but not colocalized with Cx31.3, and is most prominent in larger myelinated fibers of cerebral and spinal cord white matter, where Cx31.3 is absent. Cx47 is found on oligodendrocyte cell bodies and proximal processes in both gray and white matter. These findings match our localizations of Cx29, Cx32, and Cx47 in rodent CNS (Altevogt et al. 2002; Kleopa et al. 2004), and Cx47 in a non-human primate brain (Orthmann-Murphy et al. 2007). Oligodendrocytes have been classified according to their morphological features: Type I and II oligodendrocytes myelinate many, small axons mostly in the gray matter, whereas type III and IV oligodendrocytes myelinate few, large axons mostly in the white matter (Butt et al. 1998; Remahl and Hildebrand 1990). Similar to murine Cx29, Cx31.3 is mostly expressed in type I and II human oligodendrocytes.
The lack of Cx29/Cx31.3 in GJ plaques and its limited co-localization with other connexins indicate that Cx29/Cx31.3 does not contribute to A:O coupling (Altevogt et al. 2002; Kamasawa et al. 2005; Kleopa et al. 2004; Li et al. 2004; Nagy et al. 2003a; Nagy et al. 2003b). Rather, Cx32 and Cx47 are the primary connexins on the oligodendrocyte side of A:O GJs. Like Cx29 (Ahn et al. 2007), Cx31.3 does not form GJ plaques or functional GJs by either dye transfer or electrophysiological assays in transfected cells. How Cx29/Cx31.3 functions in oligodendrocytes is unknown, but its localization on the adaxonal membrane of small myelinated axons (facing the periaxonal space) leads to the speculation that it forms hemichannels (Altevogt et al. 2002; Kamasawa et al. 2005; Kleopa et al. 2004; Menichella et al. 2006). This hypothesis is supported by our results presented here including uptake of neurobiotin and efflux of Calcein Blue dye in Cx31.3 expressing heterologous cells that could be blocked by the hemichannel inhibitor octanol. However, we could not demonstrate Cx31.3 hemichannel currents by electrophysiologic recordings. One possible explanation is that dialysis of cell contents, which occurs during whole cell patch clamping, may inhibit channel activity. A second possibility is that hemichannel openings are too infrequent or too brief to be resolved by electrophysiologic methods (Contreras et al. 2003).
Cx32 mutants that are associated with CNS manifestations appear to be retained intracellularly, in the ER or Golgi, both in vitro (Kleopa et al. 2002) and in vivo (Jeng et al. 2006). These CNS mutants may exert dominant-negative effects on other GJ proteins that follow the same pathway to the cell membrane, but in this study they did not lead to an overt effect on the expression and trafficking of Cx31.3. Furthermore, we show that neither an ER-retained nor a Golgi-retained Cx32 mutant affect the hemichannel function of Cx31.3 as assayed by neurobiotin uptake, supporting the lack of functional effects of Cx32 mutants on Cx31.3. Although these in vitro results may not fully reflect the in vivo situation, at least one Cx32 mutant associated with CNS findings (R142W) did not affect the localization of Cx29 in myelinating Schwann cells (Jeng et al. 2006). However, a possibility not investigated in this study is that lack of one functional connexin gene could affect the expression of other genes in the CNS (Iacobas et al. 2005). Through this mechanism Cx32 mutations could have indirect intracellular consequences on other connexins without necessarily directly impeding their function.
Although it appears that Cx32 mutations do not have a functional effect on the in vitro hemi-channel formation by Cx31.3, Cx32 mutations may have more resonance in vivo on the subset of oligodendrocytes which co-express both connexins rather than the residual population which express either Cx31.3 or Cx32. In the developing brain, connexin expression follows a temporal pattern, suggesting that the cells which express both connexins may in fact be proliferating or developing glial cells rather than a third separate population. This could be important in terms of understanding if the effects of Cx32 mutations may be more toxic to developing rather than adult oligodendrocytes. Nevertheless, the prominent expression of Cx29 (Kleopa et al. 2004) and Cx31.3 (present report) in gray and not white matter does not match the observed CNS alterations in symptomatic CMT1X patients, in whom abnormalities are seen in the white matter (Kassubek et al. 2005; Kleopa et al. 2002; Paulson et al. 2002; Taylor et al. 2003). Because it overlaps with Cx32 in the white matter, Cx47 is a more likely target for functional effects of Cx32 mutants. Further studies using animal models to reproduce the CNS phenotype of CMT1X and examine possible GJ protein interactions both in myelinating oligodendrocytes and in Schwann cells in vivo are currently underway.
