

Abstract
Aberrant promoter hypermethylation is one of the major mechanisms in carcinogenesis and some critical growth regulatory genes have shown commonality in methylation across solid tumors. Twenty-six genes, 14 identified through methylation in colon and breast cancers, were evaluated using primary lung adenocarcinomas (n = 175) from current, former and never smokers. Tumor specificity of methylation was validated through comparison of 14 lung cancer cell lines to normal human bronchial epithelial cells derived from bronchoscopy of 20 cancer-free smokers. Twenty-five genes were methylated in 11–81% of primary tumors. Prevalence for methylation of TNFRSF10C, BHLHB5 and BOLL was significantly higher in adenocarcinomas from never smokers than smokers. The relation between methylation of individual genes was examined using pairwise comparisons. A significant association was seen between 138 (42%) of the possible 325 pairwise comparisons. Most notably, methylation of MMP2, BHLHB4 or p16 was significantly associated with methylation of 16–19 other genes, thus predicting for a widespread methylation phenotype. Kaplan–Meier log-rank test and proportional hazard models identified a significant association between methylation of SULF2 (a pro-growth, -angiogenesis and -migration gene) and better patient survival (hazard ratio = 0.23). These results demonstrate a high degree of commonality for targeted silencing of genes between lung and other solid tumors and suggest that promoter hypermethylation in cancer is a highly co-ordinated event.
Introduction
Genetic and epigenetic inactivation of tumor suppressor genes is probably causal for initiation and progression of human malignancies. Two recently published genome-wide screening assays discovered a large number of previously unknown mutations in breast, colon and lung cancers. Ding et al. (1) screened 623 genes in lung adenocarcinoma and detected >1000 somatic mutations. Prior to that, Sjoblom et al. (2) sequenced >13 000 genes in breast and colon cancers and identified an average of 90 mutant genes per tumor. However, the most prevalent genetic changes identified in these studies still involved mutations of TP53 and KRAS. Most of the mutations discovered in these studies occurred at prevalences <10%, suggesting that non-genetic abnormalities such as epigenetic alterations could also be responsible for loss of function during cancer development.
Aberrant promoter CpG island hypermethylation of growth regulatory genes is now a well-established alternative to genetic abnormalities. It silences the normal functions of tumor suppressor genes during carcinogenesis and is frequently observed in various human malignancies. In addition, epigenetic abnormalities may also work in harmony with genetic defects to completely inactivate a tumor suppressor gene through Knudson's two-hits, where mutation or loss of heterozygosity and methylation each inactivate one allele (3). Many of the new genes discovered to occasionally harbor somatic mutations contain a promoter CpG island that is methylated in >50% of breast and colon cancers (4,5). These findings suggest that epigenetic aberrations may dominate somatic mutations in driving initiation and progression of cancer and may commonly affect multiple genes across many solid tumors. Critical growth regulatory genes such as p16, RASSF1A, E-cadherin and adenomatosis polyposis coli (APC) are common targets for methylation in many solid tumors including lung, breast and colon.
Lung cancer, the most prevalent and the leading cause of cancer mortality worldwide (6), displays both genetic and epigenetic changes in various genes and chromosomal locations (1,7). We have recently evaluated methylation of candidate tumor suppressor genes located within one of the hot spots for genetic abnormalities in lung cancer, the long arm of chromosome 6 (8). While differences in prevalence were not observed between adenocarcinomas from smokers and never smokers, methylation of five candidate tumor suppressor genes was commonly seen in tumors.
Recent studies suggest lung cancer in lifelong never smokers, currently responsible for ∼15 000 deaths annually in the USA, is increasing and may involve some different pathways than in smokers (9,10). Some of the unique molecular and biological characteristics of lung cancer in never smokers include a shift in tumor histology to predominantly adenocarcinoma, some distinct genetic and epigenetic abnormalities and a better response to drugs targeting the epidermal growth factor receptor (11–13). These findings prompted us to investigate the methylation profile of selected candidate tumor suppressor genes in lung adenocarcinomas from different smoking groups.
The two major goals of the current study were to assess whether genes commonly methylated in other solid tumors are also methylated in lung cancer and to determine if the methylation profiles of these genes could differentiate lung tumors from smokers and never smokers. Twenty-six genes, 14 studied for the first time in lung adenocarcinoma, were evaluated using 175 primary lung adenocarcinomas from current, former and never smokers (4,5,14). The influence of smoking history, age, gender and stage of lung cancer on methylation was evaluated. The relation between methylation of each gene to patient survival and gene–gene associations were also compared.
Materials and methods
Samples
A total of 175 frozen lung adenocarcinomas from current (n = 37), former (n = 59) and never (n = 75) smokers were obtained from frozen tumor banks at Johns Hopkins and the Mayo Clinic. Demographic variables including age, gender and stage of lung cancer have been described previously (8). Each tumor was classified histologically as adenocarcinoma according to the current World Health Organization criteria and considered suitable for the study based on the presence of >80% tumor cells (15). Normal human bronchial epithelial cells (NHBEC) isolated from bronchoscopy of cancer-free smokers (n = 20) were used as controls. Fourteen lung cancer-derived cell lines (H23, H358, H1435, H1568, H1993, H2023, H2085, H2228, H2009, Calu-3, Calu-6, SKLU-1, SKMES1 and H1975) obtained from the American Type Culture Collection (Manassas, VA) and maintained in American Type Culture Collection-recommended media were studied.
DNA methylation analysis
DNA extraction and modification were done exactly as described (8) and 40 ng of modified DNA was used per polymerase chain reaction (PCR). Promoter CpG island methylation of 26 candidate genes was studied in NHBEC and lung cancer cell lines using Combined Bisulfite Modification and Restriction Analysis as described (8). Methylation of NHBEC, lung cancer cell lines and primary adenocarcinomas was evaluated using methylation-specific PCR as described (8). Combined Bisulfite Modification and Restriction Analysis and methylation-specific PCR primer sequences and amplification conditions for each gene are described in (supplementary Tables S1 and S2, respectively, are available at Carcinogenesis Online).
