

Abstract
The aging brain is characterized by a shift from the homeostatic balance of inflammatory mediators to a proinflammatory state. This increase in neuroinflammation is marked by increased numbers of activated and primed microglia, increased steady state levels of inflammatory cytokines and decreases in anti-inflammatory molecules. These conditions sensitize the aged brain to produce an exaggerated response to the presence of an immune stimulus in the periphery or following exposure to a stressor. In the brain, proinflammatory cytokines can have profound effects on behavioral and neural processes. As the aged brain is primed to respond to inflammatory stimuli, infection or stress may produce more severe detriments in cognitive function in the aged. Typically after an immune stimulus, aged animals display prolonged sickness behaviors, increased cytokine induction and greater cognitive impairments compared to adults. Additionally, aging can also augment the central response to stressors leading to exaggerated cytokine induction and increased decrements in learning and memory. This alteration in neuroinflammation and resultant sensitization to extrinsic and intrinsic stressors can have considerable effects upon the elderly’s recovery and coping during disease and stress.
Keywords: neuroinflammation, aging, cognition, microglia, infection, stress
Introduction
The aging process is defined by a slow deterioration of homeostatic functions throughout the lifespan of an organism. For example, in the adult brain there is a balance between proinflammatory and anti-inflammatory cytokines, but with increased age this balance is shifted towards a proinflammatory state. This increased state of neuroinflammation makes the aged brain more vulnerable to the disruptive effects of both intrinsic and extrinsic factors such as disease, infection or stress. Furthermore, this vulnerability may present as a short-term alteration in normal cognitive function or complicate, exacerbate or hasten the progression of underlying neuropsychological conditions. The purpose of this brief review is to discuss the changes associated with neuroinflammation in the aged brain and how they may contribute to behavioral and cognitive changes associated with infection or stress.
Neuroinflammatory consequences of aging
Microglia are the resident immune cells of the central nervous system and are important mediators of the brain’s response to trauma, disease, and infection. Normally, microglia reside in a quiescent state but stimulation with a foreign antigen activates microglia, which induces a phenotypic shift including changes in cell surface marker expression (e.g., MHC class II antigen [1–3]), retraction of processes, and release of cytokines. Increased numbers of activated microglia in the aged brain may be indicative of increased neuroinflammation and heightened reactivity, which may underlie age-related alterations in the way the brain responds to and recovers from insult.
Apart from resting microglia, a subset of microglia appear to reside in an intermediate state characterized by shortened processes and expression of cell surface markers, similar to activated microglia, but without appreciable secretion of cytokines. This microglia subset is referred to as ‘primed’ due to more rapid induction and greater cytokine release upon activation compared to normally, activated non-primed microglia [4–6] (Figure 1A). A similar state of heightened reactivity was first described in association with neurodegenerative diseases including multiple sclerosis, Alzheimer’s disease and the murine ME7 model of prion disease [4, 5]. Presumably, abnormal proteins specific to each disease may act as priming factors for microglia. Similar to the priming paradigm associated with neurodegenerative disease, increasing evidence suggests that primed microglia may also reside in the normal, aged brain. For example, Streit et al. [3] observed a 10-fold increase in microglial abnormalities in the brain of a 68-year old human compared with the brain from a 38-year old subject. Moreover, it has been proposed that microglial senescence and subsequent changes in microglial function may support the development of neurodegenerative diseases such as Alzheimer’s [3, 7] and may exacerbate cognitive changes associated with normal aging.
Figure 1.
