

Abstract
Pemphigus encompasses a group of organ specific, antibody mediated autoimmune diseases of the skin characterized by keratinocyte detachment that leads to the development of blisters and erosions, which can become life-threatening. The pathogenic autoantibodies recognize desmogleins, which are members of the desmosomal cadherin family of cell adhesion molecules. Desmoglein 3 is targeted in pemphigus vulgaris while desmoglein 1 is targeted in pemphigus foliaceus and its endemic form, fogo selvagem. This review will briefly define the salient features of pemphigus and the proposed steps in pathogenesis. We will then summarize the most recent advances in three important areas of investigation: (i) epidemiologic, genetic, and immunologic features of fogo selvagem, (ii) molecular mechanisms of injury to the epidermis, and (iii) novel therapeutic strategies targeting specific steps in disease pathogenesis. The advances in each of these three seemingly separate areas contribute to the overall understanding of the pemphigus disease model. These recent advancements also underscore the dynamic interplay between the treatment of patients in a clinical setting and basic science research, which has led to an integrative understanding disease pathogenesis and treatment and allow pemphigus to serve as a paradigm of human autoimmunity.
Keywords: Pemphigus vulgaris, pemphigus foliaceus, fogo selvagem, desmoglein, treatment
1. Introduction
The development of autoimmune disease is one of the fundamental enigmas of immunology. The triggers that direct the immune response against self-antigens are believed to be multifactorial, involving both genetic susceptibility and environmental factors. Current research is aimed at understanding the molecular details of both genetic and environmental contributions. These studies are often limited by the complex nature of such interactions and the seemingly sporadic development of disease in the general population. An ideal research model would focus on an organ specific autoimmune response directed against a well-defined autoantigen in a population of patients in which the prevalence of disease is fairly high and environmental and genetic factors could be studied over a long period of time. The rarity of such research models in today’s geographically mobile society has led many researchers to turn to animal models of disease, which have been quite instructive. The application of these findings to human clinical disease is ongoing.
Our area of interest lies in autoimmune diseases of the skin, which include pemphigus vulgaris (PV) and pemphigus foliaceus (PF). The pemphigus spectrum of diseases has many features that fulfill the criteria for an ideal research model of human autoimmune disease described above. These diseases involve autoantibody formation directed against the desmoglein (Dsg) family of cell adhesion molecules in keratinocytes, resulting in keratinocyte detachment (acantholysis) and intraepithelial blister formation. The anti-Dsg autoantibodies are pathogenic by passive transfer into mice and can be followed as markers of disease activity [1, 2]. Desmoglein 3 (Dsg3) has been identified as the target of PV autoantibodies, while desmoglein 1 (Dsg1) is the autoantigen recognized by PF autoantibodies [3]. These self-antigens have been fully characterized, allowing researchers to map epitopes throughout disease progression. Furthermore, the skin and sera of patients are easily accessible for collection and use in research settings. While the molecular features of pemphigus are well defined, the rare and sporadic development of disease in the general population is somewhat limiting to research efforts.
Although pemphigus is rare in the general population, endemic foci of disease have been identified in certain regions of Brazil. This endemic form of PF, also known as Fogo Selvagem (FS) amongst local inhabitants of rural Brazil, shares similar clinical, histological and immunologic features with the non-endemic form of PF seen in the USA and around the world. As in the non-endemic form of PF, FS is characterized by pathogenic IgG autoantibodies that recognize the Dsg1 ectodomain [4, 5] resulting in subcorneal blisters producing localized or generalized skin syndromes (Fig. 1). Novel immunologic features of this disease have been uncovered through studies of a highly endemic focus of FS in the Limao Verde Amerindian reservation in Brazil [6]. Investigations on the evolution of the anti-Dsg1 autoimmune response and the appearance of disease in the inhabitants of Limao Verde and neighboring communities provide a unique opportunity to study the development of autoimmune disease in a well-defined, geographically limited population with a high prevalence of disease.
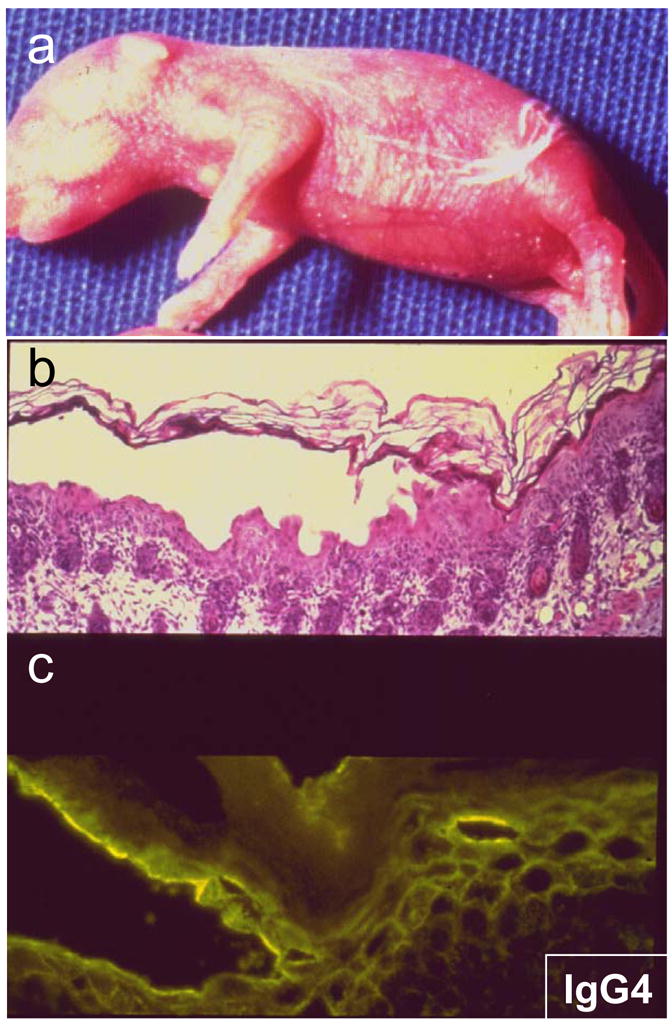
Figure 1.

Clinical presentation of Fogo Selvagem with (a) spontaneous generalized blisters and erosions. (b) Biopsies of these lesions demonstrate intra-epidermal subcorneal vesicles due to cell detachment (acantholysis). (c) Direct immunofluorescence locates the total IgG or IgG4 to the surface of detached epidermal cells.
Collectively, the current body of knowledge in the pemphigus field points to a multistep model of disease pathogenesis in which (i) a genetically susceptible individual is exposed to (ii) a triggering environmental antigen that leads to (iii) autoreactive T and B cell activation with specific autoantibody class and subclass responses and epitope spreading. Subsequent (iv) binding of desmoglein specific autoantibodies then triggers (v) desmoglein signaling and apoptotic events that may be associated with acantholysis and blister formation. This latter step of pathogenesis focusing on downstream events following autoantibody binding has proven to be an exciting and active area of research over the last several years and has led to a better understanding of the molecular mechanisms of injury to the epidermis. Not only does this multistep model of pathogenesis provide a unique example of how an ideal research model can lead to a comprehensive and global view of the development of autoimmunity, but it also serves as a springboard for novel approaches to the treatment of pemphigus aimed at interfering with each step of pathogenesis.