5-Aza-deoxycytidine and trichostatin A treatment
Lung cancer cell lines were maintained in American Type Culture Collection-recommended media and cells at log phase of growth were treated in duplicate as follows: Vehicle (0.6 μl ethanol in 10 ml medium), trichostatin A (TSA) [300 nM for 18 h (Sigma, St. Louis, MO; stock solution 5 μM in ethanol)] or 5-aza-deoxycytidine (DAC) [500 nM for 96 h with fresh medium containing the drug changed every 12 h (Sigma; stock solution 10 mM in phosphate-buffered saline)]. Cells treated with Vehicle or TSA underwent fresh media changes in parallel with DAC treatment and TSA treatment was conducted 18 h before all groups were harvested in TRI-reagent (Sigma). Cells in TRI-reagent were stored at −80°C until RNA isolation.
Gene expression analysis
RNA was isolated as described (8). Total RNA (3 μg) was reverse transcribed using the SuperScript™ First-Strand Synthesis System for reverse transcription–polymerase chain reaction (RT–PCR) (Invitrogen, Carlsbad, CA) according to the complementary DNA synthesis protocol from Invitrogen®. Transcription of selected genes was evaluated using RT–PCR and electrophoresis in 3% agarose gels. To avoid PCR products from contaminating DNA, RNA isolation was done in the presence of DNase, and large introns were included in the RT–PCR amplification product. For Reprimo, which is a single-exon gene, RT-negative PCR was done using complementary DNA synthesized in the absence of SuperScript II. RT–PCR primers and amplification conditions are described in (supplementary Table S3 is available at Carcinogenesis Online).
Chromatin immunoprecipitation assay
Methylation of histone-3 lysine residues was examined using EZ ChiP™ (Upstate Biotech, Charlottesville, VA). Antibodies specific for histone H3, H3 di-methyl K4, H3 di-methyl K9, H3 tri-methyl K27 and RNA polymerase II (Upstate Biotech) were used to capture protein–DNA complexes. Rabbit and mouse IgGs were used for isotype control. Chromatin immunoprecipitation PCR analysis was performed using 2–3 μl of DNA and primers specific for the SFRP1 promoter: sense, 5′-CCAGCTTAGGCAACAAGAGC-3′ and anti-sense, 5′-GGGGAGGAGGAAAGAGCAA-3′. A 198 bp product was PCR amplified at 94°C for 30 s followed by 40 cycles of 94°C for 30 s, 63°C for 30 s, 72°C for 30 s and a final extension at 72°C for 5 min.
Data analysis
Patient characteristics, including age, gender, smoking status, race and gene methylation, are summarized with mean and standard deviation for continuous variables and proportions for categorical variables. Fisher's exact test was used to assess the association between methylation and patient characteristics and the association between pairs of genes. Methylation of genes and patient characteristics were assessed for association with survival. Survival time was calculated from time of diagnosis until death or last follow-up visit. Individual genes and variables were assessed with Kaplan–Meier plots and the log-rank test. Proportional hazards models were used to simultaneously examine multiple genes, while adjusting for patient characteristics. All analyses were conducted using SAS version 9.1.3.
Results
Hypermethylation of multiple genes in lung adenocarcinoma
CpG island hypermethylation of 26 genes involved in various growth regulatory pathways (Table I) was examined in primary lung adenocarcinomas from current, former and never smokers. The genes were selected from genome-wide screening assays that identified abnormal methylation and/or mutations in human malignancies such as breast and colon cancers. Methylation of BOLL, TUBB4, PCDH10, MMP2, APC2, GPNMB, ICAM5, JPH3, GNB4, SULF2, AK5, XT3, BHLHB4 and BHLHB5 has not been described in lung adenocarcinoma. Methylation was first assessed in NHBEC from 20 cancer-free smokers and 14 lung tumor-derived cell lines using Combined Bisulfite Modification and Restriction Analysis. Twenty-five genes displayed tumor-specific methylation that was detected in lung cancer cell lines but not in NHBEC (Table II). GNB4 was methylated in 15% of NHBEC and 8.3% of lung cancer cell lines. In primary lung adenocarcinomas, methylation was detected in 11.4% (CHFR) to 80.6% (SULF2) of tumors. The prevalence for methylation of p16, RASSF1A and MGMT, which are often described in lung cancer, was used as standard for this collection of primary tumors and was similar to other sets of lung cancers (Tables I and II) (12,16–18).
Table I.
