

Abstract
Two recent reports provide new information on how DNA damage may generate progeroid changes at the cell and organismal level by suppressing growth hormone (GH)/insulin-like growth factor 1 (IGF1) endocrine signaling. This endocrine signaling pathway is of particular interest as it is a key regulator of metabolism and longevity in many organisms.
DNA damage and constitutional defects in DNA repair can promote mutagenesis, elevate disease risk and, in some circumstances, lead to features of premature aging. How DNA damage leads to these important, undesirable outcomes is not well understood. Two recent reports [1, 2] from collaborating investigators in the Netherlands, United States and Japan provide new information on this question, and identify a mechanism by which DNA damage may trigger progeroid changes: DNA damage-dependent suppression of growth hormone (GH)/insulin-like growth factor 1 (IGF1) endocrine signaling. This endocrine signaling system is a deeply conserved, key regulator of metabolism and longevity in many organisms.
This story began with analyses of nucleotide excision repair (NER) defects in patients and in mouse models. Niedernhofer and colleagues [1] were stimulated to re-examine a mouse model of Ercc1 deficiency [3] after studying a 15 yr old boy with sun sensitivity who shared many phenotypic similarities with Ercc1−/− mice (summarized in Supplemental Table II of [1]). These features included a NER defect and progressive progeroid features that led to multi-organ system failure and death at age 16. The clinical and pathologic findings in this patient were distinct from classical xeroderma pigmentosum (XP), Cockayne syndrome (CS) or XP/CS, and appeared to result from a previously unreported homozygous XPF mutation that leads to a R153P substitution at a highly conserved residue in the N-terminal XPF helicase motif. XPF, together with its protein partner ERCC1, constitute the ERCC1-XPF endonuclease that is a core component in both the NER and DNA cross link repair pathways (reviewed in [4–6]). Fibroblasts from this patient had reduced levels of XPF and ERCC1 proteins, were UV-sensitive as measured by both UV survival and the recovery of RNA synthesis after UV radiation and were exquisitely mitomycin-C sensitive. This unusual constellation of clinical and cellular features was designated XFE (for XPF-ERCC1) progeroid syndrome, to distinguish this patient and phenotype from other XPF mutation carriers who typically have subtle biochemical defects, retain partial NER and display a mild XP clinical phenotype [4, 7, 8].
Mouse mutants deficient for Ercc1 or Xpf had been previously generated in order to better understand the role of each protein in NER. Mutant mice in each instance were viable, but displayed phenotypes more severe than previously observed in other mouse models of NER deficiency [3, 9, 10]. For example, Ercc1−/− mice display a profound growth arrest beginning 2 weeks after birth, followed by the appearance of cellular progeroid changes and progressive multi-system organ dysfunction. Virtually all Ercc1−/− mice die within 4 weeks of birth. Niedernhofer et al. took advantage of this mouse mutant to gain insight into the pathogenesis of XFE progeroid syndrome by performing liver gene expression analyses using 15 day old Ercc1−/− mice, litter mate controls, and young or aged (130 week old) mice. Fifteen day-old Ercc1−/− mice were chosen for these analyses by determining the age of maximum weight gain prior to the onset of widespread, potentially confounding pathologic changes.
A total of 1,675 genes, or 4.9% of those represented on the Affymetrix 430 v2.0 array that was used, displayed significant expression differences in Ercc1−/− mice as compared with litter mate controls. Many of these genes fell into a small number of functionally interrelated Gene Ontology biological process categories (www.geneontology.org; [11](Figure 1). Genes whose expression was suppressed were involved in endocrine-mediated somatotroph, lactotroph and thyrotroph growth and metabolic regulatory pathways, or in oxidative and carbohydrate metabolism. In contrast, genes whose expression was upregulated in mutant mice were associated with glycogen and fatty acid synthesis, β-oxidation, or with anti-oxidant and toxicant defense, DNA repair or the upregulation of apoptosis. Expression changes in a small subset of these genes were verified by qRT-PCR, and shown to become progressively more marked between 15 and 21 days when half of Ercc1−/− mice have died.
Figure 1. Gene expression changes in repair-deficient Ercc1−/− and Csbm/m/Xpa−/− mice compared with aged 130 week-old control mice.