In this review we will summarize recent findings in three exciting areas of investigation, including (i) epidemiologic and immunologic features of FS, (ii) mechanisms of immunologic injury to the epidermis, and (iii) novel therapeutic strategies targeting specific steps in disease pathogenesis. Advances in each of these areas contribute to the overall understanding of pemphigus at different steps of the multistep model.
2. Advances in the Epidemiologic and Immunologic Features of FS
2.1 The endemic regions of FS in Brazil and the Limao Verde Reservation
FS occurs in Brazilian states located between 45° to 60° west longitude and 5° to 25° south latitude in regions with an altitude between 500–800 meters (1,600–2,600 feet) [7–9]. The weather in endemic regions of FS is subtropical and supports coffee, sugarcane and cacao in the northern regions, while corn, soybeans and cotton are the predominant crops in southern regions.
Young adults and children are typically affected and there is no reported sex or racial predisposition [7–12]. FS patients are outdoor workers, usually farmers or family members of a farmer. The daily activities of a family include agriculture, care of livestock and home chores (i.e. cooking, caring for small animals, or washing laundry in nearby rivers or streams). Many wives take part in farming activities as well. The family shares a common bedroom and personal hygiene is poor. The diet primarily consists of high carbohydrates and low protein intake.
The houses are usually built of reed walls and thatched roofs with open doors and windows. They commonly harbor rodents and other small wild animals and are usually infested with blood-feeding arthropods such as bedbugs and Reduvid bugs. A variety of other insects are found in nearby rivers and streams, including Simuliids (black fly, also known as “borrachudo” in Portuguese) and sandflies. Epidemiological studies have shown that the number of new cases of FS is greatest at the end of the rainy season (September to March) and least during the dry summer (April to August), suggesting that insect multiplication and increase in the number of FS patients are related phenomena [7–9]. Interestingly, the same ecological systems found in the “pemphigus country” overlap with those described in Chagas disease [13–15], Leishmaniasis [16–18] and Onchocerciasis [19, 20]. These diseases, as well as FS, were initially reported in Brazil during the first decade of the last century affecting the newly arrived pioneers to previously unconquered lands [13, 18, 21]. The diseases were common among workers laboring in the construction of railroads and highways in the interior of the endemic states, i.e. Sao Paulo and Minas Gerais. Interestingly, these diseases tend to disappear once the working and living conditions of the settlers improve. In recent years the frequency of cases of Chagas disease, Leishmaniasis and FS in the endemic states, as compared with those seen in the middle part of the 20th century, has dramatically decreased [11, 22–28] and this decrease has been associated with modernization in agricultural techniques [29]. It is clear that the ecological systems of FS share similarities with these insect vector borne diseases.
The Amerindian reservation of Limao Verde, located in the state of Mato Grosso do Sul, Brazil, is the home of 1,351 members of the Terena tribe of Amerindians, and is an active focus of FS, exhibiting a ~3 % prevalence of disease [6, 30] (Fig. 2). The people of this reservation are heavily exposed to hematophagous insects inside and outside of their homes [31] and these insects are considered risk factors for FS [32, 33]. For the last 14 years, we have systematically collected clinical and serological data from FS patients as well as clinically normal individuals residing in and around this settlement. Interestingly, a significant number of normal individuals (55%) living on the reservation possess anti-Dsg1 antibodies [30]. The autoantibody response against Dsg1 in healthy individuals from Limao Verde and neighboring communities is common, and directly related to physical distance from this reservation [6, 30, 34]. Follow up studies of this settlement have shown an average of 1–3 new cases of FS per year. Over the course of our investigation, we have also observed the clinical and serological conversion from normal to disease state, initially in 5 cases [30, 35], and recently to include another 6 individuals [36].
Figure 2.

The endemic regions of FS. The endemic regions of FS are located in the central planes of Brazil, comprising the following states: Sao Paulo, Parana, Minas Gerais, Goias, Mato Grosso and Mato Grosso do Sul and the Distrito Federal de Brasilia. Hospitals specialized to treat FS are located in cities of Sao Paulo, Goiania and Campo Grande. The Limao Verde Amerindian reservation is one of six Terena Indians settled in the state of Mato Grosso do Sul. The Limao Verde reservation shows a 3% prevalence of FS and is under investigation by our group since 1993.
2.2 FS: An environmentally triggered autoimmune disease of the skin
Although FS was originally described, and is still most frequently found in Brazil [6–11], reports exist of other foci of endemic PF in Colombia and Tunisia [37, 38]. FS, however, shows several unique and remarkable features such as the geographic and temporal clustering of cases, the increased frequency of cases among young adults and children, the increased frequency of familial cases, and an association with certain distinct HLA-DR alleles [39]. HLA susceptibility genes for endemic pemphigus have already been identified, namely HLA DRB1 0102, 0404, 1402, 1406. These alleles all share a consensus sequence LLEQRRAA at position 67–74 in the third hypervariable domain of the DRB1 gene.
The temporal and geographic clustering has also led to the hypothesis of a triggering environmental antigen resulting in molecular mimicry with Dsg1. Early studies in FS have demonstrated that the autoantibody response exhibits a limited heterogeneity, consisting of oligoclonal IgG banding, when tested with epidermal antigens by affinity immunoblotting [40]. Recent analysis of the variable regions of heavy and light chains from FS anti-Dsg1 autoantibodies suggests that the response is indeed antigen selected [41]. In this study, a total of seventy-seven hybridomas were generated from the B cells of eight FS patients and of one patient four years prior to the onset of clinical disease. Among the hybridomas analyzed, there were multiple clonally related sets of anti-Dsg1 antibodies within individual patients. Heavy and light chain V gene use was biased within IgG hybridomas and most hybridomas exhibited a bias favoring CDR amino acid replacement mutations, both of which are signs of antigen selection. Interestingly, hybridomas analyzed from the patient during pre-clinical phase also exhibited evidence of antigen selection and displayed significant overlap in VH gene usage with FS hybridomas. The pre-FS and FS hybridomas shared multiple replacement mutations suggesting selection by the same or similar antigen. These novel findings suggest that antigen selection is a process that occurs well before the onset of disease [41]. Current studies are underway to further investigate the nature of the environmental trigger. Certainly, the link to hemaphagous insects is intriguing and provides a new avenue of investigation.
2.3 B cell responses in FS
2.3.1 The IgG Subclasses in FS
Once the proposed environmental antigen has triggered the immune system, the B cell response can be studied in terms of IgG subclass response giving further insight into the nature of the pathogenesis. Our studies have shown that the IgG autoantibodies in FS are isotype-restricted and the bulk of pathogenic anti-Dsg1 autoantibodies are IgG4 [36, 42–44] (Fig. 3). For instance, studies on IgG subclasses of the anti-Dsg1 response in FS patients and normal subjects from Limao Verde show that FS patients have IgG4 levels 19.3 fold higher than ELISA positive normal subjects [44]. Similarly, patients with active disease have similar levels of IgG1, but 74.3 fold higher IgG4 levels compared to patients in clinical remission. Most importantly, IgG subclass restriction was followed in the 5 patients from whom we had blood samples both before and after the onset of clinical disease. Levels of IgG1 increased 3.45 fold with onset of disease, whereas levels of IgG4 increased 103.08 fold with onset of clinical disease (both values represent the mean of 5 patients). These results may indicate that progression from a pre-clinical to a clinical phase of the disease, and also the transition from remission to active disease, is closely associated with subclass switching from IgG1 to IgG4. These results also suggest that IgG1 anti-Dsg1 autoantibodies from FS patients alone are incapable of triggering disease. Indeed, studies using the murine passive transfer model show that IgG1 subclass antibodies from a normal subject (who only has an IgG1 response) are incapable of mediating disease at 15 mg/kg. In contrast, IgG4 antibodies from a patient were capable of mediating disease at 1.5mg/kg, a concentration 10 times lower [35]. These studies suggest that the autoantibody response in FS exhibits an early IgG1 response followed by a sustained IgG4.