Functions and methylation prevalences of selected genes in lung adenocarcinoma
| Gene | Location | Functions of encoded proteins | Methylation in lung ADC |
| p16 | 9p21 | Cyclin-dependent kinase inhibitor that blocks cell cycle progression | 31–98 (12,16,17) |
| RASSF1A | 3p21.3 | Modulates apoptotic and cell cycle checkpoint pathways | 47–69 (12,16,17) |
| MGMT | 10q26 | DNA repair enzyme | 30–51 (18,19) |
| Reprimo | 2q23.3 | Involved in the p53-induced G2 cell cycle arrest | 32–33 (20,21) |
| TNFRSF10C | 8p22–p21 | A TNF receptor family member that modulates apoptosis | 12 (22) |
| TNFRSF10D | 8p21 | A TNF receptor family member that modulates apoptosis | 15 (22) |
| CHFR | 12q24.33 | Mitotic checkpoint protein that delays transition to metaphase | 10–19 (23–25) |
| BOLL | 2q33 | RNA-binding protein regulating meiotic G2/M transition | NR |
| TSLC1 | 11q23.2 | Intercellular adhesion molecule | 46 (26) |
| TUBB4 | 19p13.3 | A major component of microtubules | NR |
| PCDH10 | 4q28.3 | A-cadherin family gene involved in cell–cell adhesion | NR |
| MMP2 | 16q13–q21 | Degrades extracellular matrix | NR |
| GPNMB | 7p15 | A transmembrane protein suppressing growth and metastasis | NR |
| ICAM5 | 19p13.2 | A member of the intercellular adhesion molecule | NR |
| JPH3 | 16q24.3 | A junctional protein between plasma membrane and ER | NR |
| SFRP1 | 8p12–11.1 | Antagonist of the transmembrane frizzled receptor that is component of the Wnt signaling | 34–76 (17,27) |
| SFRP2 | 4q31.3 | Antagonist of the transmembrane frizzled receptor that is component of the Wnt signaling | 52–84 (17,27) |
| APC2 | 19p13.3 | Depletes the beta-catenin pool and inhibits Wnt signaling | NR |
| GNB4 | 3q26.32–33 | A guanine nucleotide-binding protein that integrate signals between receptors and effector proteins | NR |
| SULF2 | 20q12–13.2 | Heparin sulfate 6-O-endosulfatases that selectively remove 6-O-sulfate groups from heparin sulfate | NR |
| IGFBP3 | 7p13–p12 | A carrier of IGFs that negatively regulates IGF-1 bioavailability | 62–76 (28,29) |
| DAB2IP | 9q33.1–33.3 | Cytosolic adapter protein for LDL endocytosis and also suppresses mitogenic activity of Ras | 37 (30) |
| AK5 | 1p31 | Synthesis of adenine nucleotides for cellular metabolism | NR |
| XT3 | 3p21.3 | Transport of hydrophilic solutes across cell membrane | NR |
| BHLHB4 | 20q13.33 | Basic helix-loop-helix-containing transcriptional regulator | NR |
| BHLHB5 | 8q13 | Inhibitor of a neuronal differentiation gene BETA2 | NR |
APC, adenomatosis polyposis coli; ER, endoplasmic reticulum; IGF, insulin-like growth; LDL, low density lipoprotein; NR, no report of DNA methylation in lung adenocarcinoma or lung cancer in general. TNF, tumor necrosis factor.
Table II.
Prevalence of DNA methylation in lung cancer cell lines and primary tumors
| Gene | CpG island |
Lung cancer cell lines (%) (n = 14) | Primary lung cancer (%) |
||
| Locationa | No. of CpGs | Smokers (n = 100) | Never smokers (n = 75) | ||
| p16 | Within | 63 | 42.8b | 66.7 | 64.0 |
| RASSF1A | Within | 84 | 33.3 | 46.9 | 40.0 |
| MGMT | Within | 75 | 33.3 | 31.3 | 37.3 |
| Reprimo | Within | 106 | 50.0 | 72.9 | 65.3 |
| TNFRSF10Cc | Within | 50 | 66.7 | 44.8 | 72.0 |
| TNFRSF10D | Within | 53 | 16.7 | 9.4 | 16.0 |
| CHFR | Within | 91 | 8.3 | 11.5 | 9.3 |
| BOLLcd | Within | 166 | 50.0 | 66.3 | 80.0 |
| TSLC1 | Within | 176 | 0.0 | 54.2 | 53.3 |
| TUBB4d | Within | 25 | 33.3 | 33.3 | 32.0 |
| PCDH10d | Within | 90 | 41.7 | 70.8 | 62.7 |
| MMP2d | Within | 42 | 50.0 | 72.9 | 77.3 |
| GPNMBd | +906 bp | 34 | 75.0 | 59.4 | 69.3 |
| ICAM5d | Within | 407 | 16.7 | 52.1 | 58.7 |
| JPH3d | Within | 78 | 33.3 | 70.8 | 77.3 |
| SFRP2 | Within | 112 | 58.3 | 68.8 | 65.3 |
| SFRP1 | Within | 147 | 41.7 | 77.1 | 78.7 |
| APC2d | Within | 44 | 83.3 | 66.7 | 74.7 |
| GNB4d | Within | 122 | 8.3 | 27.1 | 21.3 |
| SULF2d | Within | 143 | 33.3 | 81.3 | 80.0 |
| IGFBP3 | Within | 139 | 8.3 | 65.6 | 70.7 |
| DAB2IP | +131 kb | 71 | 58.3 | 17.7 | 10.7 |
| AK5d | Within | 103 | 16.7 | 24.0 | 21.3 |
| XT3d | Within | 88 | 16.7 | 31.3 | 25.3 |
| BHLHB4d | Within | 272 | 58.3 | 71.9 | 73.3 |
| BHLHB5dc | Within | 143 | 25.0 | 22.9 | 37.0 |
APC, adenomatosis polyposis coli
Location of transcriptional start site relative to the CpG islands analyzed.
p16 is inactivated in 93% (13/14) of lung cancer cell lines; deleted in seven and methylated in six.
Significantly different methylation prevalence between smokers and never smokers (P < 0.05).
Methylation is reported for the first time in lung adenocarcinoma.
Cigarette smoking and aberrant CpG island methylation
The effect of smoking on promoter methylation was evaluated for each gene. Primary lung adenocarcinomas from never smokers showed significantly higher prevalence for methylation of TNFRSF10C, BHLHB5 and BOLL than smokers (current and former) (Table II). Methylation of at least one of these three genes was detected in 97.3% (73/75) of tumors from never smokers compared with 78.9% (75/95) in current and former smokers (P < 0.001). Methylation of two of three, or all three, genes was also more common in never smokers than smokers (P = 0.045, P = 0.02, respectively). None of the comparisons of this three-gene panel were different between former and current smokers. The remaining genes displayed similar methylation patterns regardless of smoking history.