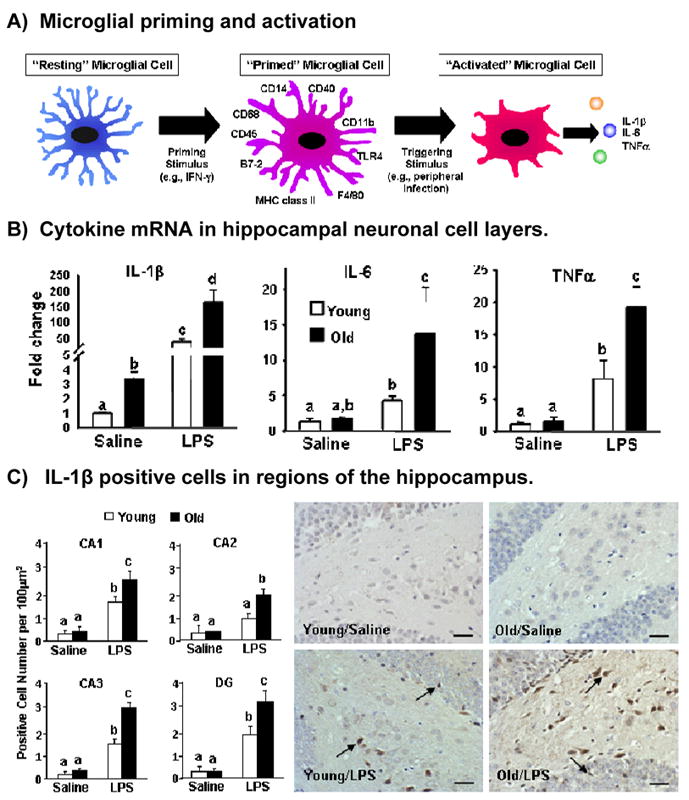
The aged brain is characterized by increased numbers of activated and primed microglia which sensitize the brain to produce increased levels of proinflammatory cytokines following peripheral immune activation. A) Microglial cells in the aged brain may be primed as characterized by increased expression of cell-surface markers and shorter processes. In response to stimulation, they produce an exaggerated amount of cytokines compared to stimulation of resting microglia (modified from [Dilger & Johnson, submitted]). B) Cytokine mRNA production in the hippocampal neuronal layer 4 h after LPS treatment in young and old mice (modified from [16]). C) IL-1β positive cells in the CA1, CA2, CA3 and dentate gyrus regions of the hippocampus 4 h LPS treatment in adult and aged mice (modified from [16]).
In addition to increased numbers of activated and primed microglia, the aged brain exhibits increased steady-state levels of inflammatory cytokines. Normally, interleukin(IL)-6 is upregulated in the brain following peripheral immune activation or injury; however, it also appears to be upregulated in normal aging and may be a key mediator of aged-related neuroinflammation. For example, circulating levels of IL-6 have been shown to be consistently increased in the elderly population, and in longitudinal studies, increased plasma IL-6 levels were associated with increased chances of cognitive impairment later in life [8]. Kiecolt-Glaser et al [9] examined longitudinal changes in circulating IL-6 production in spousal Alzhemier’s caregivers (a population at risk for increased stress) and noncaregivers and saw that the caregivers had increases in IL-6 production that were four times greater than noncaregivers. Additionally, increased IL-6 production has been demonstrated in the hippocampus, cerebral cortex, and cerebellum of aged mice compared to juvenile mice [10], and 10-month old senescence-accelerated mice (SAMP8) had increased IL-6 levels in the cerebral cortex and hippocampus compared to aged-matched control mice [11]. While circulating levels of IL-6 may have diagnostic and predictive value, the increases in central IL-6 levels seem to be a function of increased production in the CNS rather than a product of increased circulating levels. For example, coronal sections of the cerebral cortex taken from aged mice spontaneously secreted more IL-6 than did sections taken from adult animals [12, 13]. This upregulation of IL-6 production in the age brain seems to be mediated by glial cells. Mixed astrocyte and microglial cultures demonstrated that glial cultures from aged mice produced higher steady-state levels of IL-6 mRNA and spontaneously secreted higher levels of IL-6 protein compared to both adult and neonate cultures [10, 13]. Utilizing flow cytometric analysis, it was demonstrated that the proportion of IL-6 positive microglia was greater in aged animals [10]. In addition, endothelial cells, which form the blood-brain barrier, may contribute to increased levels of IL-6 in the aged brain. Endothelial cells cultured from the brains of aged rhesus monkeys spontaneously secreted more IL-6 protein than those cultured from adult or neonatal/fetal brains [14]. Furthermore, a study demonstrated that nuclear factor kappaB (NFkappaB) binding to the IL-6 promoter is increased in the brain of aged mice leading to increased IL-6 production [15]. Importantly, the increase in cytokine levels may support microglial priming and, in turn, lead to increased neuroinflammatory responses, excessive production of cytokines and maladaptive alterations in cognition and behavior especially when the peripheral immune system is activated [16, 17].
In addition to increases in proinflammatory cytokines, the aged brain can be characterized by a general reduction in levels of anti-inflammatory mediators, such as IL-10 [13, 18]. Following exposure to immunological stimulus there is normally an upregulation of proinflammatory mediators that is accompanied by a subsequent release of anti-inflammatory mediators; however, levels of IL-10 are significantly reduced in the aged brain [13]. Of consequence for the aging animal, this decrease in IL-10 may predispose the CNS to increased neuroinflammation and neuronal damage. For example, recombinant IL-10 protected primary cortical neuron cultures from NMDA or glutamate induced damage [19]. Additionally, IL-10 knockout mice demonstrated an increased vulnerability to behavioral pathologies associated with prion disease [20]. Also, an increased vulnerability to neurodegenerative diseases, including Alzheimer’s, has been associated with decreased IL-10 levels in humans [21]. Therefore, reduction of anti-inflammatory molecules in the brain may exacerbate the neuroinflammatory status within the CNS and predispose aged individuals to a discordant inflammatory response following peripheral immune activation or stress.
