

Abstract
Mutations in the VHL tumor suppressor gene result in constitutive expression of many hypoxia-inducible genes, at least in part because of increases in the cellular level of hypoxia-inducible transcription factor HIF1α, which in normal cells is rapidly ubiquitinated and degraded by the proteasome under normoxic conditions. The recent observation that the VHL protein is a subunit of an Skp1-Cul1/Cdc53-F-box (SCF)-like E3 ubiquitin ligase raised the possibility that VHL may be directly responsible for regulating cellular levels of HIF1α by targeting it for ubiquitination and proteolysis. In this report, we test this hypothesis directly. We report development of methods for production of the purified recombinant VHL complex and present direct biochemical evidence that it can function with an E1 ubiquitin-activating enzyme and E2 ubiquitin-conjugating enzyme to activate HIF1α ubiquitination in vitro. Our findings provide new insight into the function of the VHL tumor suppressor protein, and they provide a foundation for future investigations of the mechanisms underlying VHL regulation of oxygen-dependent gene expression.
The von Hippel-Lindau (VHL) tumor suppressor gene on chromosome 3p25.5 is mutated in most sporadic clear cell renal carcinomas and in VHL disease, an autosomal dominant familial cancer syndrome that predisposes affected individuals to a variety of tumors including clear cell renal carcinomas, cerebellar hemangioblastomas and hemangiomas, retinal angiomas, and pheochromocytomas (1, 2). A striking feature of clear cell renal carcinomas and tumors seen in VHL disease is their high vascularity, which is believed to result at least in part from high expression of vascular endothelial growth factor (VEGF).
VEGF and other hypoxically regulated genes are repressed in normal cells under normoxic growth conditions but are strongly induced in cells deprived of oxygen. Studies carried out in several laboratories led to the discovery that clear cell renal carcinoma cells lacking a functional VHL gene constitutively express hypoxia-inducible genes like VEGF, which is believed to play a role in promoting vascularization of tumors (3–5). These studies demonstrated that reintroduction of the wild-type VHL gene into clear cell renal carcinoma cells is sufficient to repress VEGF gene expression under normoxic conditions and to restore its normal regulation by hypoxia.
Efforts to understand how the VHL protein regulates expression of hypoxia-inducible genes have revealed that the cellular levels of hypoxia-inducible transcription factors HIF1α and HIF2α are elevated in clear cell renal carcinoma cells lacking a functional VHL gene (6). HIF1α and HIF2α positively regulate VEGF and other hypoxia-inducible genes. Hypoxic activation of gene expression is known to result at least in part from increases in the cellular levels of HIF1α and HIF2α, which are rapidly ubiquitinated and degraded by the proteasome under normoxic conditions (7).
The recent observation that the VHL protein is a subunit of a multiprotein complex possessing associated E3 ubiquitin ligase activity raised the possibility that the VHL protein may be directly responsible for regulating cellular levels of hypoxia-inducible transcription factors and may target them for ubiquitination and proteolysis (8, 9). The VHL complex resembles the well characterized SCF (Skp1-Cul1/Cdc53-F-box) ubiquitin ligase complexes. Known subunits of the VHL complex include Cul2, Elongins B and C, and the RING-H2 finger protein Rbx1 (also referred to as ROC1 or Hrt1) (10–15). Cul2 is a member of the Cullin protein family, which includes SCF subunit Cul1/Cdc53; Elongin C is a Skp1-like protein; Rbx1 is a subunit of both the VHL and SCF complexes, where it potently activates ubiquitination by the E1/E2 ubiquitin-activating and -conjugating enzymes; and the VHL protein has been proposed to function similarly to F-box proteins, which bind to and recruit ubiquitination substrates to SCF complexes (16, 17).
In this report, we test the hypothesis that the VHL complex is capable of supporting ubiquitination of hypoxia-inducible transcription factors. We report the development of methods for production of the purified, recombinant 5-subunit VHL complex and present direct biochemical evidence that the VHL complex is capable of potently activating HIF1α ubiquitination in vitro. Our findings shed new light on the function of the VHL tumor suppressor protein, and they provide a framework for future investigations of the mechanisms underlying oxygen-dependent regulation of gene expression.
Materials and Methods
Antibodies.
Anti-VHL monoclonal antibody Ig32 was purchased from PharMingen. Anti-Cul2 and anti-Elongin C monoclonal antibodies were obtained from Transduction Laboratories (Lexington, KY). Anti-myc monoclonal antibody 9E10 was from Boehringer Mannheim. Anti-HIF1α monoclonal antibody H1α67 was purchased from Novus (Littleton, CO). Anti-Flag monoclonal antibody M2 was obtained from Sigma. Anti-HPC4 monoclonal antibody (18) was provided by C. T. Esmon. Anti-Elongin B rabbit polyclonal antibody was described previously (19).
Expression of Recombinant Proteins in Escherichia coli.