Significant association between methylation of genes
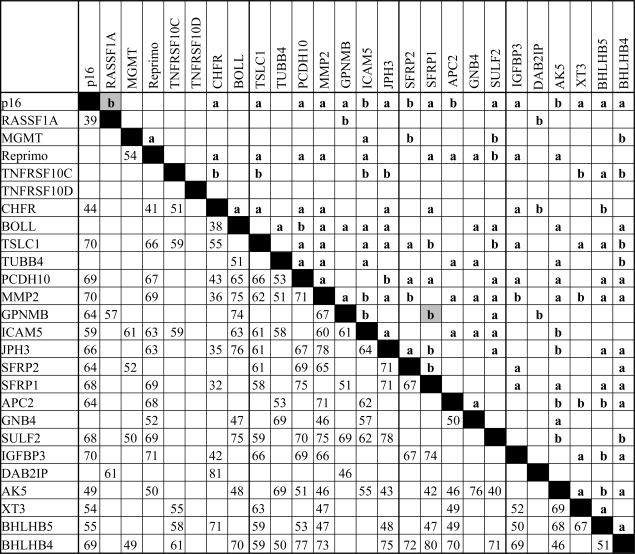
Pairwise comparisons to define the relation between methylation of genes identified significant associations for 138 (42.5%) of the possible 325 comparisons (P < 0.05) (Table III). Positive agreement, indicating presence or absence of methylation in paired genes, was seen in 136 (98.6%) of these associations (Table III). The largest number of significant and positive associations were observed for MMP2 (19/25), BHLHB4 (16/25) and p16 (16/25) suggesting that methylation of these genes could be used as indicator of a widespread methylation phenotype in lung cancer. For example, among a panel of 19 genes, adenocarcinomas with methylated MMP2 on average showed 4.7 (3.5–5.9, 95% confidence interval) more methylated genes than those with unmethylated MMP2 (Table IV). Similarly, two separate 16-gene panels had an average of 4.2 and 3.5 more methylated genes in tumors with methylated BHLHB4 and p16, respectively, than those without methylation of these two genes.
Table III.
Pairwise association between gene methylation
 |
APC, adenomatosis polyposis coli
Upper right half of matrix: P-values from Fisher's exact test, (a) P < 0.01, (b) P < 0.05; letters in gray boxes indicate negative associations. Lower left half of matrix: numbers indicate percent agreement for significantly associated genes.
Table IV.
Summary of the relationship between methylation of MMP2, BHLHB4 and p16 with the remaining genes
| Gene | No. of genes in the panela | Methylation status | No. of primary tumors | Mean number of positive genes in panel | Difference in means (methylated to unmethylated) |
|
| Difference | 95% CI | |||||
| MMP2 | 19 | + | 129 | 11.5 | 4.7 | 3.5–5.9 |
| − | 42 | 6.8 | ||||
| BHLHB4 | 16 | + | 126 | 10.6 | 4.2 | 3.2–5.1 |
| − | 45 | 6.5 | ||||
| p16 | 16 | + | 113 | 10.4 | 3.5 | 2.6–4.5 |
| − | 59 | 6.9 | ||||
Genes included in panel were significantly (P < 0.05) and positively associated with MMP2, BHLHB4 or p16.
DNA methylation and expression
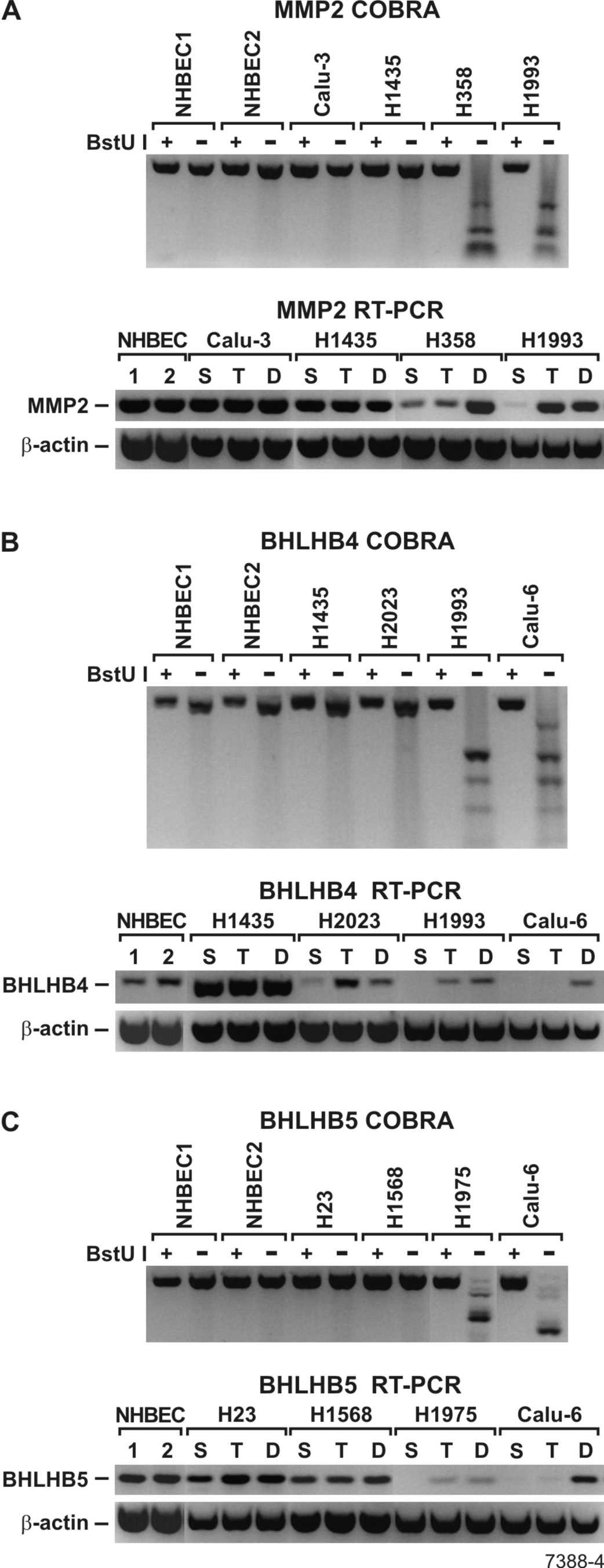
The effect of methylation and histone modification on transcription was characterized using a subset of genes methylated in >50% of primary tumors. Gene expression in DAC-, TSA- and vehicle-treated cells was compared using semiquantitative RT–PCR. Silencing of the SFRP1 gene in lung cancer cell lines occurred by methylation and histone modification or histone modification alone. SFRP1 was readily expressed in NHBEC, H1568 and Calu-3 cells where its promoter was unmethylated, and DAC- and TSA-treatments had no effect (Figure 1A). In contrast, SFRP1 was silenced in H358, SKLU1 and Calu-6 due to dense promoter methylation and treatment with DAC but not TSA restored expression to normal levels. On the other hand, SFRP1 expression in H1435 cells was solely regulated by chromatin remodeling. This gene was silenced despite a completely unmethylated promoter, and suggestive of chromatin remodeling, expression was restored by TSA but not DAC treatment (Figure 1A). A marked enrichment in the H3 di-methyl-K9/di-methyl-K4 ratio was seen in SFRP1 promoter of silenced cells (H1435) and TSA treatment leads to a 44.6% reduction of this ratio (supplementary Figure S1 is available at Carcinogenesis Online). Similarly, TSA treatment also induced a 54.5% increase in RNA polymerase II binding to the promoter substantiating chromatin remodeling as the mechanism for silencing of SFRP1 in this cell line (supplementary Figure S1 is available at Carcinogenesis Online).
Fig. 1.