Immune-to-Brain Communication is Altered in Aged Animals
Cytokines can act on the CNS in a wide variety of ways. Because the brain receives signals from an activated immune system it has been suggested that the immune system itself may act as an additional “sensory pathway” that renders a central representation of the peripheral immune status which, in turn, influences and directs behavior [22]. Changes in behavior associated with increased central cytokine levels include reduced food and water intake, decreased exploration, decreased social interactions, somnogenesis and global effects on mood and cognition. These behavioral alterations in the animals with an infection have been collectively characterized as “sickness behavior”[23].
Rather than being a simple reflection of the passive response of a debilitated organism, sickness behavior is characterized by alterations in the motivational state of the organism, orchestrated by interactions between the CNS and the peripheral innate immune system [23]. While these changes in behavior may be adaptive in the short term, this response may turn pathological in cases in which the responsiveness of the CNS has been altered such as neurodegenerative diseases or aging [23].
While the CNS was once thought to be an immune privileged organ, we now know that the peripheral immune system can influence the CNS through a number of different pathways. One such means for cytokine entry into the brain is passive diffusion at the circumventricular organs (areas of the brain lacking a blood brain barrier) through which diffusion readily occurs [24]. However, passive diffusion accounts only for a small amount of cytokines entry into the brain, and certain cytokines are actively transported across the blood-brain barrier [25, 26]. In addition, endothelial cells that form the blood-brain barrier are capable of secreting immune-related molecules, including cytokines, in response to peripheral immune signals [14]. Therefore, it is possible that cytokines circulating in the periphery may elicit the secretion of cytokines in the brain by binding to receptors on endothelial cells on the peripheral side of the blood-brain barrier that, in turn, secrete cytokines into the CNS [14]. Additionally, studies employing subdiaphragmatic vagotomy show that vagal afferents carry visceral sensory information to the CNS following peripheral immune stimulation, thereby altering central gene expression and neural firing patterns [27]. Therefore, cytokines that are produced in the periphery activate humoral and neural communication pathways that induce glial cells in discrete brain regions to produce the same inflammatory cytokines that in turn produce neurophysiological and behavioral changes [27, 28].
Although, cytokines represent an important communication pathway for the brain and immune system, they are also intimately involved in the stress response. For example, proinflammatory cytokine production in the brain is upregulated following exposure to physiological or psychological stressors [6]. These alterations in cytokine signaling may have important implication for the animal’s behavior and may be modified by the neuroinflammatory status of the CNS.
Sickness Behavior and the Aged Brain
Once transported to or produced in the brain, proinflammatory cytokines can have profound effects on behavior. Given that the aged brain contains more activated and primed microglia and higher steady state levels of cytokines, it may be hyper responsive and more vulnerable to the neurobehavioral effects associated with peripheral infection.
A number of studies have examined the effects of a peripheral immune stimulus on behavior and subsequent cytokine induction in adult and aged mice. Consistently, adult and aged mice demonstrate similar declines in locomotor activity and social interactions during the initial period; however, aged mice have a prolonged duration of these behavioral deficits compared to adult animals [29]. Additionally, a number of studies have demonstrated that peripheral lipopolysaccharide treatment (LPS; a bacterial cell wall fragment) led to increased proinflammatory cytokines (primarily IL-1β, IL-6 and TNFα) in the hippocampus, cerebral cortex, and cerebellum as well as whole brain in aged mice compared to adult animals (Figure 1B&C) [29]. Micro-array analysis demonstrated significantly greater expression of genes associated with inflammation and oxidative stress following LPS injection in brains of aged animals compared to adults [29].
In order to better elucidate the role of the of the peripheral innate immune system and the resident immune cells of the CNS in the aged animal, Huang et al [30] injected LPS into the lateral ventricle of aged and adult mice. Their results demonstrated that aged mice had protracted reductions in food intake, locomotor activity and social exploration compared to adult mice. Aged mice also have been shown to have prolonged reductions in locomotor activity following central administration of IL-1β. Furthermore, central administration of IL-1 receptor antagonist (IL-1ra) attenuated reductions in locomotor activity associated with peripheral immune activation in old but not young mice. (Abraham et al, submitted). These results support the theory that changes within the CNS are important mediators of the dysregulated behavioral responses to immune stimuli observed in aged animals (Figure 2A).