Human Ubc5a (hUbc5a) cDNA was provided by Dr. A. Weissman. Mouse E2-21K cDNA (AI326684), mouse E2-35K cDNA (AA124105), and mouse E2-24K cDNA (AA798039) were from Research Genetics (Huntsville, AL) and were subcloned into pRSET B (Invitrogen) with an N-terminal 6-histidine tag and a C-terminal Flag tag. Saccharomyces cerevisiae Uba1 containing an N-terminal myc tag and a C-terminal 6-histidine tag (20) and human Ubc3 with an N-terminal 6-histidine tag (9) were prepared as described. Mammalian ubiquitin was subcloned into pGEX4T-2 (Amersham Pharmacia). Proteins were expressed in E. coli strain BL21 (DE3) and purified by Ni2+-agarose or glutathione-Sepharose affinity chromatography. After dialysis against 40 mM Hepes-NaOH (pH 7.9), 60 mM potassium acetate, 2 mM DTT, 1 mM MgCl2, 0.5 mM EDTA (pH 7.9), and 10% (vol/vol) glycerol, proteins were stored at −80°C. hUbc5a, hUbc3, E2-21K, E2-35K, and E2-24K were similarly active in accepting ubiquitin in the presence of Uba1, glutathione S-transferase (GST)-ubiqutin, and ATP (data not shown).
Expression of Recombinant Proteins in Sf21 Insect Cells.
Human wild-type VHL and VHL mutants VHL[1–155] and VHL[L158P], human Cul2, human Elongin B, human Elongin C, human wild-type VHL containing an N-terminal 6-histidine tag (His-VHL), human wild-type VHL and VHL [117–213] containing an N-terminal 6-histidine tag and a C-terminal FLAG tag (His-VHL-FLAG), mouse Rbx1 containing an N-terminal myc tag (myc-Rbx1), and human HIF1α containing N-terminal 6-histidine and HPC4 tags (His-HPC4-HIF1α) were subcloned into pBacPAK8. Human HIF1α containing an N-terminal Flag tag (Flag-HIF1α) was subcloned into pBacPAK6. Recombinant baculoviruses were generated with the BacPAK baculovirus expression system (CLONTECH). Baculoviruses encoding mouse Rbx1 containing N-terminal 6-histidine and myc tags (His-myc-Rbx1) (15), S. cerevisiae Cln2 containing N-terminal 6-histidine and HA tags, S. cerevisiae Cdc28 containing N-terminal 6-histidine and myc tags, and S. cerevisiae Cks1 containing N-terminal 6-histidine and T7 tags (20) were described previously.
Sf21 cells were cultured at 27°C in Sf-900 II SFM with 5% FCS, penicillin (100 units/ml), and streptomycin (100 μg/ml). Sf21 cells were infected with the recombinant baculoviruses indicated in the figures. Sixty hours after infection, cells were collected and lysed in ice-cold buffer containing 40 mM Hepes-NaOH (pH 7.9), 150 mM NaCl, 1 mM DTT, 0.5% (vol/vol) Triton X-100, 10% (vol/vol) glycerol, 5 μg/ml leupeptin, 5 μg/ml antipain, 5 μg/ml pepstatin A, and 5 μg/ml aprotinin. In some experiments, cells were resuspended in ice-cold buffer containing 40 mM Hepes-NaOH (pH 7.9), 150 mM NaCl, 20 mM imidazole (pH 7.9), 5 μg/ml leupeptin, 5 μg/ml antipain, 5 μg/ml pepstatin A, and 5 μg/ml aprotinin and lysed by French press (American Instruments).
Purification of the Recombinant VHL Complex from Lysates of Sf21 Insect Cells.
Sf21 cells were coinfected with baculoviruses indicated in the legends to Figs. 1B and 5B. Cells were harvested and lysed by French press as described above. After centrifugation at 10,000 × g for 20 min at 4°C, the resulting supernatant was mixed with 1 ml of Ni2+-agarose preequilibrated in buffer containing 40 mM Hepes-NaOH (pH 7.9), 150 mM NaCl, and 20 mM imidazole (pH 7.9). After 2 h, the Ni2+-agarose was washed three times with the same buffer and packed into a 0.8-cm-diameter column. The column was eluted stepwise with buffer containing 40 mM Hepes-NaOH (pH 7.9), 50 mM NaCl, 300 mM imidazole (pH 7.9), and 10% (vol/vol) glycerol. A peak fraction containing the recombinant VHL complex was diluted with 40 mM Tris⋅HCl (pH 7.9), 1 mM DTT, 0.5 mM EDTA, and 10% (vol/vol) glycerol and brought to a conductivity equivalent to that of buffer containing 40 mM KCl. After centrifugation at 10,000 × g for 20 min at 4°C, the resulting supernatant was applied to a TSK DEAE-NPR HPLC column (4.6 mm by 35 mm; Toso-Haas) preequilibrated in buffer containing 40 mM Tris⋅HCl (pH 7.9), 40 mM KCl, 1 mM DTT, 0.5 mM EDTA, and 10% (vol/vol) glycerol. The column was eluted at 0.2 ml/min with a 3-ml linear gradient of 40 mM to 350 mM KCl, and 0.1-ml fractions were collected.
Figure 1.