Supplementary Material
Cx31.3 immunostaining of transfected cells. These are images of untransfected HeLa cells (A&D), or HeLa cells stably expressing Cx31.3 (B&E) or Cx32 (C&F). All cells were stained with a combination of a mouse monoclonal antibody against Cx32 (green) and one of two rabbit antisera (red), called 726 (A-C) and 727 (D-F) against a peptide from the C-terminus of Cx31.3, and counterstained with DAPI (blue). Note that both antisera specifically label Cx31.3, which is localized at the cell membrane (arrows) but does not form GJ plaques; parental cells (A&D) or cells expressing Cx32 (C&F) are not labeled. The Cx32 antibody labels GJ plaques (arrowheads) only in Cx32-expressing cells. Scale bar: 10 μm.
Cx31.3 does not form functional channels in transfected cells. These are representative images of HeLa cells that stably express Cx32 (A&D), Cx31.3 (B&E) or parental cells (C&F) scrape-loaded in the presence of Lucifer Yellow or neurobiotin. In Cx32-expressing cells, both tracers diffuse into cells well away from the scrape line, whereas in Cx31.3-expressing cells and parental cells, there is uptake only immediately adjacent to the scrape line. Scale bar: 20 μm. Mean ± SEM numbers of labeled cells in at least 10 fields in each of 3 independent experiments are shown on the right. The number of labeled cells was significantly higher in Cx32-expressing cells (p<0.005 for both tracers) but did not differ between Cx31.3-expressing and parental cells (p=0.255 for Lucifer Yellow and p=0.489 for neurobiotin), indicating that Cx31.3 does not form functional GJs.
Electrophysiological properties of Cx31.3 expressing cells. A. Input resistances of Cx31.3 expressing cells and controls. As shown, the input resistances of Cx31.3 expressing cells are similar to those for controls (CTR) (columns one and two). On the other hand expression of Cx26, known to form functional hemichannels, does reduce input resistance. Columns four through seven show that metabolic inhibition (MI), either through pre-incubation (columns four and five) or perfusion (columns six and seven) of already patched cells, does substantially reduce input resistance. However, no differences are seen between Cx31.3 expressing or control cells. B. Activation curves for Neuro2a cells expressing Cx31.3 and control cells. Single cells were pulsed from -60 mV to voltages between -80 mV and +80 mV in 20 mV increments. As shown the curves for the control and Cx31.3 are virtually superimposable below +60 mV. The larger average currents in the Cx31.3 expressing cells at +60 and +80 mV are due to large currents in about 15% of the Cx31.3 expressing cells.
Cx47 co-localizes with an oligodendrocyte marker in human brain. These are images of frozen sections of human brain immunostained with a rabbit antiserum against aspartoacylase (ASPA; red) and a mouse monoclonal antibody against Cx47 (green), and counterstained with DAPI. Note that Cx47-immunoreactivity is found on the perimeter of the ASPA-positive cell, including one possible GJ plaque (arrowhead). Scale bar: 10 μm.
Specific binding of the Cx31.3 antiserum to small myelinated fibers in the human cortex. These are images of human cortex stained with the rabbit anti-Cx31.3 antiserum 726 showing labeling of thin fibers (A). Staining after preincubation of the antiserum with the immunogenic peptide for 1 hr (B) shows only background reactivity. Cell nuclei are visualized with DAPI. Scale bar: 10 μm.