Figure 2.
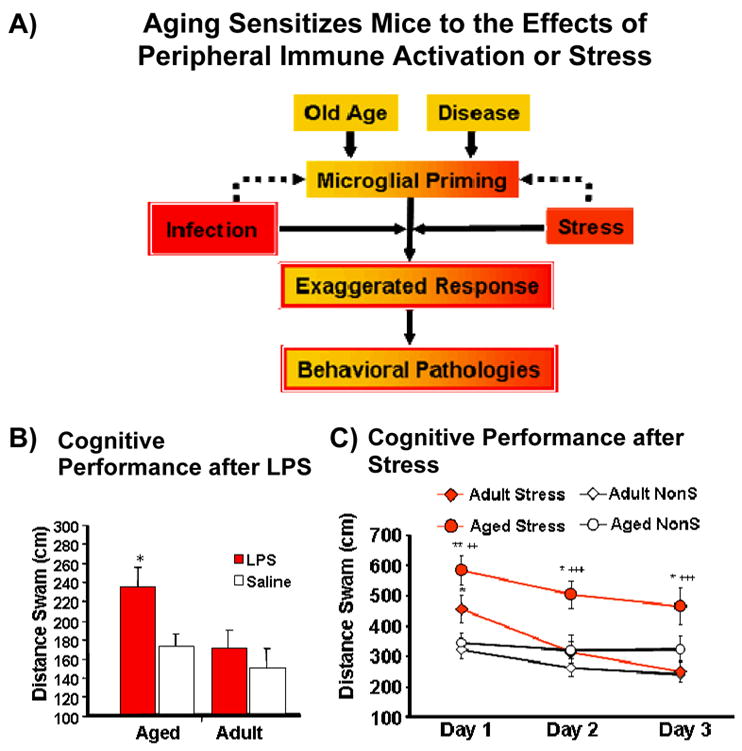
A) Aging sensitizes mice to the effects of peripheral immune activation or stress leading to exaggerated neuroinflammation and increased cognitive deficits in the aged animal. B) Cognitive performance of aged and adult mice following LPS treatment. C) Cognitive performance of aged and adult mice exposed to repeated mild psychological stressor.
Neuroinflammation Exacerbates Cognitive Dysfunction in Aged Animals
Even more insidious than the behavioral alterations associated with peripheral immune activation are the potential cognitive deficits that may accompany these changes. Although cytokine receptors are diffusely located throughout the brain, the highest levels of cytokine binding have been demonstrated in certain areas associated with learning and memory, including regions of the hippocampus [31]. Furthermore, increased levels IL-1β has been shown to disrupt long-term potentiation (LTP), a cellular mechanism believed to be important for certain types of memory. In fact, IL-1β blocks the expression of LTP in areas of the hippocampal formation [32]. Rachal Pugh and colleagues [33] demonstrated that post-training injections of LPS or IL-1β impaired hippocampal-dependent memory consolidation. However, the action of IL-1β in the brain is not entirely straight forward. More recently, research has demonstrated an inverted U-shaped curve in which basal levels of IL-1β support learning and memory while perturbations that increase or decrease these levels may result in cognitive impairment [34]. Additionally, the Morris water maze, a hippocampal-dependent test of spatial learning, has been shown to be sensitive to disruption by infection or peripheral immune activation and subsequent upregulation of proinflammatory cytokines [17, 35].
Given that aging is associated with an exaggerated central cytokine response associated with peripheral immune stimulation, which can have deleterious effects upon neuronal function, it should not be surprising that neuroinflammation is associated with increased cognitive dysfunction in the aged. For example, infection is a major risk factor for dementia in Alzheimer’s patients [36]. Additionally, many elderly patients admitted to emergency rooms with dementia symptoms are found to have underlying infections that when cleared alleviate the dementia [37].
In order to examine the cognitive consequences of peripheral immune stimulation in aged mice, Chen et al [16] trained mice to locate a hidden platform in a 5-arm radial swim maze in which the position of the platform was varied across day. Although aged mice took longer to acquire the task, they achieved the same level of proficiency by day 8. On day 9, both aged and adult animals were treated with LPS or saline and tested in the maze 4 h later. Results demonstrated that of the 4 groups, aged mice treated with LPS were significantly less effective in their ability to locate the platform. There were no differences for distance swam between saline-treated aged animals, LPS- or saline-treated adults (Figure 2B). The results suggests that while aged animals have the ability to perform the task at the same efficiency as adult animals after training, they are more vulnerable to deficits associated with peripheral immune activation [16].