Figure 3.
Anti-Dsg1 IgG4 antibodies from FS patients are pathogenic in neonatal mice. IgG4 purified from FS patients was injected intraperitoneally to neonatal Blab/c mice. (a) In the course of 8–24 hours, these animals develop superficial vesicles and erosions. (b) Histologic examination reveals subcorneal vesicles and (c) direct IF shows the human IgG4 bound to the epidermal intercellular spaces.
In an effort to identify early sensitive and specific serological markers of FS in healthy individuals, we tested the IgG subclass anti-Dsg1 autoantibody response by ELISA in a large number of patients (n= 214) and normal donors (n= 261) [36]. Analysis shows that IgG4 is a novel classifier/predictor that identifies donors with immunologic features of FS (values of IgG4 anti-Dsg1 > 6.43 index value units). The IgG4 classifier is highly sensitive [92% (95% CI: 82–95)] and specific [97% (95% CI: 89–100)]. In a population with a prevalence of 3%, this classifier has a positive predictive value (PPV) of 49% and a negative predictive value (NPV) of 99.7%. This instrument has been validated in two patient populations, a group of 60 Japanese patients with PF and PV and a group of 11 FS patients from Limao Verde where preclinical stage sera were available. The use of this classifier tool may facilitate the identification of individuals during the preclinical stage of FS, thus enhancing the chances of disclosing the etiological agent(s) triggering this human autoimmune disease. Since IgG anti-Dsg1 autoantibodies are detected in a large number of normal individuals from endemic areas of FS [30, 44, 45], the high specificity of the IgG4 anti-Dsg1 classifier will be of greater use than the total IgG assays when used in these seroepidemiological studies.
Restriction of IgG4 antibody response in humans has been reported in patients with parasitosis [46], individuals undergoing hyposensitization therapy for allergies [47, 48], individuals exposed to bee stings [49] and patients with autoimmune pancreatitis [50]. IgG4 restriction of the autoimmune response has also been reported in other autoimmune blistering diseases, including PV [51–55]. However, there is limited information about their pathogenic role in skin disease [55]. For instance, Kricheli et al. [54] identified antibodies to Dsg-3 in the serum of 91% of PV patients by immunoblot, and also in 49% of those related to patients. The distribution of IgG1 through IgG3 was similar in patients and relatives, however IgG4 was seen in 62% of patients, but fewer than 2% of the relatives and none of the normal subjects. Bhol et al. found a predominantly IgG1 response to Dsg3 in relatives of patients and patients in remission, while patients with clinical disease had both an IgG1 and IgG4 response [51]. When considering the pathogenic role of IgG4 antibodies, it is possible that these antibodies recognize a different epitope than the non-pathogenic IgG1 antibodies. Alternatively, IgG4 may have an effector function that is not present in IgG1, or a higher in vivo binding affinity for Dsg1.
2.3.2 Relevance of the IgM autoantibody response in FS
We have recently described a relatively high prevalence of IgM anti-Dsg1 autoantibodies in sera from both FS patients and clinically normal donors residing in rural settings in or near an endemic area such as Limao Verde [45]. Patients with PF, other autoimmune bullous diseases, as well as healthy individuals from more urban settings, did not demonstrate a significant IgM anti-Dsg1 response. Strikingly, over 50% of sera from healthy donors between the ages of 5 and 20 from Limao Verde possess IgM anti-Dsg1 autoantibodies. This IgM response is acquired sometime during early infancy, as neonates from mothers with FS do not possess IgM anti-Dsg1 antibodies. These novel findings support the hypothesis that an environmental antigen(s) sensitizes individuals living in these rural endemic areas of FS. Further, these results strengthen our findings that B cell repertoire in FS is antigen driven during the preclinical stage.
While the observed seroepidemiological studies of FS suggest that the IgM anti-Dsg1 autoantibody response may be secondary to exposure to an environmental antigen(s), there is also the possibility that they may represent natural IgM antibodies. These natural antibodies are known to be polyreactive and represent the first barrier against infection, eliminating bacteria by complement activation, thus bridging innate and adaptive immunity [56]. Many of these natural IgM antibodies recognize single or multiple self-antigens [57] and have been detected from early childhood throughout life [58]. They are often an indicator of a recent infection or re-infection with viral diseases such as rubella [59], infectious mononucleosis [60] or hepatitis B [61], bacterial infections such as Lyme disease [62], or parasitosis such as toxoplasmosis [63, 64]. Natural IgM antibodies against self-antigens have been reported in autoimmune diseases such as lupus erythematosus [65–68] autoimmune hemolytic anemia [69], and autoimmune thrombocytopenia [70]. Some have proposed that anti-Ro IgM autoantibodies in lupus may represent part of the natural IgM repertoire that leads to production of pathogenic IgG autoantibodies by epitope spreading in genetically predisposed individuals [65]. Certainly, it is intriguing that polyreactive IgM or the IgG/IgM ratio of anti-dsDNA antibodies in systemic lupus erythematosus may modulate the disease and prognosis in experimental murine models and SLE patients alike [66, 68, 71].
In summary, these seroepidemiological observations suggest that the IgM response in FS patients from Limao Verde likely arises from recurrent and persistent exposure to an environmental antigen(s) that shares similar epitopes to Dsg1 harbored in this and other Amerindian reservations. Sensitization begins in early childhood and continues throughout life in these individuals, resulting in the production of non-pathogenic IgM and IgG anti-Dsg1 autoantibodies. Pathogenic IgG anti-Dsg1 autoantibodies and clinical disease subsequently occur in only a small fraction of genetically predisposed individuals. It is likely that serum concentrations of IgM anti-Dsg1 autoantibodies from FS patients referred to metropolitan hospitals, away from their native environment, decrease due to elimination of the environmental antigenic stimuli.
2.3.3 Dsg1 epitopes recognized by IgG anti-Dsg1 autoantibodies during preclinical stages of FS
Just as the transition from preclinical FS to clinical disease is marked by an IgG isotype switch, different epitopes are known to be targeted during this conversion [35]. Our studies have shown that anti-Dsg1 antibodies from normal individuals and individuals in the preclinical stage of FS all recognize the EC5 domain (residues 453–496) of the molecule. Significantly, transition from the pre-clinical to clinical stage of FS is accompanied by emergence of autoantibodies specific for the EC1 (residues 1–108) and EC2 (residues 109–221) domains. Although the majority of patients with active disease possess a mixture of anti-EC1 and anti-EC2 antibody populations, a few sera possess antibodies to EC1 without EC2, or to EC2 without EC1. This observation indicates that autoantibodies against EC1 and EC2 recognize at least two distinct epitopes, and either one alone may be capable of causing acantholysis, and therefore, disease [35].