Cx31.3 and Cx32 expression in the human spinal cord. These are transverse sections of fixed cervical spinal cord, immunostained with a mouse monoclonal antibody against myelin oligodendrocyte glycoprotein (MOG; green) and rabbit antisera against Cx31.3 or against Cx32 (red). Merged images are shown in the right column. In the gray matter (A-F), Cx31.3 (red arrows in A) colocalizes with MOG (green arrows in B) along small myelinated fibers, but Cx32 (D) is not expressed along these fibers. In the white matter (G-L), Cx31.3 is expressed along some thin transverse fibers colocalizing with MOG (arrows in G and H), but not in large longitudinal myelinated fibers (green arrowheads in H). In contrast, Cx32 is expressed in large fibers (arrowheads in J and K) but not along thin transverse fibers (green arrows in K). Scale bar: 10 μm.
Cellular expression of Cx32 mutants with CNS manifestations. These are images of HeLa cells expressing E102del (A), R164W (B) or C168Y (C) mutants, double-labeled with a mouse monoclonal antibody (A) or a rabbit antiserum (B&C) against Cx32, and either a rabbit antiserum against COPII (a marker for vesicles that shuttle between the ER and the Golgi), or mouse monoclonal antibodies against a 58 kDa protein (a Golgi marker) or calnexin (an ER marker), as indicated. Merged images are shown in the right column. The E102del mutant is partly co-localized in COPII-positive vesicles (A); the R164W mutant is co-localized with the 58 kDa protein (B); the C168Y mutant is co-localized with calnexin (C). Scale bar: 10 μm.
Acknowledgments
This work was supported by the National Multiple Sclerosis Society (Grant RG3457A2/1 to K.A.K.), Telethon and the NIH (RO1 NS43560 and NS42878 to S.S.S. and NS50345 and NS050705 to C.K.A.). We thank the UK Multiple Sclerosis Brain Tissue Bank for postmortem human brain sections, Dr. Klaus Willecke for HeLa cells, Drs. James Garbern, Elior Peles and Sarah Piddlesden for antibodies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrams C, Freidin M, Bukauskas F, Dobrenis K, Bargiello T, Verselis V, Bennett M, Chen L, Sahenk Z. Pathogenesis of X-linked Charcot-Marie-Tooth disease: differential effects of two mutations in connexin 32. J Neurosci. 2003;23:10548–58. doi: 10.1523/JNEUROSCI.23-33-10548.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn M, Lee J, Gustafsson A, Enriquez A, Lancaster E, Sul JY, Haydon PG, Paul DL, Huang Y, Abrams CK, et al. Cx29 and Cx32, two connexins expressed by myelinating glia, do not interact and are functionally distinct. J Neurosci Res. 2007 Oct 30; doi: 10.1002/jnr.21561. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altevogt BM, Kleopa KA, Postma FR, Scherer SS, Paul DL. Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J Neurosci. 2002;22:6458–6470. doi: 10.1523/JNEUROSCI.22-15-06458.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altevogt BM, Paul DL. Four classes of intercellular channels between glial cells in the CNS. J Neurosci. 2004;24:4313–23. doi: 10.1523/JNEUROSCI.3303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergoffen J, Scherer SS, Wang S, Oronzi-Scott M, Bone L, Paul DL, Chen K, Lensch MW, Chance P, Fischbeck K. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- Butt AM, Ibrahim M, Berry M. Axon-myelin sheath relations of oligodendrocyte unit phenotypes in the adult rat anterior medullary velum. J Neurocytol. 1998;27(4):259–269. [PubMed] [Google Scholar]