A similar study utilizing a reference memory version of the water maze demonstrated that aged rats inoculated with Escherichia coli prior to the start of training showed deficits in long-term memory for the quadrant location, although they perform equally as well as infected- or saline-inoculated young or saline-inoculated aged animals in acquisition and measures of short-term memory [35]. Together these results suggest that aged animals are more vulnerable to cytokine-related disruptions of hippocampal-associated behavioral processes and consolidation of long-term memories.
As steady state levels of proinflammatory cytokines, notably IL-6, are altered in the aged brain, and have been implicated in human studies of dementia in the aged, it is useful to examine the role of this cytokine in cognitive disturbances associated with neuroinflammation. Sparkman et al [17] demonstrated that IL-6 deficient mice were recumbent to LPS-induced deficits in spatial working memory. Notably, this was accompanied by an attenuation of proinflammatory cytokine induction in the neuronal layer of the hippocampus of IL-6 deficient animals that received LPS compared to LPS-treated wild-type mice. Therefore, IL-6 seems to be an important central mediator of neuroinflammation associated with peripheral immune activation and may be an important determinant of the glial microenvironment. These results suggest that the increased levels of IL-6 observed in aged animals may exacerbate cognitive deficits associated with neuroinflammation and could serve to support increased proinflammatory cytokine levels with the CNS following an immune stimulus.
Additionally in a complimentary study (Richwine et al, unpublished data), IL-10 deficient mice demonstrated an LPS-induced deficit in the water maze, while LPS and saline-treated wild type and IL-10 knockout animals did not. Furthermore, IL-10 deficient animals showed increased levels of proinflammatory cytokines after LPS-treatment and demonstrated protracted sickness behaviors compared to wild type animals. This study demonstrates that IL-10 deficiency, similar to the reduction in the aged brain, leads to increased vulnerability to behavioral and cognitive deficits associated with peripheral immune stimulation.
Taken together, the above studies demonstrate that the aged brain is more susceptible to immune-related changes in cognitive function. Furthermore, they suggest that this age-related vulnerability may be tied to key increases in levels of proinflammatory mediators and a concurrent reduction of anti-inflammatory molecules in the aged brain.
Neuroinflammatory Consequences of Stress in the Aged Brain
As stressors have been shown to induce proinflammatory cytokines within the CNS [24] and prior exposure to a stressor can sensitize the immune response to an immunological stimulus [38], it as been suggested that stress may “prime” microglia leading to a sensitization of the CNS and exacerbation of neuroinflammation [6]. Stressors are capable of activating resident microglia and upregulating MHC class II [6] and certain stressors are associated with increased IL-1β production; a cytokine associated with disruption of learning and memory and increased behavioral deficits in aged animals. Furthermore, stress-induced disruptions of learning and memory have been shown to be abrogated after central infusion of IL-1ra or implantation of IL-1ra producing neural precursor cells into the dentate gyrus region of the hippocampus [39] [40]. The studies demonstrate an important role of IL-1 in regulating behavioral and cognitive changes associated with stress. Importantly, these findings have consequences for the aged brain, as it may be possible for microglial priming associated with aging to exacerbate the effects of stressors to produce profound effects upon the neuroinflammatory status of the CNS in ways that may impede normal cognitive functions.
It is well known that stress can disrupt learning and memory [41]. As systemic immunological stressors (e.g., LPS) have been shown to have an exaggerated impact on the aged brain and provoke similar responses in the CNS as psychological stressors, it may be possible that effects of psychological stressors are also exacerbated in the aged brain. For example, elderly patients are more likely to report memory failures on days that they experience stress [41].