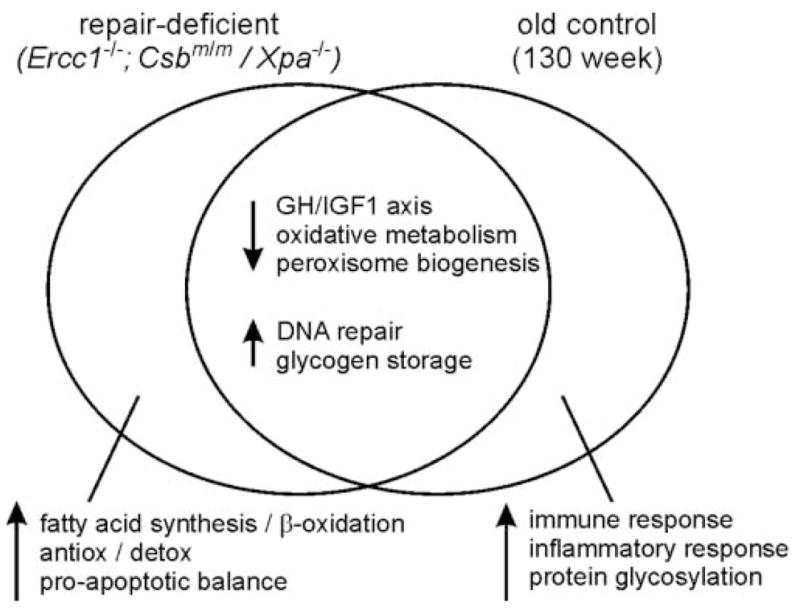
Expression analyses were performed using RNA prepared from each mutant genotype isolated at post-natal day 15 (mutant mice) or at 130 weeks of age (C57BL/6J controls). Four or 6 mice were used for each analysis. Groups of genes with expression changes that were shared across all three genotypes are shown in the overlap region (center). Gene groups selectively up- or down-regulated in either mutant or 130 week old control mice are shown in non-overlap regions to the left (mutant) or right (aged control). Up-and down-regulation are indicated, respectively, by up or down arrows. Gene groups were identified and designated by biological process as identified by a Gene Ontology analysis (see [1,2,11] for additional details.
The identification of IGF1 endocrine pathway suppression in Ercc1−/− mice is of particular interest as this pathway is a major determinant of both metabolism and life span in a wide range of organisms [12–15]. IGF1 suppression could also provide a ready explanation for a lack of post-natal weight gain in Ercc1−/− mice, and persistent suppression of this pathway could explain the development of progeroid features. In order to more fully characterize IGF1 pathway suppression the authors demonstrated that gene expression changes and phenotype were progressive and did not have a substantial, overt developmental component; that IGF1 suppression was peripheral and secondary, and not the result of a central pituitary defect; that IGF1 suppression preceded the development of frank liver pathology; and that a similar IGF1 suppression could be induced in otherwise normal 10 week old mice by the chronic administration of subtoxic doses of the DNA cross linking agent mitomycin-C (0.1 mg kg−1 for 10 weeks).
Additional gene expression analyses were also performed as part of this series of experiments in order to identify gene expression changes attributable to Ercc1 deficiency alone or to advanced chronological age. These analyses used control juvenile mice (8 weeks old), young adult mice (16 weeks old) or aged mice (130 weeks old). Many of the expression changes observed in Ercc1−/− mice were also observed in aged (130 week old) control mice, though not in control mice of earlier ages. Gene expression changes limited to Ercc1−/− mice included upregulation of apoptosis-promoting and anti-oxidant defense genes. In contrast, the upregulation of immune response, inflammatory response and protein glycosylation gene expression was limited to aged control mice (Figure 1).
A parallel story was recently reported by the collaborating group of van der Pluijm and colleagues [2] provides additional information that confirms and extends these analyses. van der Pluijm and colleagues analyzed hepatic gene expression in Csbm/m/Xpa−/− (or/Xpc−/−) mice. Csbm/m mice are deficient in transcription-coupled repair (TCR), whereas Xpa- or Xpc-deficient mice are NER-deficient [2]. Csbm/m/Xpa−/− double-mutant mice were shown to be completely NER-deficient, and resembled Ercc1−/− mice in having a global growth defect after birth and widespread cell- and tissue-specific defects. These mice also develop a progeroid appearance and die within 4 weeks of birth.