- Contreras JE, Sáez JC, Bukauskas FF, Bennett MV. Gating and regulation of connexin 43 (Cx43) hemichannels. Proc Natl Acad Sci U S A. 2003;100:11388–93. doi: 10.1073/pnas.1434298100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciani V, Mikalsen SO. The vertebrate connexin family. Cell Mol Life Sci. 2006;63:1125–40. doi: 10.1007/s00018-005-5571-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschênes SM, Walcott JL, Wexler TL, Scherer SS, Fischbeck KH. Altered trafficking of mutant connexin32. J Neurosci. 1997;17:9077–9084. doi: 10.1523/JNEUROSCI.17-23-09077.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman SD, Chen TH, Falk MM, Mendelson TC, Iovine MK. Phylogenetic analysis of three complete gap junction gene families reveals lineage-specific duplications and highly supported gene classes. Genomics. 2006;87:265–74. doi: 10.1016/j.ygeno.2005.10.005. [DOI] [PubMed] [Google Scholar]
- el-Fouly MH, Trosko JE, Chang CC. Scrape-loading and dye transfer. A rapid and simple technique to study gap junctional intercellular communication. Exp Cell Res. 1987;168:422–30. doi: 10.1016/0014-4827(87)90014-0. [DOI] [PubMed] [Google Scholar]
- González D, Gómez-Hernández JM, Barrio LC. Species specificity of mammalian connexin-26 to form open voltage-gated hemichannels. FASEB J. 2006;20:2329–38. doi: 10.1096/fj.06-5828com. [DOI] [PubMed] [Google Scholar]
- Hahn AF, Ainsworth PJ, Naus CCG, Mao J, Bolton CF. Clinical and pathological observations in men lacking the gap junction protein connexin 32. Muscle Nerve. 2000:S39–S48. [PubMed] [Google Scholar]
- Hanemann CO, Bergmann C, Senderek J, Zerres K, Sperfeld A. Transient, recurrent, white matter lesions in X-linked Charcot-Marie-Tooth disease with novel connexin 32 mutation. Arch Neurol. 2003;60:605–9. doi: 10.1001/archneur.60.4.605. [DOI] [PubMed] [Google Scholar]
- Iacobas DA, Iacobas S, Urban-Maldonado M, Spray DC. Sensitivity of the brain transcriptome to connexin ablation. Biochim Biophys Acta. 2005;1711:183–96. doi: 10.1016/j.bbamem.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Jeng LJ, Balice-Gordon RJ, Messing A, Fischbeck KH, Scherer SS. The effects of a dominant connexin32 mutant in myelinating Schwann cells. Mol Cell Neurosci. 2006;32:283–98. doi: 10.1016/j.mcn.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Kamasawa N, Sik A, Morita M, Yasumura T, Davidson K, Nagy J, Rash J. Connexin-47 and connexin-32 in gap junctions of oligodendrocyte somata, myelin sheaths, paranodal loops and Schmidt-Lanterman incisures: Implications for ionic homeostasis and potassium siphoning. Neuroscience. 2005;136:65–86. doi: 10.1016/j.neuroscience.2005.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassubek J, Bretschneider V, Sperfeld AD. Corticospinal tract MRI hyperintensity in X-linked Charcot-Marie-Tooth Disease. J Clin Neurosci. 2005;12:588–9. doi: 10.1016/j.jocn.2004.07.020. [DOI] [PubMed] [Google Scholar]
- Kleopa KA, Orthmann JL, Enriquez A, Paul DL, Scherer SS. Unique distribution of gap junction proteins connexin29, connexin32, and connexin47 in oligodendrocytes. Glia. 2004;47:346–357. doi: 10.1002/glia.20043. [DOI] [PubMed] [Google Scholar]
- Kleopa KA, Scherer SS. Molecular genetics of X-linked Charcot-Marie-Tooth disease. Neuromolecular Med. 2006;8:107–122. doi: 10.1385/nmm:8:1-2:107. [DOI] [PubMed] [Google Scholar]
- Kleopa KA, Yum SW, Scherer SS. Cellular mechanisms of connexin32 mutations associated with CNS manifestations. J Neurosci Res. 2002;68:522–534. doi: 10.1002/jnr.10255. [DOI] [PubMed] [Google Scholar]
- Kleopa KA, Zamba-Papanicolaou E, Alevra X, Nicolaou P, Georgiou DM, Hadjisavvas A, Kyriakides T, Christodoulou K. Phenotypic and cellular expression of two novel connexin32 mutations causing CMT1X. Neurology. 2006;66:396–402. doi: 10.1212/01.wnl.0000196479.93722.59. [DOI] [PubMed] [Google Scholar]