Furthermore, anti-EC1 and anti-EC2 autoantibodies disappear with disease remission and reappear with disease relapses. EC5-specific antibodies are present mainly in patients during disease onset or clinical relapse, and persist during remission. The immunologic mechanism responsible for the elimination of anti-EC1/EC2 antibodies in FS patients during remission and recurrence during relapses is not clear. It is possible that the reappearance these antibodies during disease relapse may result from reactivation of memory B and T cells by re-exposure to self-Dsg1 [35].
Moreover, by passive transfer experiments we have demonstrated that anti-EC1/EC2 autoantibodies are pathogenic in neonatal mice, whereas the IgG fraction containing anti-EC5 autoantibodies are incapable of inducing blisters. These data suggest that the anti-Dsg1 autoimmune response in FS is initially raised against nonpathogenic epitopes located in the EC5 domain and later against pathogenic epitopes mapped to the EC1 and EC2 domains of the molecule [35]. It is likely that anti-EC1/EC2 autoantibodies are generated through the mechanism of intramolecular epitope spreading [72, 73] from the initial anti-EC5 autoantibodies rather than through cross-reactivity, since there is no significant sequence homology between the EC5 domain and the cadherin-like domains (EC1 to EC4) (Fig. 4). While IgG1 autoantibodies target the EC5 domain of Dsg1 during the pre-clinical stage, a predominantly IgG4-mediated autoantibody response against the Dsg1 EC1-2 domains is seen with the onset of clinically active disease [35, 36, 44].
Figure 4.

Molecular homology between Dsg1, Dsg3 and E-cadherin. The desmogleins 1 and 3 are desmosomal transmembrane proteins sharing the same cadherin-like domain structure of E-cadherin. The EC1-EC2 domains of Dsg1 are recognized by pathogenic autoantibodies of FS patients whereas Dsg3 is bound by autoantibodies from pemphigus vulgaris patients. Autoantibodies against E-cadherin have also been described in mucocutaneous PV, PF and FS [95].
Finally, a recent study shows that a significant number of patients with diseases that are transmitted by hematophagous vectors, [i.e., onchocerciasis (black flies), leishmaniasis (sand flies) and Chagas disease (kissing bugs)] possess antibodies against the EC5 domain of Dsg1 [74]. Onchocerciasis is unknown in the Limao Verde reservation, but leishmaniasis, Chagas disease and FS are all prevalent in this settlement. We hypothesize that a salivary antigen(s) from these blood-feeding insects may contain a cross-reactive molecule(s) that triggers the anti-Dsg1 EC5 antibody response.
Based on the current findings, we propose a two-phase model to encompass the immunopathogenic mechanisms of FS. In the first phase, an environmental antigen(s), bearing sequence homology with the EC5 domain of Dsg1, triggers an initial non-pathogenic antibody response to the EC5 domain of Dsg1. At this stage, individuals remain free of skin disease. In the second phase, in certain genetically susceptible individuals, intramolecular epitope spreading occurs, which leads to the production of pathogenic anti-Dsg1 antibodies against the EC1 and EC2 domains, inducing skin blistering. According to our recent studies the incubation time between the first and the second phase of FS could last as long as 10 years [36].
2.4 T cell responses in FS
Although pemphigus pathogenesis is mediated by autoreactive antibodies produced by Dsg specific B cells, autoreactive T cells are necessary for B cell activation. Several groups have identified CD4+ T cell proliferative responses to the Dsg3 ectodomain in PV patients [75–77]. PV patients that exhibit combined autoantibody responses to Dsg3 and Dsg1 also possess T cells that proliferate in response to either Dsg3 or Dsg1 peptides [78]. Recognition of Dsg3 peptides by Th1 and Th2 cells is restricted by pemphigus associated MHC alleles DRB1-0402 and DQB1-0503 as the T cell proliferative responses to Dsg3 peptides can be inhibited by anti-DR and anti-DQ antibodies [76, 79–84]. PV Th1 and Th2 cells recognize a limited set of Dsg3 peptides characterized by a similar positive charge at position 4, which is necessary for maintaining an ideal fit within the MHC binding pockets [77, 85]. Interestingly, T cells from healthy controls sharing the same PV associated HLA alleles demonstrated similar T cell responses to the same set of Dsg3 peptides, suggesting that loss of tolerance in PV is within the B cell compartment [85].
T cell clones from patients with FS also recognize epitopes of the Dsg1 ectodomain as T cells from 13 of 15 FS patients responded to recombinant Dsg1 [76]. The Dsg1 response of T cell clones derived from 5 patients was blocked by anti-DR antibodies, but not by anti-DQ or -DP antibodies, suggesting that the Dsg1-specific response of FS T cells is restricted to HLA-DR. The T cell clones derived from FS patients produce IL-4, IL-5, and IL-6, but not γ-IFN, suggesting that they secrete a Th2-like cytokine profile. The type II cytokines such as IL-4 might modulate the IgG subclass response in these patients. Analysis of the TCR regions from anti-Dsg T cell clones from PV and FS patients revealed an oligoclonal response with a limited number of TCRs utilized [86, 87].
Both in vitro and in vivo studies have shown that these Dsg specific T cells are necessary for autoreactive B cell activation. Anti-Dsg3 B cells were induced to secrete antibody when PV patient peripheral lymphocytes were stimulated with Dsg3. Depletion of CD4+ cells or addition of anti-DR/DQ antibodies abolished this response suggesting that the T cell/MHC interaction is critical for Dsg3 specific B cell activation and antibody secretion [88]. In vivo evidence supporting the critical role of Dsg specific T cells in B cell activation was provided by Tsunoda et al. using an active murine model of pemphigus first described by Amagai et al. [89]. In this model, splenocytes from Dsg3 immunized Dsg3−/− mice are transferred into immunodeficient Rag2−/− mice recapitulating the oral erosions and suprabasilar acantholysis found in PV [89]. Tsunoda et al. showed that pathogenic anti-Dsg3 antibodies were only produced in Rag2−/− recipients when both B and T cell splenocyte fractions were transferred [90].
More recently, several groups have studied the importance of regulatory T cells in PV. Regulatory T cells help to maintain tolerance and control autoimmune disease in the T cell compartment. In addition to naturally occurring CD25+ T regulatory cells produced continuously by the thymus, antigen induced Type 1 regulatory T cells have been described [91]. Type 1 regulatory T cells secrete IL-10 and TGF-beta, cytokines that are felt to be immunosuppressive and critical in maintaining T cell tolerance. Both types of regulatory T cells seem to be involved in PV. Sugiyama and colleagues have shown that CD4+CD25hi cells are markedly decreased in PV patients peripheral blood compared to healthy controls [92]. Recent studies have also shown that IL-10 secreting Dsg3 specific T cells are present in the majority of healthy controls, but less than 20% of PV patients, suggesting that loss of regulatory T cell function may play a role in disease development [93]. These Dsg3 specific Type 1 regulatory T cells inhibit activation of Dsg3 specific Th1 and Th2 cells through secretion of IL-10 and TGF-beta and express Foxp3, a transcriptional repressor known to be important in regulatory T cell development and function [94]. Inactivation of Foxp3 using antisense technology induced a phenotypic loss of Dsg3 regulatory T cell markers and function. These cells instead developed a robust proliferative response to Dsg3 and secreted a cytokine profile similar to that of Th2 cells [94]. Thus, regulatory T cells seem to play a crucial role in maintaining T cell tolerance and may also represent a target for potential novel therapies.