The purified recombinant VHL tumor suppressor complex activates HIF1α ubiquitination in vitro. (A) An aliquot of recombinant His-HPC4-HIF1α used as substrate in the ubiquitination reactions of C and D was subjected to 8% SDS/PAGE, and proteins were visualized by Coomassie staining. His-HPC4-HIF1α was expressed in Sf21 cells and purified by Ni2+-agarose chromatography. (B) The recombinant VHL complex was purified as described in Materials and Methods from lysates of insect cells infected with baculoviruses encoding His-VHL, Cul2, Elongin B, Elongin C, and myc-Rbx1. Aliquots of TSK DEAE-NPR column fractions were subjected to 13% SDS/PAGE, and proteins were visualized by Coomassie staining (Top). Aliquots of TSK DEAE-NPR column fractions were subjected to 8% or 13% SDS/PAGE, and proteins were transferred to Hybond P membranes and visualized by Western blotting as described in Materials and Methods, using the antibodies indicated in the figure (Bottom). EloB, Elongin B; EloC, Elongin C. (C) Aliquots of TSK-DEAE-NPR column fractions indicated in the figure were assayed as described in Materials and Methods for the ability to activate HIF1α ubiquitination. Reactions contained ≈50 ng of the His-HPC4-HIF1α shown in A. Reaction products were subjected to 8% SDS/PAGE, and GST-ubiquitin-HIF1α conjugates (GST-Ub-HIF1α) were transferred to Hybond P membranes and visualized by Western blotting as described in Materials and Methods, using anti-HPC4 antibodies. (D) Aliquots of TSK-DEAE-NPR column fraction 25 were assayed as described in Materials and Methods for the ability to activate HIF1α or Cln2 ubiquitination activity in the presence and absence of Uba1, hUbc5a, GST-ubiquitin (GST-Ub), and ATP. Reactions contained ≈50 ng of either the His-HPC4-HIF1α shown in A or about 50 ng of the phosphorylated Cln2 complex. Reaction products were subjected to 8% SDS/PAGE and transferred to Hybond P membranes and visualized by Western blotting as described in Materials and Methods, using anti-HPC4 or anti-HA antibodies. (E) Aliquots of TSK-DEAE-NPR column fraction 25 were assayed as described in Materials and Methods for the ability to activate HIF1α ubiquitination activity in the presence of Uba1, GST-ubiquitin (GST-Ub), ATP, and approximately equimolar amounts of the E2 ubiquitin-conjugating enzymes shown in the figure (≈100 ng hUbc5a, ≈200 ng hUbc3, ≈100 ng mE2-21K, ≈200 ng mE2-35K, ≈150 ng mE2-24K). Reactions contained ≈50 ng of the His-HPC4-HIF1α shown in A. Reaction products were subjected to 8% SDS/PAGE, and GST-ubiquitin-HIF1α conjugates (GST-Ub-HIF1α) were transferred to Hybond P membranes and visualized by Western blotting as described in Materials and Methods, using anti-HPC4 antibodies.
Figure 5.

A VHL mutant lacking a portion of the β domain is defective in HIF1α binding and ubiquitination. (A) Total cell lysates or anti-HIF1α immunoprecipitates from the indicated baculovirus-infected Sf21 cells were immunoblotted with anti-Flag antibody. (B) Sf21 cells were coinfected with baculoviruses encoding Cul2, Elongin B, Elongin C, His-myc-Rbx1, and either His-VHL-Flag or His-VHL-Flag[117–213]. Recombinant VHL complexes were purified as described in Materials and Methods. After TSK DEAE-NPR HPLC, aliquots of peak fractions from were subjected to 13% SDS/PAGE, and proteins were visualized by Coomassie staining. (C) Aliquots of the TSK DEAE-NPR fractions shown in B were assayed as described in Materials and Methods for the ability to activate HIF1a ubiquitination. Reactions contained ≈50 ng of the His-HPC4-HIF1a shown in Fig. 1A. The reaction products were subjected to 8% SDS/PAGE, transferred to Hybond P membranes, and visualized by Western blotting using anti-HPC4 antibody. H, His; Fl, Flag.
Immunoprecipitations and Western Blotting.
Sf21 cells were infected with the baculoviruses indicated in the figures. After 60 h, cells were harvested and lysed as described above. Cell lysates were incubated with antibody and protein A Sepharose for 2 h at 4°C. Protein A Sepharose was washed three times in buffer containing 40 mM Hepes-NaOH (pH 7.9), 150 mM NaCl, 1 mM DTT, 0.5% (vol/vol) Triton X-100. Immunoprecipitated proteins were separated by SDS/PAGE and transferred to Hybond P membranes (Amersham Pharmacia) and visualized by Western blotting with either SuperSignal West Pico or SuperSignal West Dura chemiluminescent reagent (Pierce).
Assay of Ubiquitination in Vitro.
To assay the purified VHL complex for its ability to activate HIF1α or Cln2 ubiquitination, aliquots of the indicated TSK DEAE-NPR HPLC column fractions were mixed with ≈50 ng of Uba1, ≈100 ng of hUbc5a, ≈3 μg of GST-ubiquitin, and either ≈50 ng of purified His-HPC4-HIF1α or ≈50 ng of phosphorylated Cln2/Cdc28/Cks1 complex in a 10-μl reaction containing 40 mM Hepes-NaOH (pH 7.9), 60 mM potassium acetate, 2 mM DTT, 5 mM MgCl2, 0.5 mM EDTA (pH 7.9), 10% (vol/vol) glycerol, and 1.5 mM ATP. Reaction mixtures were incubated for 1 h at 30°C. Phosphorylated Cln2/Cdc28/Cks1 complex was prepared as described (20).