A recent experiment [42] examined the possibility that a repeated mild psychological stressor (short-term, 30-min restraint) would lead to increased cytokine production in the aged brain and exacerbate cognitive disruptions in aged mice compared to adult animals. After the first day of stress, aged and adult mice demonstrated an impaired ability to locate the platform compared to unstressed aged and adult mice. However, on subsequent days only aged stressed mice demonstrated the impairment while adult stressed animals had returned to levels comparable to the unstressed animals (Figure 2C). Restraint stress failed to induce IL-1β in the adult brain; however, aged-stressed animals demonstrated increased levels of IL-1β in the hippocampus compared to adult animals and aged non-stressed animals. Interestingly, the level of cognitive impairment (i.e., distance swam) in the water maze was positively correlated with the levels of IL-1β in aged mice but there was no such correlation in adult mice. Furthermore, it was shown that adult mice had a stress-related decrease in MHC II expression, while aged mice had a stressed-related increase in MHC II expression in the hippocampus. As cortisol has been shown to affect learning and memory [43]), plasma corticosterone (CORT) levels in adult and aged mice that had been exposed to the stressor was also examined. Both ages demonstrated an increase in CORT across sessions of stress; however, this did not correspond with the cognitive effects as stressed adult animals showed no impairment after the initial session of stress. Together, the results suggest that a mild repeated stressor was able to exacerbate neuroinflammation in aged mice and was more disruptive to learning and memory compared to adults. These results support the theory that the aged brain may be primed to react to a variety of stressors (psychological or immunological) in a way that leads to more profound and longer lasting behavioral alterations compared to the adult brain.
Conclusions
The aged brain is characterized by a homeostatic shift in which neuroinflammation is increased and may serve to sensitize the brain to effects of infection or stress. In aged animals, including humans there are increased numbers of activated and primed microglia that may lead to increased production of cytokines following induction of proinflammatory mediators within the CNS. Additionally, the reduction of anti-inflammatory mediators may make the aged brain less able to effectively regulate perturbations to the neuroinflammatory status of the CNS. As the responses to immune stimulation and stressors share many of the same characteristics and pathways, it is possible that the factors associated with increased neuroinflammation in the aged brain may underlie the behavioral responses to each of these stimuli. Therefore, aged animals display prolonged behavioral deficits following peripheral immune activation and are sensitized to the cognitive effects associated an immune stimulus or stress. Importantly, these alterations in neuroinflammation and sensitization to extrinsic and intrinsic stressors may have profound effects upon the aged patient’s recovery and coping during disease and stress.
Acknowledgments
Supported by NIH grants AG016710, AG023580 and MH069148
References
- 1.Perry VH, Matyszak MK, Fearn S. Altered antigen expression of microglia in the aged rodent CNS. Glia. 1993;7(1):60–7. doi: 10.1002/glia.440070111. [DOI] [PubMed] [Google Scholar]
- 2.Sheffield LG, Berman NE. Microglial expression of MHC class II increases in normal aging of nonhuman primates. Neurobiol Aging. 1998;19(1):47–55. doi: 10.1016/s0197-4580(97)00168-1. [DOI] [PubMed] [Google Scholar]
- 3.Streit WJ, Sammons NW, Kuhns AJ, Sparks DL. Dystrophic microglia in the aging human brain. Glia. 2004;45(2):208–12. doi: 10.1002/glia.10319. [DOI] [PubMed] [Google Scholar]
- 4.Perry VH, Newman TA, Cunningham C. The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci. 2003;4(2):103–12. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- 5.Boche D, Cunningham C, Gauldie J, Perry VH. Transforming growth factor-beta 1-mediated neuroprotection against excitotoxic injury in vivo. J Cereb Blood Flow Metab. 2003;23(10):1174–82. doi: 10.1097/01.WCB.0000090080.64176.44. [DOI] [PubMed] [Google Scholar]