3. Mechanisms of immunological injury to the epidermis in pemphigus
The mechanisms of autoantibody-induced epidermal detachment (acantholysis) in PF/FS remain under intense investigation in different laboratories around the world. The sera of patients with all clinical variants of pemphigus, like that of patients with other autoimmune diseases, contain autoantibodies against multiple self-antigens. Some of the best characterized antigens are expressed on keratinocyte cell-cell adhesion organelles such as the desmosomes, i.e. Dsg1, Dsg3, Dsg4, desmocollins [3] or interdesmosomal domains of the keratinocyte membrane, i.e. E-cadherin [95]. In PV, PF and FS, anti-Dsg1 and anti-Dsg3 autoantibodies are pathogenic and serve as diagnostic markers of disease [1–3]. Anti-Dsg3 autoantibodies are markers of the mucosal variant of PV; whereas, the mucocutaneous variant of PV is characterized by a mixed population of anti-Dsg1 and anti-Dsg3 autoantibodies [96]. The pathogenic relevance of other autoantibodies detected in PV, PF or FS sera remains unknown. Given our current understanding, we must conclude that pemphigus is a group of diseases in which desmosomal antigens are targeted by pathogenic and non-pathogenic autoantibodies. The resulting disease phenotypes are epidermal-specific (PF/FS), mucosal-specific (mPV), or epidermal and mucosal specific (mcPV). Only mucosal surfaces that possess squamous epithelium are involved in mPV and mcPV.
Desmosomes mediate cell-cell adhesion and can be found not only in skin, but also in cardiomyocytes, brain arachnoidal cells and lymph node dendritic reticulum cells. Desmosomes are cellular organelles that structurally are formed by a central core and two intracellular plaques. While the core is composed of transmembrane glycoproteins (the desmosomal cadherins), the intracellular plaque contains large molecular weight protein members of the plakin family, i.e. desmoplakins, plakoglobin, and plakophillin [97]. The desmosomal cadherins comprise Dsg1, Dsg2, Dsg3 and desmocollins. Dsg1 and Dsg3 are restricted to stratified squamous epithelia, while Dsg2 is expressed in all tissues expressing desmosomes. Similar to E cadherin, the extracellular domain of all desmosomal cadherins, except Dsg1, show 5 domains which participate in interactions with desmogleins on opposing cells, thereby establishing adhesion [98]. Through linker proteins, the intracellular (or cytoplasmic) domains of E cadherin, desmogleins and desmocollins are associated with the keratinocyte cytoskeleton.
The distribution and density of Dsg1 and Dsg3 varies within the layers of the stratified epidermis and mucosal epidermis. As the desmogleins are the target of autoantibodies in pemphigus, their differential distribution is thought to be responsible for the different histological sites of blister formation in PF and PV. This concept is known as the compensation theory [99, 100]. For example, Dsg1 is expressed throughout the epidermis, but its expression is increased in the superficial epidermis. Dsg3 is expressed in an inverse pattern with increased expression in the basal layers of the epidermis. In mucosal epithelium, Dsg1 is expressed in the superficial squamous epithelium, while Dsg3 is expressed throughout. It is hypothesized that in PF, the anti-Dsg1 autoantibodies target Dsg1, leading to blister formation in the superficial epidermis as Dsg3 expression in basal cells compensates for the loss of Dsg1 mediated adhesion. The mucosa is protected by expression of Dsg3 throughout all layers, and therefore PF patients do not exhibit mucosal involvement. In PV, where the target is Dsg3, blister formation is suprabasilar as superficial epidermal adhesion is maintained by Dsg1. Mucosal involvement is common as there is no expression of Dsg1 in the basal layers of mucosal epithelium to compensate for the loss of Dsg3 function [99, 100].
In addition to the distribution of Dsg within the epithelia, i.e. compensation theory, we propose that the epithelial damage in all forms of pemphigus is modulated by the epitope-specificity of the anti-desmoglein autoantibodies. As previously shown, healthy individuals and FS patients during the preclinical stages of FS recognize epitopes on Dsg1 or Dsg3 that are not pathogenic. Once an individual mounts an autoimmune response against epitopes located on the EC1 and EC2 domain of Dsg1 or Dsg3 the autoantibodies lead to pathogenic damage of the epithelia. Importantly, these pathogenic autoantibodies in FS are IgG4 restricted. It is possible that the availability of pathogenically relevant epitopes on Dsg1 and Dsg3 in vivo to react with PV and PF/FS autoantibodies would determine the level of tissue damage. The histological phenotype of pemphigus therefore, would be determined by the fine specificity of the anti-Dsg1 or anti-Dsg3 autoantibodies as well as the distribution of the respective desmosomal antigens in the epithelia.
To understand the complexity of the humoral autoimmune response in all forms of pemphigus we hypothesize that the autoantibody response against “unique epitopes” on Dsg1 and Dsg3 in humans is associated with a spectrum of clinical phenotypes (Table 1). They include (i) normal individuals who exhibit normal skin and have non-pathogenic anti-Dsg1/Dsg3 autoantibodies, (ii) PV patients with pure mucosal disease and pathogenic anti-Dsg3 autoantibodies (oral PV), (iii) PV patients exhibiting combined mucosal and cutaneous disease with pathogenic anti-Dsg3 and anti-Dsg1 autoantibodies (mucocutaneous PV). The subset of pemphigus showing a pure cutaneous blistering eruption and having pathogenic anti-Dsg1 autoantibodies includes PF/FS patients. Importantly, our studies on FS in Brazil are allowing us to investigate the transition of individuals from a preclinical state (possessing non-pathogenic anti-Dsg1 autoantibodies) to individuals with classic FS, exhibiting pathogenic anti-Dsg1 autoantibodies. Moreover, we have recently described normal individuals possessing non-pathogenic anti-Dsg3 autoantibodies and a subset of patients with an endemic form of mucocutaneous PV [101]. We believe that the clinical phenotypes of all forms of pemphigus may result, aside from the Dsg1/Dsg3 distribution in the target tissues, from the epitope specificity of the autoantibodies harbored in normal individuals and patients, and this in turn, from the immunogenetic make up of the host.
Table 1.
Diversity of anti-desmoglein autoantibodies present in normal individuals and patients exhibiting the different disease phenotypes of pemphigus.