To assay immunoprecipitated VHL complexes for their ability to activate HIF1α ubiquitination, Sf21 cells infected with the baculoviruses indicated in the figures were lysed with ice-cold buffer containing 40 mM Hepes-NaOH (pH 7.9), 150 mM NaCl, 1 mM DTT, 0.5% (vol/vol) Triton X-100, 10% (vol/vol) glycerol, 5 μg/ml leupeptin, 5 μg/ml antipain, 5 μg/ml pepstatin A, and 5 μg/ml aprotinin. After centrifugation at 10,000 × g for 20 min at 4°C, the supernatants were immunoprecipitated with 2 μg of anti-VHL (Ig32) antibody and 10 μl of protein A-Sepharose. The beads were mixed with ≈50 ng of Uba1, ≈100 ng of hUbc5a, ≈3 μg of GST-ubiquitin, and an aliquot of lysate of Sf21 cells expressing Flag-HIF1α in a 20-μl reaction containing 40 mM Hepes-NaOH (pH 7.9), 60 mM potassium acetate, 2 mM DTT, 5 mM MgCl2, 0.5 mM EDTA (pH 7.9), 10% (vol/vol) glycerol, 1.5 mM ATP, 10 mM creatine phosphate, and 10 μg of creatine phosphokinase. Reaction mixtures were incubated for 1 h at 30°C.
Results
The Purified Recombinant VHL Complex Activates HIF1α Ubiquitination in Vitro.
To investigate the possibility that the VHL complex supports ubiquitination of HIF1α, we reconstituted the recombinant, 5-subunit VHL complex in Sf21 cells, purified it to near homogeneity, and assayed it for its ability to stimulate HIF1α ubiquitination in vitro. Sf21 insect cells were coinfected with baculoviruses encoding N-terminal 6-histidine-tagged VHL (His-VHL), N-terminal myc-tagged Rbx1 (myc-Rbx1), and untagged Cul2, Elongin B, and Elongin C. The VHL complex was purified from cell lysates by consecutive Ni2+-agarose chromatography and TSK DEAE-NPR HPLC. As shown in the Coomassie-stained SDS/polyacrylamide gel and Western blots of Fig. 1B, apparently stoichiometric amounts of Cul2, Rbx1, and Elongins B and C cofractionated closely with VHL during gradient elution from TSK DEAE-NPR. To determine whether the purified VHL complex supports HIF1α ubiquitination, aliquots of the DEAE-NPR column fractions were assayed in the experiment of Fig. 1C for the ability to activate HIF1α ubiquitination in the presence of ATP and purified recombinant GST-ubiquitin, E1 ubiquitin-activating enzyme Uba1, and the E2 ubiquitin-conjugating enzyme hUbc5a, which was previously shown to be activated by the VHL complex (9). The HIF1α used as substrate for ubiquitination assays contained N-terminal 6-histidine- and HPC4-tags, and it was expressed in Sf21 cells and purified by Ni2+-agarose chromatography (Fig. 1A). HIF1α and lower electrophoretic mobility GST-ubiquitin-HIF1α conjugates were detected by Western blotting with anti-HPC4 antibodies. As shown in Fig. 1C, HIF1α ubiquitination activity closely copurified with the VHL complex. Activation of HIF1α ubiquitination by the VHL complex was strongly dependent on ATP, GST-ubiquitin, E1 ubiquitin-activating enzyme Uba1, and E2 ubiquitin-conjugating enzyme hUbc5a (Fig. 1D). In addition, the VHL complex failed to activate ubiquitination of phosphorylated Cln2, a substrate for ubiquitination activated by the SCFGrr1 complex (Fig. 1D).
The VHL complex has been shown previously to activate formation of polyubiquitin chains by the E2s hUbc3 (Cdc34) and hUbc5a, hUbc5b, and hUbc5c (8, 9). To characterize further the requirements for HIF1α ubiquitination in our reconstituted system, we compared the abilities of several E2s to catalyze conjugation of GST-ubiquitin to HIF1α. Of the E2s tested, only hUbc5a supported maximal levels of HIF1α ubiquitination, whereas hUbc3 (Cdc34) supported very low, but detectable, levels of HIF1α ubiquitination (Fig. 1E). Whether Ubc5 group enzymes and/or Ubc3 are the major E2s activated by the VHL complex in cells remains to be determined.
Activation of HIF1α Ubiquitination Depends on VHL, Cul2, Rbx1, and the Elongin BC Complex.