Effect of promoter methylation on gene expression. Digestion of PCR fragments with BstUI was used to assess the methylation state of each gene promoter. Samples were amplified in duplicate to compare digested to undigested products. Unmethylated alleles in the BstUI+ lanes are resistant to enzymatic digestion and are the same size as the undigested product (BstUI−). In contrast, methylated alleles in the BstUI+ lanes are digested into smaller fragments proportional to the number of methylated BstUI sites within the amplified promoter sequence. The presence of methylation in all templates referred to as complete methylation is detected by the presence of only smaller PCR products compared with the undigested control. (A) Combined Bisulfite Modification and Restriction Analysis (COBRA) results reveal the promoter CpG island of SFRP1 was not methylated in NHBEC1, NHBEC2 and lung cancer cell lines (H1435, H1568 and Calu‑3) but completely methylated in H358, SKLU-1 and Calu-6. In NHBEC and lung cancer cell lines with unmethylated promoter (H1568 and Calu-3), SFRP1 was expressed in vehicle-treated (S) cells but silenced in vehicle-treated (S) H1435, H358, SKLU1 and Calu-6 cells. Consistent with the methylation data, TSA (T) but not DAC treatment (D) restored SFRP1 expression in H1435 and DAC but not TSA treatment restored SFRP1 expression in H358, SKLU1 and Calu-6. As shown for H1568 and Calu-3, neither drugs affected SFRP1 expression in cell lines that normally express the gene. For all RT–PCR assays, β-actin expression was used as the internal control. (B) Transcription of BOLL was completely silenced in NHBEC and all vehicle-treated (S) lung cancer cell lines regardless of methylation at the promoter CpG island. However, expression of the two major transcripts (BOLL-1 and BOLL-2) was restored in all cell lines by DAC treatment. TSA treatment partially restored expression of BOLL-1 only in cell lines with a unmethylated CpG island. (C) JPH3 was unmethylated in NHBEC1, NHBEC2 and some lung cancer cell lines such as H2023 and H2085 (upper panel) and was expressed. However, JPH3 was also expressed in lung cancer cell lines that are methylated (H1993 and Calu-6). In H358, JPH3 was completely methylated and silenced. With exception of a slight induction by TSA, neither drugs could restore normal expression. (D) Reprimo was expressed in NHBEC and cell lines with an unmethylated promoter CpG island such as H1568 and H2228, but it was also equally expressed in cell lines with completely methylated islands (H1435 and Calu-6). Because Reprimo is a single-exon gene, RT–PCR was done in the absence of the reverse transcribing enzyme (SuperScript II) to make sure that PCR products are not coming from DNA contamination.
As expected, promoter CpG island methylation was directly involved in silencing or strongly downregulating gene expression. Examples of normal gene expression in NHBEC and lung cancer cell lines with an unmethylated gene promoter as well as gene silencing due to promoter methylation that could be restored primarily with DAC treatment are shown in the supplementary Figure S2 (available at Carcinogenesis Online). However, we have also observed an indirect effect of DNA methylation on gene silencing, which refers to the re-expression by DAC of a gene whose promoter CpG island is not methylated. All BOLL transcripts were silenced in all samples without (all NHBEC, Calu-3, H1435 and H2023) or with (H23, H2228 and Calu-6) methylation in their CpG island (Figure 1B). DAC treatment restored the two most common BOLL transcripts (BOLL-1 and BOLL-2) in all samples. TSA treatment restored BOLL-1 only in cells with an unmethylated CpG island, suggesting that expression was regulated by changes in the histone code. In contrast, JPH3 (Figure 1C) and Reprimo (Figure 1D) were expressed even in the presence of complete methylation of their respective promoter CpG islands. JPH3 is silenced in H358 and except for a slight induction after TSA treatment; neither DAC nor TSA could restore its expression.