| Disease Phenotype | Autoantibodies | Antigens | Pathogenicity: (Mouse model) | |
|---|---|---|---|---|
| Pemphigus Vulgaris | Mucosal type | IgG Autoantibodies (common) | Dsg3 alone | Non-pathogenic |
| IgG Autoantibodies (Rare) | Dsg3 alone | Pathogenic | ||
| Mucocutaneous type | IgG Autoantibodies (Common) | Dsg3 & Dsg1 | Pathogenic | |
| IgG Autoantibodies (Common) | Dsg3 alone | Pathogenic | ||
| Non-Endemic Pemphigus Foliaceus | Cutaneous | IgG Autoantibodies (Common) | Dsg1 alone | Pathogenic |
| IgG Autoantibodies (Rare) | Dsg1 & Dsg3 | Pathogenic | ||
| Fogo Selvagem | Cutaneous | IgG Autoantibodies (Common) | Dsg1 alone | Pathogenic |
| IgM Autoantibodies (Common) | Dsg1 alone | Unknown | ||
| Normal Individuals from Endemic Areas | Normal skin:Pre-clinical stage | IgG & IgM Autoantibodies (Common) | Dsg1 alone | Unknown |
| IgG Autoantibodies (Common) | Dsg3 alone | Unknown | ||
| “Endemic” Pemphigus Vulgaris | Mucocutaneous type | IgG Autoantibodies (Common) | Dsg3 & Dsg1 | Unknown |
| IgG Autoantibodies: ? | Dsg3 alone | Unknown | ||
It has been proposed that anti-desmosomal antibodies induce acantholysis by steric hindrance, where autoantibody binding directly blocks Dsg adhesive interactions on opposing cells [102]. Steric hindrance is supported by the compensation theory as well as by findings in staphylococcal scalded skin syndrome, in which an exfoliative toxin cleaves Dsg1 within the extracellular domains and leads to an identical histological phenotype as PF [103]. In addition, Dsg3 knockout mice display blister formation as seen in PV [104]. Thus, it seems that impairing the function of these desmogleins at the extracellular level leads to histological features resembling acantholysis triggered by binding of anti-Dsg1 or anti-Dsg3 autoantibodies to their target antigen. The detachment process is complement-independent and can be triggered by Fab fragments of the pathogenic autoantibodies [105–107]. Waschke et al [108], utilizing single molecule-based micromechanical laser tweezers and atomic force microscopy, have probed Dsg1 and Dsg3 ectodomain transinteractions. They have suggested that PV anti-Dsg3 autoantibodies impair the adhesive function of Dsg3 and thus trigger acantholysis; whereas, PF autoantibodies trigger acantholysis by decreasing the amounts of Dsg1 available on the surface of keratinocytes secondary to intracellular signaling. These studies need to be extended and reproduced by other laboratories. The fate of the anti-Dsg1 or anti-Dsg3 autoantibodies following the initial binding to the ectodomain of the antigens has been studied at the ultrastructural level by Patel el al. in 1984 [109] and recently by Delva et al. [110] in cell culture. They demonstrated that PV IgG molecules are internalized via a clathrin and dynamin-independent pathway to form endocytic vesicles that fuse with lysosomes. This process is associated with widening of the intercellular spaces. Pharmacological inhibition of the internalization process may provide new therapeutic approaches for pemphigus acantholysis. In addition to these findings, other investigators [111, 112] have demonstrated that following the binding of PV IgG to keratinocytes surfaces, Dsg3 is depleted from desmosomes, which in turn may be relevant in triggering acantholysis.
Several studies have identified signaling capabilities of the desmogleins, which may also play a role in autoantibody induced acantholysis. It was initially observed that keratinocyte cell lines treated with PV sera show a transient increase in intracellular calcium and inositol 1,4,5-triphosphate, supporting the idea that autoantibody binding to desmogleins initiates intracellular signaling events [113]. These investigators extended these studies by demonstrating that PV IgG induces phosphorylation of Dsg3 and subsequent dissociation from plakoglobin in cultured primary keratinocytes, thus triggering acantholysis [114]. This conclusion was reasonable since it was well known that phosphorylation of the classical cadherins at serine and tyrosine residues, regulates cell adhesion [115, 116]. It has been proposed that the plakoglobin linker protein plays a crucial role in the signaling process mediated by desmosomes and consequently in pemphigus acantholysis. Caldelari et al. [117] showed that keratinocytes from plakoglobin knockout mice were unresponsive to the pathogenic effects of PV IgG on cell adhesion compared to keratinocytes from wild type mice. They hypothesize that the plakoglobin/c-Myc proto-oncogene axis may be relevant to the pathogenesis of pemphigus acantholysis [118].
Members of our own group have shown that primary human keratinocytes treated with PV IgG show a time and dose-dependent increase in levels of phosphorylated p38MAPK and heat shock protein HSP27 [119]. Both p38MAPK and HSP27 are known to be important in regulating cytoskeletal components such as actin and keratin intermediate filaments. Further studies have shown that inhibitors of MAPK signaling not only blocked phosphorylation of HSP27 following PV IgG stimulation of cultured primary keratinocytes, but also prevented keratin filament retraction, an early change in the cytoskeleton associated with acantholysis [119]. These findings were recapitulated in the PV passive transfer murine model [120]. Subsequent studies have identified increased phosphorylation of p38MAPK and HSP27 in skin biopsies of patients with PV and PF compared to that of controls [121]. Collectively, these data have identified an important signaling pathway in the pathogenesis of PV and PF. Studies are underway to test whether p38MAPK inhibitors might be useful in treating clinical disease.
In addition to the signaling pathways described above, in vitro cell culture studies have shown that exposure of keratinocytes to PV IgG induce apoptosis in these cells [122–126]. The pro-apoptotic changes are evident by various measurements, including annexin V binding, Hoechst 33342 staining, TUNEL labeling, DNA laddering, oligonucleosome formation, caspase activation, up-regulation of pro-apoptotic proteins (Fas, FasL, Bax, p53), and down-regulation of anti-apoptotic proteins such as Bcl-2 and FLIP-l. It is speculated that keratinocyte apoptosis results in pemphigus acantholysis [122, 124–127].
While much in vitro data has been published on the induction of apoptosis by PV IgG, little is known about the possible apoptotic effect of PF IgG. Our group [128] has evaluated the role of the biochemical response of apoptosis in PF using the IgG passive transfer mouse model of the disease [2]. We found TUNEL positive epidermal cells and increased cytosolic oligonucleosomes in epidermal cells of the diseased mice. A time course study revealed that TUNEL-positive epidermal cells appear prior to intraepidermal blisters. Western blot analysis showed that the pro-apoptotic factor Bax was upregulated at the earlier time points (2 and 4 h) while the anti-apoptotic factor Bcl-xl was downregulated at the later time points (6, 8, and 20 h) post PF IgG injection. Correspondingly, the active forms of caspase-3 and -6 were detected at the later time period (6, 8, and 20 h). Administration of Ac-DEVD-cmk, a peptide-based caspase-3 inhibitor, protected mice from developing intraepidermal blisters and clinical disease induced by PF IgG. The same protective effect was also observed for a broad-spectrum caspase inhibitor Bok-D-fmk. Collectively, the findings of this study suggest that some biochemical events of apoptosis are provoked in epidermal cells by PF autoantibodies and caspase-3 activation may contribute to acantholytic process and disease pathogenesis.
This observation may suggest that activation of caspase leads to epidermal cell injury and dysfunction, which in turn is manifested as intraepidermal blisters. Acantholysis may occur before or without the end-point of epidermal cell death. We hypothesize that activated caspase-3 is involved in the process of acantholysis and/or intraepidermal blister formation, not through epidermal cell death, but through the proteolytic cleavage of a structural protein(s) involved in epidermal cell-cell adhesion. Possible candidates may include components of the desmosome, adherens junction, and cytoskeleton. Many of cell adhesion molecules are caspase substrates [129] and are degraded during apoptosis, such as Dsg1 [130, 131], Dsg3, plakoglobin, plakophillin, plakin proteins [132–134], E-cadherin [135, 136], and β-catenin [137]. Interestingly, it has been shown that shedding of ectodomain of Dsg1 or Dsg3 during the process of apoptosis is inhibited by caspase inhibitors [130, 131]. Further studies are required to identify the potential targets for caspase-3 that may be responsible for PF blistering.
Impairment of Dsg1/Dsg3 adhesive function by autoantibody induced steric hindrance or by binding of autoantibodies triggering signaling and apoptosis that lead to acantholysis are not mutually exclusive (Fig. 5). Continued research is aimed at further delineating the exact role of these multistep processes. Certainly, it is an exciting area of investigation and likely to yield potential novel treatment options.