Gene expression in the liver of Csbm/m/Xpa−/− mice was analyzed using the same microarray platform used by Niedernhofer et al. [1]. The expression of 1,865 genes (5.5% of those represented on the array) was altered as compared with litter mate controls. Many of the same expression changes observed in Ercc1−/− mice were again seen: GH/IGF1 endocrine suppression; the upregulation of genes involved in fatty acid synthesis, fat storage, β-oxidation and enhanced glycogen synthesis; and the upregulation of antioxidant defense genes. Of note, single mutant Csbm/m or Xpa−/− mice did not display these gene expression changes or show GH/IGF1 suppression. van der Pluijm and colleagues also demonstrated that chronic exposure of 4 week old control mice to subtoxic levels of a compound that can produce oxidative DNA damage, di(2-ethylhexyl)phthalate (DEHP; 1500 ppm for 9 weeks) also suppressed GH/IGF1 endocrine signaling system gene expression.
These two series of experiments indicate that the suppression of GH/IGF1 signaling was common to two mouse models with genetically engineered defects in NER, and that comparable responses could be induced in control mice by treatment with subtoxic doses of mitomycin-C or DEHP. The experiments of van der Pluijm and colleagues also point the way to explore additional, non-canonical functions of NER proteins. This opportunity stems from the observation that Csbm/m/Xpc−/− mice deficient in TCR and the global genome repair (GGR) differ in phenotype from ostensibly equivalent, TCR- and GGR-deficient Xpa-mutant mice. These differences indicate that Xpa may have function(s) outside of its role in ‘classical’ NER. These putative non-canonical functions could include, e.g., variants of TCR or BER that selectively target specific types of oxidative DNA damage, and thus are worthy of further investigation.
The inter-related series of experiments reported by Niedernhofer, van der Pluijm and colleagues bear close scrutiny both for the wealth of data they contain, and for the ideas, discussion and experiments they will provoke. The results of these experiments provide one of the most persuasive arguments to date that senescence and the appearance of progeroid features result from active recognition and response to DNA damage, rather than passive damage accumulation. These reports also identify a common mechanistic pathway, suppression of GH/IGF1 endocrine signaling, in the generation of both early growth defects and later progeroid changes. These are not the first reports to link DNA damage to IGF1 endocrine signaling, but they are the most convincing to demonstrate how DNA damage may affect this key growth and metabolic regulatory system to promote aging and disease pathogenesis.
A very recent report of the identification of a first patient with heritable ERCC1 deficiency [16] suggests this story has one additional aspect that was not apparent from analyses of Ercc1−/− and Csbm/m/Xpa−/− mice. The ERCC1-deficient patient identified by Jaspers and colleagues had, as expected, reduced levels of XPF and ERCC1, together with moderate cellular sensitivity to both UV light and mitomycin-C. This patient also had an unexpectedly severe constellation of morphologic and developmental abnormalities consistent with cerebro-oculo-facio-skeletal (COFS) or Pena-Shokier syndrome Type II (OMIM #214150). The COFS clinical phenotype appears to result from a truncating Q158X mutation in one ERCC1 allele, together with mutation of the other allele leading to a F231L substitution in a highly conserved residue in the C-terminal half of ERCC1. The clinical phenotype of this ERCC1-deficient patient, the most severe of the clinical syndromes observed in NER-deficient patients, includes developmental defects and congenital defects arising from inter-uterine growth retardation. This suggests that ERCC1, as was suggested above for XPA, might play a role in development that is distinct from the roles played by these proteins as core components of NER. Of note, patients with clinical variants of COFS syndrome have been found to harbor mutations in several different DNA repair genes including CSB, XPD and ERCC5 in addition to ERCC1 (see OMIM #214150 for additional details).
What questions remain to be answered, and where are these stories headed? Several aspects of the link between DNA damage and IGF suppression need to be better defined to start. It would be useful, for example, to know what type(s) of DNA damage have the potential to drive IGF1 suppression in addition to DNA interstrand cross link damage and oxidative damage and whether this damage is repaired predominantly by NER or BER. A second question is whether there is a DNA damage threshold for IGF1 suppression, and if so how this threshold is set and controlled. A better understanding of this issue might explain the puzzling observation that IGF1 suppression can be induced in otherwise normal mice by chronic, subtoxic exposure to mitomycin-C or DEHP, though is apparently absent in TCR-deficient Csbm/m and in NER-deficient Xpa−/− mice. A third question focuses on the molecular details of how DNA damage recognition is transduced to suppress gene expression. One hint that this (or these) transduction pathway(s) may involve non-canonical DNA damage-dependent signaling is mentioned in passing by van der Pluijm et al., who note that mutating p53 failed to suppress the phenotype of Csbm/m/Xpa−/− mutant mice despite evidence of constitutive p53 activation in the liver of double-mutant animals.