Promoter methylation, patient survival and stage of lung cancer
After adjustment for stage, the effect of methylation on patient survival was evaluated for each gene using Kaplan–Meier log-rank test and proportional hazard models. Both tests identified a significant association between SULF2 methylation and better survival (P = 0.005)that was more profound in advanced stage patients all of whom had undergone surgical resection (P < 0.001) (Figure 2A and B). The median survival of patients without or with SULF2 methylation was 35.1 and 62.8 months, respectively. The most dramatic effect of SULF2 methylation on survival was observed in advanced stage patients where the median survival was increased to >4-fold from 8.5 months in patients with unmethylated SULF2 to 36.2 months in patients with methylated SULF2, P < 0.001 (Figure 2B). The hazard ratio for methylation of SULF2 in all lung cancer cases and advanced stage lung cancer was 0.41 and 0.23, respectively. No significant association was found between methylation of the remaining genes and stage of lung cancer or patient survival.
Fig. 2.

Methylation of SULF2 and patient survival. (A) Lung adenocarcinoma patients with methylated SULF2 have better survival than those with unmethylated SULF2. (B) Advanced lung adenocarcinoma patients (Stages II–IV) with methylated SULF2 survived significantly better than those with unmethylated SULF2.
Discussion
This study reports promoter hypermethylation of 26 candidate genes, 14 for the first time, in primary lung adenocarcinoma from current, former and never smokers. Twenty-five genes were specifically methylated in tumor cells and 16 were methylated in >50% of primary tumors indicating a likely role in tumor development. Nearly half (42.5%) of the 325 pairwise comparisons between methylation of individual genes were significantly associated suggesting a co-ordinated aberrant methylation of multiple genes in lung cancer. While the prevalence for methylation of most genes was similar between different smoking groups, TNFRSF10C, BHLHB5 and BOLL were more frequently methylated in never smokers than current and former smokers. Interestingly, this study also identified a scenario where tumor-specific methylation of a candidate oncogene (SULF2) was associated with prolonged survival of lung cancer patients irrespective of tumor stage.
Concomitant methylation of multiple growth regulatory genes is increasingly associated with human malignancies (4,5). Solid tumors show some degree of commonality in hypermethylation of genes such as p16, GATA4, GATA5, E-cadherin and PAX5. The genes methylated in this study are also methylated in colon and breast cancers and may represent common targets for silencing in epithelial tumors. The similarity of methylation prevalences between early and advanced stage tumors suggests that the aberrant gene methylation is probably established relatively early in lung carcinogenesis, making these genes candidate biomarkers for early detection. Our group has shown that methylation of specific genes detected in sputum samples from smokers could predict lung cancer up to 18 months prior to clinical diagnosis (31). Inclusion of these newly identified genes as biomarkers could greatly improve the sensitivity, specificity and predictive power of this developing screening assay.
Aberrant methylation of multiple growth regulatory genes is a hallmark of cancer. This study identified a strong association between methylated genes indicating a co-ordinated methylation defect. Overexpression of cytosine DNA methyltransferases in association with aberrant methylation of multiple genes in lung cancer has been documented and could be responsible for the concomitant methylation (32). Although what targets cytosine DNA methyltransferases to specific genes is not clear, DNA damage due to chronic exposure to tobacco and other environmental carcinogens could be one possible cause. Lung adenocarcinomas harbor extensive genetic damage (1) that could serve as loci for DNMT1 recruitment and de novo methylation (33). We have recently demonstrated an association between tobacco carcinogen-induced DNA damage and repair with methylation in vitro and in sputum from heavy smokers (32,34). The fact that de novo methylation can also occur in successfully repaired sites (35) could account in part, for the higher prevalence for methylation than mutation.
Although cigarette smoking is the overwhelming risk factor for lung cancer development, an increasing number of never smokers also develop the disease (36). Distinct genetic and epigenetic abnormalities as well as differential response to therapy between smoker and never smoker lung cancer patients led some investigators to hypothesize the two might represent different diseases (9). Although the prevalence for methylation of TNFRSF10C, BHLHB5 and BOLL in primary lung adenocarcinomas from never smokers was significantly higher than smokers, its specificity to discriminate the two populations was low. Moreover, at least with regard to candidate genes, few have shown significant difference in prevalence in tumors from smokers and never smokers, suggesting commonality for deregulation of the epigenome (8,12,16,18).
Hypermethylation of promoter CpG islands often silences gene expression. For many genes, the cause and effect relation between methylation and gene silencing has been established by restoring gene expression using demethylating drugs (37). However, this study identified a different mode of regulation for JPH3, Reprimo and BOLL. JPH3 and Reprimo were expressed regardless of densely methylated promoter CpG islands suggesting promoter activity outside the respective CpG islands. No CpG island other than those analyzed was found within 50 kb upstream of the respective transcriptional start sites. In contrast, expression of BOLL was indirectly regulated by methylation. BOLL is a highly conserved meiotic G2/M transition gene (38). In humans, it is exclusively expressed in the testis and non-genetic silencing (no mutation or polymorphism) of BOLL leads to male infertility (38–40). Although BOLL was silenced in all NHBEC and lung cancer cell lines regardless of methylation, DAC treatment restored expression in all cell lines. This suggests that expression might be regulated via binding of a transcription factor, expression of which is also regulated by promoter methylation. This scenario is seen with the PAX5beta gene that encodes for the transcriptional factor B-cell-specific-activating protein and that in turn regulates CD19, a gene shown to negatively control cell growth (41). A strong association was seen between methylation of PAX5β promoter and loss of expression of CD19. Alternatively, expression of BOLL might be regulated by a regional epigenetic suppression. In colorectal cancer, a co-ordinated epigenetic silencing of multiple genes regardless of individual promoter methylation was identified within an entire 4 Mb band of chromosome 2q14.2 (42). BOLL is located at 2q33 and might be under similar transcriptional regulation.