Figure 5.

Molecular mechanisms of acantholysis in pemphigus. Desmoglein 1 and 3 are linked to the intermediate filament keratin network of the keratinocyte whereas E-cadherin links to the actin cytoskeleton. Pathogenic autoantibodies in PV and PF bind the ectodomain of Dsg3 or Dsg1 and trigger intracellular signaling that leads to acantholysis. Keratinocyte apoptosis also results from this immunopathological process.
4. Advances in the therapy of pemphigus
Prior to the advent of corticosteroids, the diagnosis of pemphigus carried a poor prognosis; it was stated that if a patient survived for more than a year the diagnosis of pemphigus was likely incorrect. Armed with limited information regarding pathogenesis, dermatologists in the 1950’s began to use systemic steroids to treat these diseases [138]. The autoimmune nature of pemphigus was used as rationale for the introduction of additional immunosuppressive drugs and plasmapheresis, both aimed at eliminating pathogenic autoantibodies from the patient. The combined use of systemic steroid and immunosuppressive agents such as azathioprine, cyclosphosphamide, methotrexate, mycophenolate mofetil, and cyclosporine have become the standard of therapy of PV and PF with good results. However, the morbidity associated with the long-term use of these drugs remains problematic for patients, especially those with diabetes, hypertension or other medical conditions. Since the introduction of systemic steroids in the 1950’s and immunosuppressive agents in the 1960’s, the prognosis of the disease has been gradually improving. Recent reports estimate an overall 5% mortality in PV [139].
The scientific basis for the therapy of these disorders has evolved tremendously in the last 60 years; however, current treatment options are still limited. Certainly, the advances in understanding the immunopathogenesis of PV and PF/FS are prompting new potential therapies and allowing researchers to target therapy to individual steps in disease pathogenesis (Fig. 6).
Figure 6.

Rationale of pemphigus therapies. The five targeted areas where immuno-intervention may play a role in controlling disease activity in PV, PF and FS are shown in this figure. A PV patient is depicted with the targeted sites: (1) elimination of the autoantigen or triggering environmental antigen, (2) deletion of autoreactive and regulatory T cell activation, (3) deletion of autoreactive B cell responses, (4) elimination of circulating autoantibody and (5) altering the keratinocyte responses to pathogenic autoantibodies (epidermis becomes “resistant” to these autoantibodies).
4.1 Elimination of the triggering antigen(s)
Assuming that the environmental antigen(s) is identified, the elimination or avoidance of this antigen(s) by genetically susceptible people would likely prevent the onset of disease. Once disease is established, it is possible that elimination of the triggering antigen would lessen disease activity and possible even induce remission. However, it is not known if disease in PV or PF/FS is maintained by an endogenous source of Dsg1 or Dsg3.
An example of how the discontinuation of an etiological agent can interrupt an autoimmune disease would be in patients with drug-induced pemphigus or lupus where disease remission is seen following the discontinuation of the offending medication such as penicillinamine or captopryl [140–142]. FS patients also provide a useful clinical subgroup for analysis in this regard. Epidemiological data suggests that patients who relocate from their native endemic environment (and theoretically away from the triggering environmental antigen) to a more industrialized city experience disease regression, as evidenced by both clinical and immunologic criteria [11]. Ongoing studies to identify the triggering environmental antigen in FS will be critically important if we are to make significant progress in targeting therapy to this step of pathogenesis.
4.2 Targeting autoreactive T cells and antigen presenting cells
Although the disease causing autoantibodies in pemphigus are produced by terminally differentiated B cells, autoreactive T cells play an important role by providing B cell help during activation directly and by TH2 cytokine secretion [88, 91, 93, 94, 143, 144]. Preventing activation of autoreactive T cells that recognize autoantigen peptides presented on MHC class II molecules may be a useful therapeutic target. For example, myasthenia gravis (MG) is a neuromuscular autoimmune disease characterized by autoantibodies directed against the acetylcholine receptor (AchR). HLA linkage studies have shown strong association with HLA-DQB1. Studies performed in MG murine models have shown prevention of disease with monoclonal antibodies directed against the antigen binding site of the MHCII molecule or with direct immunization with MHCII peptides (generating a polyclonal antibody response). Similarly, in vitro studies with MG patients’ peripheral blood T cells showed inhibition of T cell proliferation to AchR in the presence of mAb to the DQB1 binding site [145]. Therefore, targeting disease associated MHC molecules can block T cell activation and subsequently blunt B cell responses, decreasing the production of disease causing autoantibodies. Although studies directly targeting the MCHII molecule have not been performed in PV, the HLA restrictions associated with disease makes this step of pathogenesis an exciting therapeutic target.
Another way to block the MHCII/T cell interaction is by use of peptide immunotherapy. This approach involves immunization with T cell immunodominant peptides with the goal of preventing activation of disease specific T cells. A phase I clinical trial in PV patients found no significant change in anti-Dsg3 antibodies following administration of intravenous Dsg3 peptides. Additional studies are planned with longer treatment times and higher doses [146, 147]. Peptide immunotherapy is also being investigated in the industrial sector using the immunodominant T-cell epitope of Dsg3. Phase I/II studies in stable PV patients have found the drug to be safe and well tolerated. Studies on efficacy are underway [148].
Finally, emerging treatment targeted towards anti-Dsg T cells involves harnessing T regulatory cells. There is evidence that regulatory T cells may play a role in maintaining T cell tolerance in healthy individuals and are decreased in pemphigus patients [92, 93]. Manipulation of such cells in patients may also provide a future direction for therapy [94, 149]. Finally, recent data from Eming et al. [150] suggests that rituximab, a anti-CD20 monoclonal antibody targeting B cells, also leads to a decreased frequency of Dsg3 specific Th1 and Th2 cells while total T cell counts remain unchanged. This data suggests that rituximab may play a role in controlling autoreactive T cells as well as autoreactive B cells.
4.3 Targeting B cells
In an effort to better target the cell responsible for eventual autoantibody production, B cell specific treatment has emerged as an exciting treatment alternative. Rituximab is an anti-CD20 chimeric monoclonal antibody currently FDA approved for the treatment of B-cell non-Hodgkin’s lymphoma and anti-TNFα refractory rheumatoid arthritis. This therapy has been shown to be especially useful in cases of pemphigus that are resistant to conventional therapy of steroids and immunosuppressive drugs.
Several groups have published open, prospective trials studying rituximab in patients with refractory pemphigus. One study focused on 11 patients with refractory pemphigus vulgaris who received a combination of rituximab (10 infusions) and intravenous immunoglobulin (IVIg) (6 infusions) over 6 months. Nine patients had resolution of clinical disease lasting 22–37 months post treatment [151]. Other groups have reported a similar response without concurrent use of IVIg. For example, Joly and colleagues [152] reported a prospective open trial of rituximab in PV and PF patients who (i) had not responded to prednisone at 1.5 mg/kg/day or (ii) had active disease despite systemic doses of prednisone greater than 20 mg/day, or (iii) in whom systemic steroids were contraindicated. Eighteen of 21 patients treated with 4 weekly infusions of rituximab experienced complete clinical remission by 3 months. In 8 of the 18 patients this remission was maintained without steroid or immunosuppressive therapy after a median follow up of almost 3 years. One patient died of septicemia and another developed pyelonephritis. Peripheral blood B cells decreased by 99% 3 weeks after the onset of therapy and remained undetectable for 6 months in 2 patients. While a significant decrease was seen in titers of anti-Dsg1 and anti-Dsg3 autoantibodies 3 months after the onset of therapy, there was no decrease in titers of antibodies against pneumococcal capsule polysaccaride or tetanus toxoid or total IgG values. Cianchini reported similar findings in 12 patients with PV and PF following 4 treatments with rituximab. Six months following the final infusion, 9 patients had a complete response, while the remaining 3 patients had a partial response. No adverse effects were noted following treatment and no serious infections were reported [153].