To investigate the contribution of individual subunits of the VHL complex to HIF1α ubiquitination, Sf21 cells were coinfected with various combinations of baculoviruses encoding untagged VHL, N-terminal 6-histidine- and myc-tagged Rbx1 (His-myc-Rbx1), and untagged Cul2, Elongin B, and Elongin C. VHL-containing complexes were immunoprecipitated from cell lysates with anti-VHL antibodies and analyzed (i) for the presence of individual subunits of the VHL complex by Western blotting and (ii) for their abilities to activate HIF1α ubiquitination in the presence of ATP and purified recombinant GST-ubiquitin, E1 ubiquitin-activating enzyme Uba1, and E2 ubiquitin-conjugating enzyme hUbc5a. Lysate of Sf21 cells overexpressing N-terminal Flag-tagged HIF1α was the source of HIF1α used as substrate in these assays. As shown in Fig. 2, maximal activation of HIF1α ubiquitination was observed only in the presence of VHL-containing complexes purified from cells overexpressing all five subunits of the VHL complex. A low level of HIF1α ubiquitination was observed when reactions contained complexes purified from cells that were not overexpressing Elongins B and C, most likely because of the presence of a substoichiometric amount of contaminating endogenous insect cell Elongins B and C (not detectable by immunoblotting using antibodies against the mammalian cell proteins) in immunoprecipitated VHL complexes. Consistent with this possibility, VHL complexes immunoprecipitated from lysates of insect cells that were not infected with baculoviruses encoding Elongins B and C contained Cul2 (Fig. 2B), even though entry of Cul2 into the VHL complex has been shown to depend strongly on the presence of Elongins B and C (13, 21). Finally, we note that formation of an immunoreactive species that may correspond to HIF1α conjugated to a single GST-ubiquitin is stimulated by addition of control immunoprecipitates prepared from cells that do not overexpress human VHL (Fig. 2C, lane 2). It is possible that formation of this species is due to the presence of a small amount of insect cell VHL in anti-human VHL immunoprecipitates. It seems more likely, however, that its formation is due to the presence of a contaminating activity independent of the VHL complex, because these reactions contain no detectable mammalian Cul2, Elongins B and C, or Rbx1 (Fig. 2B, lane 1), which might be expected to immunoprecipitate with the endogenous insect cell VHL.
Figure 2.

All subunits of the VHL complex are required for maximal activation of HIF1α ubiquitination. (A) Total cell lysates from Sf21 cells infected with the baculoviruses indicated in the figure were subjected to 8% or 13% SDS/PAGE, and proteins were transferred to Hybond P membranes and visualized by Western blotting as described in Materials and Methods, using the antibodies indicated in the figure. (B) The cell lysates of A were subjected to immunoprecipitations with anti-VHL antibodies as described in Materials and Methods. The immunoprecipitates were subjected to 8% or 13% SDS/PAGE, and proteins were transferred to Hybond P membranes and visualized by Western blotting as described in Materials and Methods, using the antibodies indicated in the figure. (C) The cell lysates of A were subjected to immunoprecipitations with anti-VHL antibodies as described in Materials and Methods. The immunoprecipitates were assayed for the ability to activate HIF1α ubiquitination as described in Materials and Methods. Reaction products were subjected to 8% SDS/PAGE, and GST-ubiquitin-HIF1α conjugates (GST-Ub-HIF1α) were transferred to Hybond P membranes and visualized by Western blotting as described in Materials and Methods, using anti-HIF1α antibodies. EloB, Elongin B; EloC, Elongin C.
VHL Mutants with Decreased Affinities for Elongins B and C or HIF1α Exhibit Reduced Activities in HIF1α Ubiquitination.
To investigate further the requirement for Elongin BC in stimulation of HIF1α ubiquitination, we compared the activities of wild-type VHL and two VHL mutants that are impaired in their abilities to bind Elongins B and C. VHL mutant VHL [1–155] contains VHL residues 1 to 155, which includes most of the VHL β-domain, but lacks the VHL α-domain and its Elongin BC binding site (22). VHL mutant VHL[L158P] contains a leucine to proline mutation in a critical residue in the Elongin BC binding site; VHL[L158P] is a naturally occurring mutant found in some VHL kindreds and VHL-associated tumors (2).
Sf21 cells were coinfected with various combinations of baculoviruses encoding N-terminal 6-histidine- and myc-tagged Rbx1 (His-myc-Rbx1), untagged Cul2, Elongin B, Elongin C, and untagged VHL, VHL [1–155], or VHL[L158P]. VHL-containing complexes were immunoprecipitated from cell lysates with anti-VHL antibodies and analyzed (i) for the presence of individual subunits of the VHL complex by Western blotting and (ii) for their abilities to activate HIF1α ubiquitination in the presence of ATP, and purified recombinant GST-ubiquitin, E1 ubiquitin-activating enzyme Uba1, and E2 ubiquitin-conjugating enzyme hUbc5a. Lysate of Sf21 cells overexpressing N-terminal Flag-tagged HIF1α was the source of HIF1α used as substrate in these assays. As shown in Fig. 3B, VHL mutants VHL[1–155] and VHL[L158P] were severely impaired in their abilities to bind to Elongins B and C. Consistent with the previous observation that entry of Cul2 into the VHL complex depends on the presence of Elongins B and C (13, 21), VHL mutants VHL[1–155] and VHL[L158P] also failed to bind Cul2. Furthermore, VHL-containing complexes purified from insect cells expressing VHL mutants VHL[1–155] or VHL[L158P] failed to activate HIF1α ubiquitination above the low background level observed in the absence of VHL (Fig. 3C), even though VHL[1–155] and VHL[L158P] were able to bind HIF1α. As shown in Fig. 4, both VHL[1–155] and VHL[L158] could be coimmunoprecipitated with HIF1α from lysates of Sf21 cells expressing these VHL mutants and HIF1α.