Methylation of SULF2 in lung adenocarcinomas showed a significant inverse relationship with survival. SULF2 is a heparin sulfate 6-O-endosulfatase enzyme that promotes the release of growth and angiogenic factors such as fibroblast growth factor-I, fibroblast growth factor-2, vascular endothelial growth factor and stromal cell-derived growth factor-I from heparin (43,44). SULF2 is over expressed in human breast cancer (45). In hepatocellular carcinoma, its expression increases with disease progression and patients with higher SULF2 expression have lower survival and a more rapid rate of tumor recurrence after surgery (43). Inactivation of SULF2 using short hairpin RNA also reduces cell proliferation and migration (43). Similarly, our data indicate that inactivation of SULF2 via CpG island hypermethylation—as shown for the first time in any tumor type—may predict for longer survival of lung cancer patients and thus could be a promising prognostic marker.
Supplementary material
Supplementary Figures S1–S2 and Tables S1–S3 can be found at http://carcin.oxfordjournals.org/
Funding
National Institutes of Health (R01 ES008801 to S.A.B., R01 CA043318 to S.B.B.).
Acknowledgments
Conflict of Interest Statement: S.A.B. is a consultant to Oncomethylome Sciences. Under a licensing agreement between Lovelace Respiratory Research Institute and Oncomethylome Sciences, nested methylation-specific PCR was licensed to Oncomethylome Sciences, and the author is entitled to a share of the royalties received by the Institute from sales of the licensed technology. The Institute, in accordance with its conflict of interest policies, is managing the terms of these arrangements. The commercial rights to standard methylation-specific PCR also belong to Oncomethylome Sciences. S.B.B. is a consultant to Oncomethylome Sciences and is entitled to royalties from any commercial use of this procedure.
Glossary
Abbreviations
- DAC
5-aza-deoxycytidine
- NHBEC
normal human bronchial epithelial cells
- PCR
polymerase chain reaction
- RT–PCR
reverse transcription–polymerase chain reaction
- TSA
trichostatin A
References
- 1.Ding L, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sjoblom T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 3.Knudson AG. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer. 2001;1:157–162. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- 4.Chan TA, et al. Convergence of mutation and epigenetic alterations identifies common genes in cancer that predict for poor prognosis. PLoS Med. 2008;5:e114. doi: 10.1371/journal.pmed.0050114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schuebel KE, et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet. 2007;3:e157. doi: 10.1371/journal.pgen.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parkin DM, et al. Global cancer statistics, 2002. CA Cancer J. Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 7.Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat. Rev. Cancer. 2004;4:707–717. doi: 10.1038/nrc1432. [DOI] [PubMed] [Google Scholar]
- 8.Tessema M, et al. Promoter methylation of genes in and around the candidate lung cancer susceptibility locus 6q23-25. Cancer Res. 2008;68:1707–1714. doi: 10.1158/0008-5472.CAN-07-6325. [DOI] [PubMed] [Google Scholar]
- 9.Gazdar AF, et al. Lung cancer, smoke exposure, and sex. J. Clin. Oncol. 2007;25:469–471. doi: 10.1200/JCO.2006.09.4623. [DOI] [PubMed] [Google Scholar]
- 10.Thun MJ, et al. Lung cancer death rates in lifelong nonsmokers. J. Natl Cancer Inst. 2006;98:691–699. doi: 10.1093/jnci/djj187. [DOI] [PubMed] [Google Scholar]
- 11.Subramanian J, et al. Lung cancer in never smokers: a review. J. Clin. Oncol. 2007;25:561–570. doi: 10.1200/JCO.2006.06.8015. [DOI] [PubMed] [Google Scholar]
- 12.Toyooka S, et al. Mutational and epigenetic evidence for independent pathways for lung adenocarcinomas arising in smokers and never smokers. Cancer Res. 2006;66:1371–1375. doi: 10.1158/0008-5472.CAN-05-2625. [DOI] [PubMed] [Google Scholar]
- 13.Wakelee HA, et al. Lung cancer incidence in never smokers. J. Clin. Oncol. 2007;25:472–478. doi: 10.1200/JCO.2006.07.2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyamoto K, et al. Identification of 20 genes aberrantly methylated in human breast cancers. Int. J. Cancer. 2005;116:407–414. doi: 10.1002/ijc.21054. [DOI] [PubMed] [Google Scholar]
- 15.Brambilla E, et al. The new World Health Organization classification of lung tumours. Eur. Respir. J. 2001;18:1059–1068. doi: 10.1183/09031936.01.00275301. [DOI] [PubMed] [Google Scholar]
- 16.Divine KK, et al. Multiplicity of abnormal promoter methylation in lung adenocarcinomas from smokers and never smokers. Int. J. Cancer. 2005;114:400–405. doi: 10.1002/ijc.20761. [DOI] [PubMed] [Google Scholar]
- 17.Marsit CJ, et al. Examination of a CpG island methylator phenotype and implications of methylation profiles in solid tumors. Cancer Res. 2006;66:10621–10629. doi: 10.1158/0008-5472.CAN-06-1687. [DOI] [PubMed] [Google Scholar]
- 18.Pulling LC, et al. Promoter hypermethylation of the O6-methylguanine-DNA methyltransferase gene: more common in lung adenocarcinomas from never-smokers than smokers and associated with tumor progression. Cancer Res. 2003;63:4842–4848. [PubMed] [Google Scholar]