- 6.Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun. 2007;21(1):47–59. doi: 10.1016/j.bbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Blasko I, Stampfer-Kountchev M, Robatscher P, Veerhuis R, Eikelenboom P, Grubeck-Loebenstein B. How chronic inflammation can affect the brain and support the development of Alzheimer’s disease in old age: the role of microglia and astrocytes. Aging Cell. 2004;3(4):169–76. doi: 10.1111/j.1474-9728.2004.00101.x. [DOI] [PubMed] [Google Scholar]
- 8.Weaver JD, Huang MH, Albert M, Harris T, Rowe JW, Seeman TE. Interleukin-6 and risk of cognitive decline: MacArthur studies of successful aging. Neurology. 2002;59(3):371–8. doi: 10.1212/wnl.59.3.371. [DOI] [PubMed] [Google Scholar]
- 9.Kiecolt-Glaser JK, Preacher KJ, MacCallum RC, Atkinson C, Malarkey WB, Glaser R. Chronic stress and age-related increases in the proinflammatory cytokine IL-6. Proc Natl Acad Sci U S A. 2003;100(15):9090–5. doi: 10.1073/pnas.1531903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ye SM, Johnson RW. Increased interleukin-6 expression by microglia from brain of aged mice. J Neuroimmunol. 1999;93(1–2):139–48. doi: 10.1016/s0165-5728(98)00217-3. [DOI] [PubMed] [Google Scholar]
- 11.Tha KK, Okuma Y, Miyazaki H, et al. Changes in expressions of proinflammatory cytokines IL-1beta, TNF-alpha and IL-6 in the brain of senescence accelerated mouse (SAM) P8. Brain Res. 2000;885(1):25–31. doi: 10.1016/s0006-8993(00)02883-3. [DOI] [PubMed] [Google Scholar]
- 12.Prechel MM, Halbur L, Devata S, Vaidya AM, Young MR. Increased interleukin-6 production by cerebral cortical tissue of adult versus young mice. Mech Ageing Dev. 1996;92(2–3):185–94. doi: 10.1016/s0047-6374(96)01833-7. [DOI] [PubMed] [Google Scholar]
- 13.Ye SM, Johnson RW. An age-related decline in interleukin-10 may contribute to the increased expression of interleukin-6 in brain of aged mice. Neuroimmunomodulation. 2001;9(4):183–92. doi: 10.1159/000049025. [DOI] [PubMed] [Google Scholar]
- 14.Reyes TM, Fabry Z, Coe CL. Brain endothelial cell production of a neuroprotective cytokine, interleukin-6, in response to noxious stimuli. Brain Res. 1999;851(1–2):215–20. doi: 10.1016/s0006-8993(99)02189-7. [DOI] [PubMed] [Google Scholar]
- 15.Ye S, Johnson RW. Regulation of interleukin-6 gene expression in brain of aged mice by nuclear factor kappaB. J Neuroimmunol. 2001;117(1–2):87–96. doi: 10.1016/s0165-5728(01)00316-2. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun. 2008;22(3):301–11. doi: 10.1016/j.bbi.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sparkman NL, Buchanan JB, Heyen JR, Chen J, Beverly JL, Johnson RW. Interleukin-6 facilitates lipopolysaccharide-induced disruption in working memory and expression of other proinflammatory cytokines in hippocampal neuronal cell layers. J Neurosci. 2006;26(42):10709–16. doi: 10.1523/JNEUROSCI.3376-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frank MG, Barrientos RM, Biedenkapp JC, Rudy JW, Watkins LR, Maier SF. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol Aging. 2006;27(5):717–22. doi: 10.1016/j.neurobiolaging.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 19.Grilli M, Barbieri I, Basudev H, et al. Interleukin-10 modulates neuronal threshold of vulnerability to ischaemic damage. Eur J Neurosci. 2000;12(7):2265–72. doi: 10.1046/j.1460-9568.2000.00090.x. [DOI] [PubMed] [Google Scholar]
- 20.Thackray AM, McKenzie AN, Klein MA, Lauder A, Bujdoso R. Accelerated prion disease in the absence of interleukin-10. J Virol. 2004;78(24):13697–707. doi: 10.1128/JVI.78.24.13697-13707.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Luigi A, Fragiacomo C, Lucca U, Quadri P, Tettamanti M, Grazia De Simoni M. Inflammatory markers in Alzheimer’s disease and multi-infarct dementia. Mech Ageing Dev. 2001;122(16):1985–95. doi: 10.1016/s0047-6374(01)00313-x. [DOI] [PubMed] [Google Scholar]
- 22.Blalock JE. The syntax of immune-neuroendocrine communication. Immunol Today. 1994;15(11):504–11. doi: 10.1016/0167-5699(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 23.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maier SF. Bi-directional immune-brain communication: Implications for understanding stress, pain, and cognition. Brain Behav Immun. 2003;17(2):69–85. doi: 10.1016/s0889-1591(03)00032-1. [DOI] [PubMed] [Google Scholar]