- Li J, Hertzberg EL, Nagy JI. Connexin32 in oligodendrocytes and association with myelinated fibers in mouse and rat brain. J Comp Neurol. 1997;379(4):571–591. doi: 10.1002/(sici)1096-9861(19970324)379:4<571::aid-cne8>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Li X, Ionescu AV, Lynn BD, Lu S, Kamasawa N, Morita M, Davidson KGV, Yasumura T, Rash JE, Nagy JI. Connexin47, connexin29 and connexin32 co-expression in oligodendrocytes and Cx47 association with zonula occludens-1 (ZO-1) in mouse brain. Neuroscience. 2004;126:611–630. doi: 10.1016/j.neuroscience.2004.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavarao CN, Arun P, Moffett JR, Szucs S, Surendran S, Matalon R, Garbern J, Hristova D, Johnson A, Jiang Wa, et al. Defective N-acetylaspartate catabolism reduces brain acetate levels and myelin lipid synthesis in Canavan's disease. Proc Nat Acad Sci USA. 2005;102:5221–5226. doi: 10.1073/pnas.0409184102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichella DM, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Connexins are critical for normal myelination in the CNS. J Neurosci. 2003;23:5963–5973. doi: 10.1523/JNEUROSCI.23-13-05963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichella DM, Majdan M, Awatramani R, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J Neurosci. 2006;26(48):10984–91. doi: 10.1523/JNEUROSCI.0304-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JI, Ionescu AV, Lynn BD, Rash JE. Connexin29 and connexin32 at oligodendrocyte and astrocyte gap junctions and in myelin of the mouse central nervous system. J Comp Neurol. 2003a;22:356–70. doi: 10.1002/cne.10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JI, Ionescu AV, Lynn BD, Rash JE. Coupling of astrocyte connexins Cx26, Cx30, Cx43 to oligodendrocyte Cx29, Cx32, Cx47: Implications from normal and connexin32 knockout mice. Glia. 2003b;44:205–18. doi: 10.1002/glia.10278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odermatt B, Wellershaus K, Wallraff A, Seifert G, Degen J, Euwens C, Fuss B, Bussow H, Schilling K, Steinhauser C, et al. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J Neurosci. 2003;23:4549–59. doi: 10.1523/JNEUROSCI.23-11-04549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S, Ri Y, Bennett MVL, Trexler EB, Verselis VK, Bargiello TA. Changes in permeability caused by connexin 32 mutations underlie X-linked Charcot-Marie-Tooth disease. Neuron. 1997;19(4):927–938. doi: 10.1016/s0896-6273(00)80973-3. [DOI] [PubMed] [Google Scholar]
- Omori Y, Mesnil M, Yamasaki H. Connexin 32 mutations from X-linked Charcot-Marie-Tooth disease patients: functional defects and dominant negative effects. Mol Biol Cell. 1996;7(6):907–916. doi: 10.1091/mbc.7.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthmann-Murphy JL, Enriquez AD, Abrams CK, Scherer SS. Loss-of-function connexin47 mutations cause Pelizaeus-Merzbacher-like disease. Mol Cell Neurosci. 2007;34:629–41. doi: 10.1016/j.mcn.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson HL, Garbern JY, Hoban TF, Krajewski KM, Lewis RA, Fischbeck KH, Grossman RI, Lenkinski R, Kamholz JA, Shy ME. Transient central nervous system white matter abnormality in X-linked Charcot-Marie-Tooth disease. Ann Neurol. 2002;52:429–434. doi: 10.1002/ana.10305. [DOI] [PubMed] [Google Scholar]
- Rash JE, Yasumura T, Dudek FE, Nagy JI. Cell-specific expression of connexins and evidence of restricted gap junctional coupling between glial cells and between neurons. J Neurosci. 2001;21(6):1983–2000. doi: 10.1523/JNEUROSCI.21-06-01983.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remahl S, Hildebrand C. Relation between axons and oligodendroglial cells during initial myelination. I. The glial unit. J Neurocytol. 1990;19:313–328. doi: 10.1007/BF01188401. [DOI] [PubMed] [Google Scholar]
- Richard G, White TW, Smith LE, Bailey RA, Compton JG, Paul DL, Bale SJ. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum Genet. 1998;103(4):393–399. doi: 10.1007/s004390050839. [DOI] [PubMed] [Google Scholar]