One aspect of this story may, at first glance, strike many readers as paradoxical: that the same gene expression changes observed by Niedernhofer, van der Pluijm and colleagues in NER-deficient and in mutagen-treated mice have also been observed in calorie-restricted, long-lived mice (reviewed in [15]). The authors suggest this seeming paradox is explained by proposing that the gene expression changes observed in DNA repair-deficient, in mutagen-treated and in calorie-restricted mice are part of a common, highly conserved response pathway that acts to minimize DNA damage and maintain organismal integrity. Mice that are persistently mutagen-treated or that are DNA repair-deficient thus mount the same protective response, but cannot fully counter the consequences of high levels of DNA damage. Thus ‘excess’ DNA damage and correspondingly high levels of mutagenesis, cellular senescence and cell death may conspire to promote progeroid changes and disease pathogenesis (Figure 2).
Figure 2. Model linking DNA damage to IGF1 axis suppression, cancer and aging.
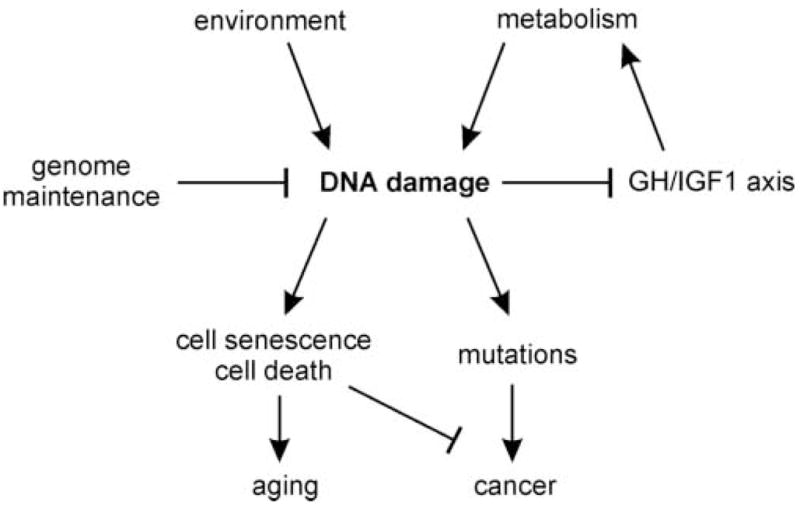
DNA damage from either environmental exposure or as a result of DNA repair defects can suppress GH/IGF1 axis signaling. This triggers a conserved protective response that suppresses the GH/IGF1 somatotroph and other axes to limit metabolism and the further accumulation of DNA damage. DNA repair-deficient mice induce the same protective response, but cannot fully counter the downstream consequences of persistently high levels of DNA damage: cell senescence, cell death and mutation accumulation that promote aging and the emergence of cancer.
How might these new findings be used to identify ways to modulate either aging or age-associated disease? One idea is to focus on gene expression changes limited to aged mice. These include genes associated with the immune response, the inflammatory response and protein glycosylation, all processes that are–or may soon be–good targets for drugs or small molecule modulators. A second, still controversial, idea is to constitutively activate DNA damage response and repair pathways to limit DNA damage accumulation. Intriguing work along these lines has already begun to appear (see, e.g., [17]). A third idea is to focus on limiting DNA damage by a combination of dietary modification and supplementation (see, e.g., [18]). van der Pluijm and colleagues close their discussion by providing us with a teaser along these lines in literally a last sentence: they describe a pilot study using Csbm/m/Xpa−/− mice in which a ‘…nutriceutical intervention…aiming at extending lifespan and delaying the onset of pathology’ [has] yielded promising results.’ These three recent reports highlight an important story that touches on many aspects of biology, medicine and disease pathogenesis. So add this story to your ‘watch’ list, and stay tuned!