- 25.Banks WA. The blood-brain barrier in psychoneuroimmunology. Neurol Clin. 2006;24(3):413–9. doi: 10.1016/j.ncl.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 26.Banks WA, Farr SA, La Scola ME, Morley JE. Intravenous human interleukin-1alpha impairs memory processing in mice: dependence on blood-brain barrier transport into posterior division of the septum. J Pharmacol Exp Ther. 2001;299(2):536–41. [PubMed] [Google Scholar]
- 27.Maier SF, Goehler LE, Fleshner M, Watkins LR. The role of the vagus nerve in cytokine-to-brain communication. Ann N Y Acad Sci. 1998;840:289–300. doi: 10.1111/j.1749-6632.1998.tb09569.x. [DOI] [PubMed] [Google Scholar]
- 28.Buttini M, Boddeke H. Peripheral lipopolysaccharide stimulation induces interleukin-1 beta messenger RNA in rat brain microglial cells. Neuroscience. 1995;65(2):523–30. doi: 10.1016/0306-4522(94)00525-a. [DOI] [PubMed] [Google Scholar]
- 29.Godbout JP, Chen J, Abraham J, et al. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. Faseb J. 2005;19(10):1329–31. doi: 10.1096/fj.05-3776fje. [DOI] [PubMed] [Google Scholar]
- 30.Huang Y, Henry CJ, Dantzer R, Johnson RW, Godbout JP. Exaggerated sickness behavior and brain proinflammatory cytokine expression in aged mice in response to intracerebroventricular lipopolysaccharide. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parnet P, Kelley KW, Bluthe RM, Dantzer R. Expression and regulation of interleukin-1 receptors in the brain. Role in cytokines-induced sickness behavior. J Neuroimmunol. 2002;125(1–2):5–14. doi: 10.1016/s0165-5728(02)00022-x. [DOI] [PubMed] [Google Scholar]
- 32.Bellinger FP, Madamba S, Siggins GR. Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993;628(1–2):227–34. doi: 10.1016/0006-8993(93)90959-q. [DOI] [PubMed] [Google Scholar]
- 33.Rachal Pugh C, Fleshner M, Watkins LR, Maier SF, Rudy JW. The immune system and memory consolidation: a role for the cytokine IL-1beta. Neurosci Biobehav Rev. 2001;25(1):29–41. doi: 10.1016/s0149-7634(00)00048-8. [DOI] [PubMed] [Google Scholar]
- 34.Goshen I, Kreisel T, Ounallah-Saad H, et al. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology. 2007;32(8–10):1106–15. doi: 10.1016/j.psyneuen.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 35.Barrientos RM, Higgins EA, Biedenkapp JC, et al. Peripheral infection and aging interact to impair hippocampal memory consolidation. Neurobiol Aging. 2006;27(5):723–32. doi: 10.1016/j.neurobiolaging.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 36.Holmes C, El-Okl M, Williams AL, Cunningham C, Wilcockson D, Perry VH. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74(6):788–9. doi: 10.1136/jnnp.74.6.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnston M, Wakeling A, Graham N, Stokes F. Cognitive impairment, emotional disorder and length of stay of elderly patients in a district general hospital. Br J Med Psychol. 1987;60 (Pt 2):133–9. doi: 10.1111/j.2044-8341.1987.tb02723.x. [DOI] [PubMed] [Google Scholar]
- 38.Johnson JD, O’Connor KA, Deak T, Stark M, Watkins LR, Maier SF. Prior stressor exposure sensitizes LPS-induced cytokine production. Brain Behav Immun. 2002;16(4):461–76. doi: 10.1006/brbi.2001.0638. [DOI] [PubMed] [Google Scholar]
- 39.Barrientos RM, Sprunger DB, Campeau S, et al. Brain-derived neurotrophic factor mRNA downregulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist. Neuroscience. 2003;121(4):847–53. doi: 10.1016/s0306-4522(03)00564-5. [DOI] [PubMed] [Google Scholar]
- 40.Ben Menachem-Zidon O, Goshen I, Kreisel T, et al. Intrahippocampal Transplantation of Transgenic Neural Precursor Cells Overexpressing Interleukin-1 Receptor Antagonist Blocks Chronic Isolation-Induced Impairment in Memory and Neurogenesis. Neuropsychopharmacology. 2007 doi: 10.1038/sj.npp.1301606. [DOI] [PubMed] [Google Scholar]
- 41.Neupert SD, Almeida DM, Mroczek DK, Spiro A., 3rd Daily stressors and memory failures in a naturalistic setting: findings from the VA Normative Aging Study. Psychol Aging. 2006;21(2):424–9. doi: 10.1037/0882-7974.21.2.424. [DOI] [PubMed] [Google Scholar]
- 42.Buchanan JBSN, Chen J, Johnson RW. Cognitive and neuroinflammatory consequences of mild repeated stress are exacerbated in aged mice. Psychoneuroendocrinology. 2008 doi: 10.1016/j.psyneuen.2008.02.013. 10.1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lupien SJ, Maheu F, Tu M, Fiocco A, Schramek TE. The effects of stress and stress hormones on human cognition: Implications for the field of brain and cognition. Brain Cogn. 2007;65(3):209–37. doi: 10.1016/j.bandc.2007.02.007. [DOI] [PubMed] [Google Scholar]