Figure 3.

VHL mutants that do not bind stably to Elongins B and C do not assemble into the VHL complex and do not detectably activate HIF1α ubiquitination. (A) Total cell lysates from Sf21 cells infected with the baculoviruses indicated in the figure were subjected to 8% or 13% SDS/PAGE, and proteins were transferred to Hybond P membranes and visualized by Western blotting as described in Materials and Methods, using the antibodies indicated in the figure. (B) The cell lysates of A were subjected to immunoprecipitations with anti-VHL antibodies as described in Materials and Methods. The immunoprecipitates were subjected to 8% or 13% SDS/PAGE, and proteins were transferred to Hybond P membranes and visualized by Western blotting as described in Materials and Methods, using the antibodies indicated in the figure. (C) The cell lysates of A were subjected to immunoprecipitations with anti-VHL antibodies as described in Materials and Methods. The immunoprecipitates were assayed for the ability to activate HIF1α ubiquitination as described in Materials and Methods. Reaction products were subjected to 8% SDS/PAGE, and GST-ubiquitin-HIF1α conjugates (GST-Ub-HIF1α) were transferred to Hybond P membranes and visualized by Western blotting as described in Materials and Methods, using anti-HIF1α antibodies. EloB, Elongin C; EloC, Elongin C.
Figure 4.
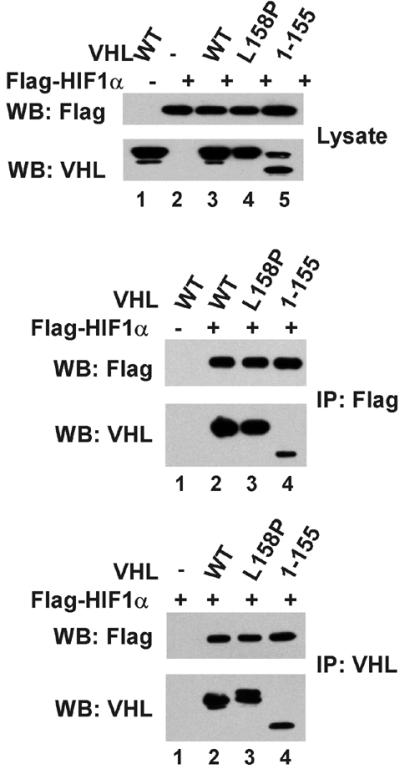
Interaction of HIF1α with VHL mutants VHL[1–155] and VHL[L158P]. (Upper) Total cell lysates from Sf21 cells infected with the baculoviruses indicated in the figure were subjected to 8% or 13% SDS/PAGE, and proteins were transferred to Hybond P membranes and visualized by Western blotting as described in Materials and Methods, using the antibodies indicated in the figure. (Middle and Lower) Cell lysates were subjected to immunoprecipitations with anti-VHL or anti-Flag antibodies as described in Materials and Methods. The immunoprecipitates were subjected to 8% or 13% SDS/PAGE, and proteins were transferred to Hybond P membranes and visualized by Western blotting as described in Materials and Methods, using the antibodies indicated in the figure.
A number of tumor-associated VHL mutations that do not significantly affect binding to Elongin B and C form a solvent-exposed patch on the VHL β domain, which has been proposed to be important for binding to proteins targeted for ubiquitination (22). A portion of the β domain is deleted in the VHL mutant VHL[117–213], and, as shown in Fig. 5A, VHL [117–213] binds poorly to HIF1α when the two proteins are coexpressed in insect cells. To determine whether VHL[117–213] is also defective in HIF1α ubiquitination, wild-type VHL or VHL[117–213] were coexpressed in insect cells with Cul2, Elongins B and C, and Rbx1, and the resulting complexes were purified from insect cell lysates by Ni2+-agarose chromatography and TSK DEAE-NPR HPLC. As shown in Fig. 5B, both wild-type VHL and VHL[117–213] assembled into chromatographically isolable complexes containing each of the VHL complex subunits; however, complexes containing VHL[117–213] were defective in their ability to support HIF1α ubiquitination (Fig. 5C).
Discussion
In summary, in this report we present direct biochemical evidence that the VHL tumor suppressor complex is capable of activating hUbc5a ubiquitination of hypoxia-inducible transcription factor HIF1α in vitro. In light of the previous observations (i) that the cellular level of HIF1α is tightly regulated by ubiquitin-dependent proteolysis (7) and (ii) that HIF1α accumulates in cells lacking a functional VHL gene (6), our findings are consistent with the model that the VHL complex is directly responsible for HIF1α ubiquitination and degradation by the proteasome.
Activation of HIF1α ubiquitination by the VHL complex depends on its individual VHL, Cul2, Elongin, and Rbx1 subunits, which appear to perform distinct tasks in recruiting HIF1α and the E2 ubiquitin-conjugating enzyme to the complex. Evidence from previous studies suggests that Cul2 and Rbx1 interact to form a Cul2/Rbx1 module that is sufficient to recruit and activate ubiquitination by members of the Ubc5 family of E2 ubiquitin-conjugating enzymes (15, 23–26). Elongins B and C form a subcomplex that functions at least in part as an adaptor that links the Cul2/Rbx1 module to the VHL protein. In light of evidence that the VHL and HIF1α proteins can be coimmunoprecipitated from mammalian and insect cells, the VHL protein may be solely responsible for binding to and recruiting HIF1α to the VHL complex, placing HIF1α in close proximity to the Cul2/Rbx1 module and its associated E2 ubiquitin-conjugating enzyme.