Acknowledgments
Work in the author’s lab is supported by grants from the U.S. National Institutes of Health, the U.S. National Cancer Institute and the Gates Grand Challenges in Global Health program. I thank colleagues Larry Loeb and Matt Kaeberlein for insightful comments on a draft of this manuscript, and an anonymous reviewer for suggesting the possibility of non-canonical functions of Xpa/XPA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GTJ, Meinecke P, Kleijer WJ, Vijg J, Jaspers NGJ, Hoeijmakers JHJ. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–1043. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- 2.van der Pluijm I, Garinis GA, Brandt RMC, Gorgels TGMF, Wijnhoven SW, Diderich KEM, de Wit J, Mitchell JR, van Oostrom C, Beems R, Niedernhofer LJ, Velasco S, Friedberg EC, Tanaka K, van Steeg H, Hoeijmakers JHJ, van der Horst GTJ. Impaired Genome Maintenance Suppresses the Growth Hormone-Insulin-Like Growth Factor 1 Axis in Mice with Cockayne Syndrome. PLoS Biology. 2007;5:e2. doi: 10.1371/journal.pbio.0050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ, Nigg A, van Steeg H, Bootsma D, Hoeijmakers JHJ. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Current Biology. 1997;7:427–439. doi: 10.1016/s0960-9822(06)00190-4. [DOI] [PubMed] [Google Scholar]
- 4.Bootsma D, Kraemer KH, Cleaver JE, Hoeijmakers JHJ. Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic & Molecular Basis of Inherited Disease. McGraw-Hill; New York: 2001. pp. 677–703. [Google Scholar]
- 5.Cleaver JE. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat Rev Cancer. 2005;5:564–573. doi: 10.1038/nrc1652. [DOI] [PubMed] [Google Scholar]
- 6.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. ASM Press; Washington, DC: 2006. pp. 267–378.pp. 865–918. [Google Scholar]
- 7.Matsumura Y, Nishigori C, Yagi T, Imamura S, Takebe H. Characterization of molecular defects in xeroderma pigmentosum group F in relation to its clinically mild symptoms. Hum Mol Genet. 1998;7:969–974. doi: 10.1093/hmg/7.6.969. [DOI] [PubMed] [Google Scholar]
- 8.Sijbers AM, van Voorst Vader PC, Snoek JW, Raams A, Jaspers NGJ, Kleijer WJ. Homozygous R788W Point Mutation in the XPF Gene of a Patient with Xeroderma Pigmentosum and Late-Onset Neurologic Disease. J Invest Dermatol. 1998;110:832–836. doi: 10.1046/j.1523-1747.1998.00171.x. [DOI] [PubMed] [Google Scholar]
- 9.McWhir J, Selfridge J, Harrison DJ, Squires S, Melton DW. Mice with DNA repair gene (ERCC-1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nat Genet. 1993;5:217–224. doi: 10.1038/ng1193-217. [DOI] [PubMed] [Google Scholar]
- 10.Tian M, Shinkura R, Shinkura N, Alt FW. Growth Retardation, Early Death, and DNA Repair Defects in Mice Deficient for the Nucleotide Excision Repair Enzyme XPF. Mol Cell Biol. 2004;24:1200–1205. doi: 10.1128/MCB.24.3.1200-1205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carter CS, Ramsey MM, Sonntag WE. A critical analysis of the role of growth hormone and IGF-1 in aging and lifespan. Trends in Genetics. 2002;18:295–301. doi: 10.1016/S0168-9525(02)02696-3. [DOI] [PubMed] [Google Scholar]
- 13.Bartke A. Minireview: Role of the Growth Hormone/Insulin-Like Growth Factor System in Mammalian Aging. Endocrinology. 2005;146:3718–3723. doi: 10.1210/en.2005-0411. [DOI] [PubMed] [Google Scholar]
- 14.Vijg J, Suh Y. Genetics of longevity and aging. Annual Review of Medicine. 2005;56:193–212. doi: 10.1146/annurev.med.56.082103.104617. [DOI] [PubMed] [Google Scholar]
- 15.Kenyon C. The Plasticity of Aging: Insights from Long-Lived Mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 16.Jaspers NGJ, Raams A, Silengo MC, Wijgers N, Niedernhofer LJ, Robinson AR, Giglia-Mari G, Hoogstraten D, Kleijer WJ, Hoeijmakers JHJ, Vermeulen W. First Reported Patient with Human ERCC1 Deficiency Has Cerebro-Oculo-Facio-Skeletal Syndrome with a Mild Defect in Nucleotide Excision Repair and Severe Developmental Failure. Am J Hum Genet. 2007;80:457–466. doi: 10.1086/512486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider JG, Finck BN, Ren J, Standley KN, Takagi M, Maclean KH, Bernal-Mizrachi C, Muslin AJ, Kastan MB, Semenkovich CF. ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metabolism. 2006;4:377–389. doi: 10.1016/j.cmet.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Melov S, Ravenscroft J, Malik S, Gill MS, Walker DW, Clayton PE, Wallace DC, Malfroy B, Doctrow SR, Lithgow GJ. Extension of Life-Span with Superoxide Dismutase/Catalase Mimetics. Science. 2000;289:1567–1569. doi: 10.1126/science.289.5484.1567. [DOI] [PubMed] [Google Scholar]