- 19.Liu Y, et al. Aberrant promoter methylation of p16 and MGMT genes in lung tumors from smoking and never-smoking lung cancer patients. Neoplasia. 2006;8:46–51. doi: 10.1593/neo.05586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki M, et al. Aberrant methylation of Reprimo in lung cancer. Lung Cancer. 2005;47:309–314. doi: 10.1016/j.lungcan.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi T, et al. Aberrant methylation of Reprimo in human malignancies. Int. J. Cancer. 2005;115:503–510. doi: 10.1002/ijc.20910. [DOI] [PubMed] [Google Scholar]
- 22.Shivapurkar N, et al. Aberrant methylation of trail decoy receptor genes is frequent in multiple tumor types. Int. J. Cancer. 2004;109:786–792. doi: 10.1002/ijc.20041. [DOI] [PubMed] [Google Scholar]
- 23.Corn PG, et al. Frequent hypermethylation of the 5′ CpG island of the mitotic stress checkpoint gene Chfr in colorectal and non-small cell lung cancer. Carcinogenesis. 2003;24:47–51. doi: 10.1093/carcin/24.1.47. [DOI] [PubMed] [Google Scholar]
- 24.Mizuno K, et al. Aberrant hypermethylation of the CHFR prophase checkpoint gene in human lung cancers. Oncogene. 2002;21:2328–2333. doi: 10.1038/sj.onc.1205402. [DOI] [PubMed] [Google Scholar]
- 25.Takeshita M, et al. CHFR expression is preferentially impaired in smoking-related squamous cell carcinoma of the lung, and the diminished expression significantly harms outcomes. Int. J. Cancer. 2008;123:1623–1630. doi: 10.1002/ijc.23673. [DOI] [PubMed] [Google Scholar]
- 26.Fukami T, et al. Promoter methylation of the TSLC1 gene in advanced lung tumors and various cancer cell lines. Int. J. Cancer. 2003;107:53–59. doi: 10.1002/ijc.11348. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki M, et al. Synchronous alterations of Wnt and epidermal growth factor receptor signaling pathways through aberrant methylation and mutation in non small cell lung cancer. Clin. Cancer Res. 2007;13:6087–6092. doi: 10.1158/1078-0432.CCR-07-0591. [DOI] [PubMed] [Google Scholar]
- 28.Chang YS, et al. Correlation between insulin-like growth factor-binding protein-3 promoter methylation and prognosis of patients with stage I non-small cell lung cancer. Clin. Cancer Res. 2002;8:3669–3675. [PubMed] [Google Scholar]
- 29.Chang YS, et al. Mechanisms underlying lack of insulin-like growth factor-binding protein-3 expression in non-small-cell lung cancer. Oncogene. 2004;23:6569–6580. doi: 10.1038/sj.onc.1207882. [DOI] [PubMed] [Google Scholar]
- 30.Yano M, et al. Aberrant promoter methylation of human DAB2 interactive protein (hDAB2IP) gene in lung cancers. Int. J. Cancer. 2005;113:59–66. doi: 10.1002/ijc.20531. [DOI] [PubMed] [Google Scholar]
- 31.Belinsky SA, et al. Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort. Cancer Res. 2006;66:3338–3344. doi: 10.1158/0008-5472.CAN-05-3408. [DOI] [PubMed] [Google Scholar]
- 32.Damiani LA, et al. Carcinogen-induced gene promoter hypermethylation is mediated by DNMT1 and causal for transformation of immortalized bronchial epithelial cells. Cancer Res. 2008;68:9005–9014. doi: 10.1158/0008-5472.CAN-08-1276. [DOI] [PubMed] [Google Scholar]
- 33.Cuozzo C, et al. DNA damage, homology-directed repair, and DNA methylation. PLoS Genet. 2007;3:e110. doi: 10.1371/journal.pgen.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leng S, et al. Double-strand break damage and associated DNA repair genes predispose smokers to gene methylation. Cancer Res. 2008;68:3049–3056. doi: 10.1158/0008-5472.CAN-07-6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Hagan HM, et al. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4:e1000155. doi: 10.1371/journal.pgen.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun S, et al. Lung cancer in never smokers–a different disease. Nat. Rev. Cancer. 2007;7:778–790. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
- 37.Jones PA, et al. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu EY, et al. Human BOULE gene rescues meiotic defects in infertile flies. Hum. Mol. Genet. 2003;12:169–175. doi: 10.1093/hmg/ddg017. [DOI] [PubMed] [Google Scholar]
- 39.Kostova E, et al. Association of three isoforms of the meiotic BOULE gene with spermatogenic failure in infertile men. Mol. Hum. Reprod. 2007;13:85–93. doi: 10.1093/molehr/gal101. [DOI] [PubMed] [Google Scholar]
- 40.Luetjens CM, et al. Association of meiotic arrest with lack of BOULE protein expression in infertile men. J. Clin. Endocrinol. Metab. 2004;89:1926–1933. doi: 10.1210/jc.2003-031178. [DOI] [PubMed] [Google Scholar]
- 41.Palmisano WA, et al. Aberrant promoter methylation of the transcription factor genes PAX5 alpha and beta in human cancers. Cancer Res. 2003;63:4620–4625. [PubMed] [Google Scholar]
- 42.Frigola J, et al. Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band. Nat. Genet. 2006;38:540–549. doi: 10.1038/ng1781. [DOI] [PubMed] [Google Scholar]
- 43.Lai JP, et al. Sulfatase 2 up-regulates glypican 3, promotes fibroblast growth factor signaling, and decreases survival in hepatocellular carcinoma. Hepatology. 2008;47:1211–1222. doi: 10.1002/hep.22202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uchimura K, et al. HSulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparin-bound growth factors and chemokines: effects on VEGF, FGF-1, and SDF-1. BMC Biochem. 2006;7:2. doi: 10.1186/1471-2091-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morimoto-Tomita M, et al. Sulf-2, a proangiogenic heparan sulfate endosulfatase, is upregulated in breast cancer. Neoplasia. 2005;7:1001–1010. doi: 10.1593/neo.05496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}