- Rouan F, White T, Brown N, Taylor A, Lucke T, Paul D, Munro C, Uitto J, Hodgins M, Richard G. trans-dominant inhibition of connexin-43 by mutant connexin-26: implications for dominant connexin disorders affecting epidermal differentiation. J Cell Sci. 2001;114:2105–2113. doi: 10.1242/jcs.114.11.2105. [DOI] [PubMed] [Google Scholar]
- Sáez JC, Retamal MA, Basilio D, Bukauskas FF, Bennett MV. Connexin-based gap junction hemichannels: gating mechanisms. Biochim Biophys Acta. 2005;1711:215–24. doi: 10.1016/j.bbamem.2005.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Deschênes SM, Xu YT, Grinspan JB, Fischbeck KH, Paul DL. Connexin32 is a myelin-related protein in the PNS and CNS. J Neurosci. 1995;15:8281–8294. doi: 10.1523/JNEUROSCI.15-12-08281.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Xu YT, Nelles E, Fischbeck K, Willecke K, Bone LJ. Connexin32-null mice develop a demyelinating peripheral neuropathy. Glia. 1998;24:8–20. doi: 10.1002/(sici)1098-1136(199809)24:1<8::aid-glia2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Sohl G, Eiberger J, Jung YT, Kozak CA, Willecke K. The mouse gap junction gene connexin29 is highly expressed in sciatic nerve and regulated during brain development. Biol Chem. 2001;382:973–8. doi: 10.1515/BC.2001.122. [DOI] [PubMed] [Google Scholar]
- Taylor RA, Simon EM, Marks HG, Scherer SS. The CNS phenotype of X-linked Charcot-Marie-Tooth disease: more than a peripheral problem. Neurology. 2003;61:1475–1478. doi: 10.1212/01.wnl.0000095960.48964.25. [DOI] [PubMed] [Google Scholar]
- Trexler EB, Bennett MV, Bargiello TA, Verselis VK. Voltage gating and permeation in a gap junction hemichannel. Proc Natl Acad Sci U S A. 1996;93:5836–41. doi: 10.1073/pnas.93.12.5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiunas V. Biophysical properties of connexin-45 gap junction hemichannels studied in vertebrate cells. J Gen Physiol. 2002;119:147–64. doi: 10.1085/jgp.119.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiunas V, Weingart R. Electrical properties of gap junction hemichannels identified in transfected HeLa cells. Pflugers Arch. 2000;440:366–79. doi: 10.1007/s004240000294. [DOI] [PubMed] [Google Scholar]
- VanSlyke JK, Deschênes SM, Musil LS. Intracellular transport, assembly, and degradation of wild-type and disease-linked mutant gap junction proteins. Mol Biol Cell. 2000;11:1933–1946. doi: 10.1091/mbc.11.6.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willecke K, Eiberger J, Degen J, Eckardt D, Romualdi A, Guldenagel M, Deutsch U, Sohl G. Structural and functional diversity of connexin genes in the mouse and human genome. Biol Chem. 2002;383:725–37. doi: 10.1515/BC.2002.076. [DOI] [PubMed] [Google Scholar]
- Yum SW, Kleopa KA, Shumas S, Scherer SS. Diverse trafficking abnormalities of Connexin32 mutants causing CMTX. Neurobiol Dis. 2002;11:43–52. doi: 10.1006/nbdi.2002.0545. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cx31.3 immunostaining of transfected cells. These are images of untransfected HeLa cells (A&D), or HeLa cells stably expressing Cx31.3 (B&E) or Cx32 (C&F). All cells were stained with a combination of a mouse monoclonal antibody against Cx32 (green) and one of two rabbit antisera (red), called 726 (A-C) and 727 (D-F) against a peptide from the C-terminus of Cx31.3, and counterstained with DAPI (blue). Note that both antisera specifically label Cx31.3, which is localized at the cell membrane (arrows) but does not form GJ plaques; parental cells (A&D) or cells expressing Cx32 (C&F) are not labeled. The Cx32 antibody labels GJ plaques (arrowheads) only in Cx32-expressing cells. Scale bar: 10 μm.