In these respects, the VHL complex bears a striking resemblance to the well-characterized SCF E3 ubiquitin ligase complexes (16, 17, 27). As subunits of SCF complexes, Cul1 (or Cdc53 in S. cerevisiae) and Rbx1 interact to form a Cul1/Rbx1 module that is capable of recruiting and activating ubiquitination of target proteins by the E2 ubiquitin-conjugating enzyme Cdc34 (23, 26). The Elongin C-like Skp1 protein functions as an adaptor that links the Cul1/Rbx1 module to one of several F-box proteins that bind to and recruit ubiquitination substrates to the complex (27, 28). VHL and F-box proteins may perform similar functions as subunits of their respective VHL and SCF complexes. Notably, just as Cul1/Skp1-containing SCF complexes rely on multiple F-box proteins to target their diverse collection of ubiquitination substrates, it is possible that at least some of the more than 20 identified Elongin BC-binding proteins will function as substrate recognition subunits (29, 30). Interestingly, the Elongin BC-binding protein suppressor of cytokine signaling (SOCS)-1, initially identified as an inhibitor of Janus kinase (Jak) kinases (31–33), has been shown to bind the hematopoietic-specific guanine nucleotide exchange factor Vav and target it for ubiquitin-dependent proteolysis in cells (34). In light of this observation, it is tempting to speculate that SOCS-1 serves as the substrate recognition subunit of a ubiquitin ligase complex. Consistent with this possibility, we have observed that, like VHL, SOCS-1 and several other members of the SOCS–box family of Elongin BC-binding proteins can assemble into multiprotein complexes containing Cul2, Elongins B and C, and Rbx1 (T.K., R.C.C., and J.W.C., unpublished results).
Finally, we note that, in independent lines of research, Cockman et al. (35) and Ohh et al. (36) have recently shown that HIF1α and HIF2α can be ubiquitinated in a VHL-dependent manner in crude mammalian cell lysates. Together with our findings, the results of these studies should provide a solid foundation for future efforts to understand the biochemical events that underlie oxygen-dependent signal transduction via the VHL tumor suppressor complex.
Acknowledgments
We thank A. Weissman for Ubc5 clones, C. Esmon for anti-HPC4 monoclonal antibodies, and D. Haque and J. Mollica for expert technical assistance. This work was supported in part by National Institutes of Health Grant GM41628 and by funds provided to the Oklahoma Medical Research Foundation by the H. A. and Mary K. Chapman Charitable Trust. J.W.C. is an Associate Investigator of the Howard Hughes Medical Institute.
Abbreviations
- VEGF
vascular endothelial growth factor
- SCF
Skp1-Cul1/Cdc53-F-box
- GST
glutathione S-transferase
- SOCS
suppressor of cytokine signaling
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.190332597.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.190332597
References
- 1.Latif F, Tory K, Gnarra J, Yao M, Duh F M, Orcutt M L, Stackhouse T, Kuzmin I, Modi W, Geil L, et al. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 2.Gnarra J R, Duan D R, Weng Y, Humphrey J S, Chen D Y, Lee S, Pause A, Dudley C F, Latif F, Kuzmin I, et al. Biochim Biophys Acta. 1996;1242:201–210. doi: 10.1016/0304-419x(95)00012-5. [DOI] [PubMed] [Google Scholar]
- 3.Gnarra J R, Zhou S, Merrill M J, Wagner J R, Krumm A, Papavassiliou E, Oldfield E H, Klausner R D, Linehan W M. Proc Natl Acad Sci USA. 1996;93:10589–10594. doi: 10.1073/pnas.93.20.10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iliopoulos O, Levy A P, Jiang C, Kaelin W G, Goldberg M A. Proc Natl Acad Sci USA. 1996;93:10595–10599. doi: 10.1073/pnas.93.20.10595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siemeister G, Weindel K, Mohrs K, Barleon B, Martiny-Baron G, Marme D. Cancer Res. 1996;56:2299–2301. [PubMed] [Google Scholar]
- 6.Maxwell P H, Wiggener M S, Chang G W, Clifford S C, Vaux E C, Cockman M E, Wykoff C C, Pugh C W, Maher E R, Ratcliffe P J. Nature (London) 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 7.Semenza G L. Annu Rev Cell Dev Biol. 2000;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 8.Lisztwan J, Imbert G, Wirbelauer C, Gstaiger M, Krek W. Genes Dev. 1999;13:1822–1833. doi: 10.1101/gad.13.14.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iwai K, Yamanaka K, Kamura T, Minato N, Conaway R C, Conaway J W, Klausner R D, Pause A. Proc Natl Acad Sci USA. 1999;96:12436–12441. doi: 10.1073/pnas.96.22.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duan D R, Pause A, Burgess W H, Aso T, Chen D Y T, Garrett K P, Conaway R C, Conaway J W, Linehan W M, Klausner R D. Science. 1995;269:1402–1406. doi: 10.1126/science.7660122. [DOI] [PubMed] [Google Scholar]