Cx31.3 does not form functional channels in transfected cells. These are representative images of HeLa cells that stably express Cx32 (A&D), Cx31.3 (B&E) or parental cells (C&F) scrape-loaded in the presence of Lucifer Yellow or neurobiotin. In Cx32-expressing cells, both tracers diffuse into cells well away from the scrape line, whereas in Cx31.3-expressing cells and parental cells, there is uptake only immediately adjacent to the scrape line. Scale bar: 20 μm. Mean ± SEM numbers of labeled cells in at least 10 fields in each of 3 independent experiments are shown on the right. The number of labeled cells was significantly higher in Cx32-expressing cells (p<0.005 for both tracers) but did not differ between Cx31.3-expressing and parental cells (p=0.255 for Lucifer Yellow and p=0.489 for neurobiotin), indicating that Cx31.3 does not form functional GJs.
Electrophysiological properties of Cx31.3 expressing cells. A. Input resistances of Cx31.3 expressing cells and controls. As shown, the input resistances of Cx31.3 expressing cells are similar to those for controls (CTR) (columns one and two). On the other hand expression of Cx26, known to form functional hemichannels, does reduce input resistance. Columns four through seven show that metabolic inhibition (MI), either through pre-incubation (columns four and five) or perfusion (columns six and seven) of already patched cells, does substantially reduce input resistance. However, no differences are seen between Cx31.3 expressing or control cells. B. Activation curves for Neuro2a cells expressing Cx31.3 and control cells. Single cells were pulsed from -60 mV to voltages between -80 mV and +80 mV in 20 mV increments. As shown the curves for the control and Cx31.3 are virtually superimposable below +60 mV. The larger average currents in the Cx31.3 expressing cells at +60 and +80 mV are due to large currents in about 15% of the Cx31.3 expressing cells.
Cx47 co-localizes with an oligodendrocyte marker in human brain. These are images of frozen sections of human brain immunostained with a rabbit antiserum against aspartoacylase (ASPA; red) and a mouse monoclonal antibody against Cx47 (green), and counterstained with DAPI. Note that Cx47-immunoreactivity is found on the perimeter of the ASPA-positive cell, including one possible GJ plaque (arrowhead). Scale bar: 10 μm.
Specific binding of the Cx31.3 antiserum to small myelinated fibers in the human cortex. These are images of human cortex stained with the rabbit anti-Cx31.3 antiserum 726 showing labeling of thin fibers (A). Staining after preincubation of the antiserum with the immunogenic peptide for 1 hr (B) shows only background reactivity. Cell nuclei are visualized with DAPI. Scale bar: 10 μm.
Cx31.3 and Cx32 expression in the human spinal cord. These are transverse sections of fixed cervical spinal cord, immunostained with a mouse monoclonal antibody against myelin oligodendrocyte glycoprotein (MOG; green) and rabbit antisera against Cx31.3 or against Cx32 (red). Merged images are shown in the right column. In the gray matter (A-F), Cx31.3 (red arrows in A) colocalizes with MOG (green arrows in B) along small myelinated fibers, but Cx32 (D) is not expressed along these fibers. In the white matter (G-L), Cx31.3 is expressed along some thin transverse fibers colocalizing with MOG (arrows in G and H), but not in large longitudinal myelinated fibers (green arrowheads in H). In contrast, Cx32 is expressed in large fibers (arrowheads in J and K) but not along thin transverse fibers (green arrows in K). Scale bar: 10 μm.
Cellular expression of Cx32 mutants with CNS manifestations. These are images of HeLa cells expressing E102del (A), R164W (B) or C168Y (C) mutants, double-labeled with a mouse monoclonal antibody (A) or a rabbit antiserum (B&C) against Cx32, and either a rabbit antiserum against COPII (a marker for vesicles that shuttle between the ER and the Golgi), or mouse monoclonal antibodies against a 58 kDa protein (a Golgi marker) or calnexin (an ER marker), as indicated. Merged images are shown in the right column. The E102del mutant is partly co-localized in COPII-positive vesicles (A); the R164W mutant is co-localized with the 58 kDa protein (B); the C168Y mutant is co-localized with calnexin (C). Scale bar: 10 μm.