- 11.Kibel A, Iliopoulos O, DeCaprio J A, Kaelin W G. Science. 1995;269:1444–1446. doi: 10.1126/science.7660130. [DOI] [PubMed] [Google Scholar]
- 12.Kishida T, Stackhouse T M, Chen F, Lerman M I, Zbar B. Cancer Res. 1995;20:4544–4548. [PubMed] [Google Scholar]
- 13.Pause A, Lee S, Worrell R A, Chen D Y T, Burgess W H, Linehan W M, Klausner R D. Proc Natl Acad Sci USA. 1997;94:2156–2161. doi: 10.1073/pnas.94.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lonergan K M, Iliopoulos O, Ohh M, Kamura T, Conaway R C, Conaway J W, Kaelin W G. Mol Cell Biol. 1998;18:732–741. doi: 10.1128/mcb.18.2.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamura T, Koepp D M, Conrad M N, Skowyra D, Moreland R J, Iliopoulos O, Lane W S, Kaelin W G, Elledge S J, Conaway R C, Harper J W, Conaway J W. Science. 1999;284:657–661. doi: 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- 16.Patton E E, Willems A R, Tyers M. Trends Biochem Sci. 1998;14:236–243. doi: 10.1016/s0168-9525(98)01473-5. [DOI] [PubMed] [Google Scholar]
- 17.Deshaies R J. Annu Rev Cell Dev Biol. 1999;15:435–467. doi: 10.1146/annurev.cellbio.15.1.435. [DOI] [PubMed] [Google Scholar]
- 18.Stearns D J, Kurosawa S, Sims P J, Esmon N L, Esmon C T. J Biol Chem. 1988;263:826–832. [PubMed] [Google Scholar]
- 19.Garrett K P, Aso T, Bradsher J N, Foundling S I, Lane W S, Conaway R C, Conaway J W. Proc Natl Acad Sci USA. 1995;92:7172–7176. doi: 10.1073/pnas.92.16.7172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamura T, Conrad M N, Yan Q, Conaway R C, Conaway J W. Genes Dev. 1999;13:2928–2933. doi: 10.1101/gad.13.22.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pause A, Peterson B, Schaffar G, Stearman R, Klausner R D. Proc Natl Acad Sci USA. 1999;96:9533–9538. doi: 10.1073/pnas.96.17.9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stebbins C E, Kaelin W G, Pavletich N P. Science. 1999;284:455–461. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- 23.Skowyra D, Koepp D M, Kamura T, Conrad M N, Conaway R C, Conaway J W, Elledge S J, Harper J W. Science. 1999;284:662–665. doi: 10.1126/science.284.5414.662. [DOI] [PubMed] [Google Scholar]
- 24.Ohta T, Michel J J, Schottelius A J, Xiong Y. Mol Cell. 1999;3:535–541. doi: 10.1016/s1097-2765(00)80482-7. [DOI] [PubMed] [Google Scholar]
- 25.Tan P, Fuchs S Y, Chen A, Wu K, Gomez C, Ronai Z, Pan Z Q. Mol Cell. 1999;3:527–533. doi: 10.1016/s1097-2765(00)80481-5. [DOI] [PubMed] [Google Scholar]
- 26.Seol J H, Feldman R M, Zachariae W, Shevchenko A, Correll C C, Lyapina S A, Chi Y, Galova M, Claypool J, Sandmeyer S, Nasmyth K, Deshaies R J. Genes Dev. 1999;13:1614–1626. doi: 10.1101/gad.13.12.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper J W, Elledge S J. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- 28.Skowyra D, Craig K L, Tyers M, Elledge S J, Harper J W. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- 29.Kamura T, Sato S, Haque D, Liu L, Kaelin W G, Conaway R C, Conaway J W. Genes Dev. 1998;12:3872–3881. doi: 10.1101/gad.12.24.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J G, Farley A, Nicholson S E, Willson T A, Zugaro L M, Simpson R J, Moritz R L, Cary D, Richardson R, Hausmann G, et al. Proc Natl Acad Sci USA. 1999;96:2071–2076. doi: 10.1073/pnas.96.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Starr R, Willson T A, Viney E M, Murray L J L, Rayner J R, Jenkins B J, Gonda T J, Alexander W S, Metcalf D, Nicola N A, Hilton D J. Nature (London) 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 32.Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, Nishimoto N, Kajita T, Taga T, Yoshizaki K, Akira S, Kishimoto T. Nature (London) 1997;387:924–928. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- 33.Endo T, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H, et al. Nature (London) 1997;387:921–924. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- 34.De Sepulveda P, Ilangumaran S, Rottapel R. J Biol Chem. 2000;275:14005–14008. doi: 10.1074/jbc.c000106200. [DOI] [PubMed] [Google Scholar]
- 35.Cockman M E, Masson N, Mole D R, Jaakola P, Chang G W, Clifford S C, Maher E R, Pugh C W, Ratcliffe P J, Maxwell P H. J Biol Chem. 2000;275:10.074. doi: 10.1074/jbc.M002740200. /jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- 36.Ohh M, Park C W, Ivan M, Hoffman M A, Kim T Y, Huang L E, Pavletich N, Chau V, Kaelin W G. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]