Since the clinical remission induced by rituximab in PV and PF patients is associated with a decrease in anti-Dsg3 and anti-Dsg1 autoantibodies without changes in the titers of anti-pneumococcal and anti-tetanus antibodies, it can be concluded that rituximab specifically targets Dsg1- and Dsg3-specific B cell precursors. It is thought that long-lived plasma cells produce antigen-specific autoantibodies that show stable titers in the serum of patients, while short-lived plasma cells and plasmablasts produce antigen-specific autoantibodies that fluctuate with disease activity [154]. Neither long-lived nor short lived plasma cells express the CD20 antigen and consequently are not likely eliminated by rituximab therapy. Instead, rituximab may be eliminating Dsg1 and Dsg3 plasmablasts or precursors of short-lived plasma cells. Further studies are needed to delineate the molecular and cellular mechanisms of action of rituximab. Currently, rituximab is restricted to a limited number of PV and PF patients that are resistant to or unable to tolerate conventional therapy (systemic steroids and immunosuppressive agents). The potential for serious short-term complications such as viral and bacterial infections and the potential for yet unknown long-term complications exist with rituximab treatment.
Given the success of rituximab in targeting autoreactive B cells in pemphigus, it is likely that other B cell directed therapies will emerge. While rituximab eliminates virtually all B cell populations, B cell stimulatory pathways such as the CD40/CD40L may provide an even more specific target for breakthrough therapies aimed at eliminating autoreactive B cells in pemphigus. For example, early studies have shown that CD40 positive cells were present in the basal and suprabasal layers of the epidermis in pemphigus patients. Furthermore, CD40L mRNA was present in all pemphigus patient skin samples as compared to healthy controls where levels were undetectable. Serum levels of CD40L was also significantly higher in pemphigus patients compared to healthy controls [155]. Thus, CD40/CD40L pathways are upregulated in pemphigus patients’ sera and skin. Targeting this and other B cell costimulatory pathways may provide additional therapeutic options in directing treatment to autoreactive B cells in pemphigus.
4.4 Elimination of circulating autoantibodies (Increasing IgG catabolism, antigen specific immunoadsorption or by using anti-idiotypic antibodies)
As the anti-Dsg1 and anti-Dsg3 autoantibodies are known to be pathogenic, much effort has been aimed at eliminating the antibodies from circulation, thereby preventing subsequent acantholysis and clinical disease.
Administration of intravenous immunoglobulin (IVIg) has been reported to control disease in pemphigus patients who have failed conventional therapy and may even induce remission in some patients with longstanding disease [156, 157]. Response time and duration varies among patients. The exact mechanism of action is unknown, but recent studies have suggested that IVIg increases the catabolism of immunoglobulins in an FcR dependent manner, thereby resulting in decreased serum levels of antibodies [67, 158]. More importantly, IVIg seems to selectively decrease the autoantibodies, as normal antibodies do not seem to be affected [158, 159]. Aside from its effects on autoantibody catabolism, IVIg also upregulates endogenous caspace and calpain inhibitors, thus protecting target cells from apoptotic and oncotic effectors, and preventing acantholysis [122]. Understanding the mechanism of action behind IVIg treatment may in turn lead to a better understanding of relevant disease pathways.
Immunoadsorption (IA) also aims at eliminating the circulating autoantibodies. IA relies upon affinity binding of autoantibodies and immune complexes to antigen immobilized on an adsorber. IA is preferred over plasmapheresis, as it does not result in nonspecific removal of other plasma proteins such as clotting factors, hormones, or albumin. The data describing effectiveness of IA in pemphigus is limited to case series of patients refractory to conventional therapy. All series describe an immediate reduction in anti-Dsg antibodies and rapid clinical improvement when IA was used as adjuvant therapy along with pulse administration of more conventional immunosuppressive medications. Adsorbers and treatment regimens varied between reports. It seems that IA may be beneficial as adjuvant therapy in patients failing conventional treatment [160].
The goal of anti-idiotype antibody therapy is to generate antibodies to the variable region of pathogenic autoantibodies, which would bind and thus neutralize only the pathogenic antibodies. Alvarado-Flores et al. reported use of anti-Dsg1 from a PF patient to induce anti-idiotype antibodies by rabbit immunization. Rabbit sera was then used to prevent blister formation in response to PF IgG in the passive transfer murine model system [161]. Similarly, antibodies have been generated to the variable region genes used in PV autoantibodies. Payne et al. showed that this approach would be feasible by characterizing the variable region genes used in PV IgG by phage display studies [162]. Dsg reactive monoclonal antibodies showed usage of a limited number of variable region genes and unique gene usage between pathogenic and nonpathogenic antibodies. Rabbit antisera generated against pathogenic anti-Dsg3 monoclonal antibodies inhibited pathogenicity of monoclonal antibodies in cultured human keratinocytes. Inhibition was effective against other anti-Dsg antibodies using the same heavy chain variable gene, but different light chain variable genes. Furthermore, anti-sera was able to adsorb pathogenic antibodies from multiple PV patients’ sera, suggesting shared heavy chain variable gene usage among patients [162]. Collectively, these studies illustrate that antibodies generated against the pathogenic anti-Dsg antibodies would likely be effective and feasible, however, more studies are needed.
4.5 Drug-induced resistance of the epidermis to pathogenic autoantibodies (inhibitors of signaling and apoptosis)
The final step in anti-Dsg induced acantholysis is the response of the keratinocyte to autoantibody binding via downstream signaling events and eventual keratin filament retraction and apoptosis. As detailed above, many signaling pathways have been implicated in anti-Dsg induced acantholysis. Studies targeting each of these many steps may be useful in directing novel therapies. For example, p38MAPK and HSP27 phosphorylation in response to PV IgG is dose dependent [120]. P38MAPK inhibitors abrogate PV IgG induced p38MAPK phosphorylation and keratin filament retraction in both cell culture and murine model systems [121]. Studies are currently underway to test the safety and efficacy of p38MAPK inhibitors in PV patients.
Similarly, PV IgG induces changes in both PLC/PKC and c-myc pathways. Inhibitors of PLC/PKC and c-myc were shown to prevent PV IgG induced blister formation in the neonatal passive transfer murine model [118]. These pathways also provide possible therapeutic targets aimed at inhibiting acantholysis.
Finally, members of our group have shown that apoptotic regulator p53 is increased in lesional and nonlesional skin from PV patients compared to normal controls. Neonatal mice injected with PV IgG showed sequential expression of p53 upstream of apoptosis and acantholysis. Neonatal mice pre-treated with p53 inhibitor pifithrin-alpha were resistant to PV or PF IgG induced blister formation [163]. Furthermore, p53 knockout mice were protected from PV IgG induced disease [164].
Among all of the novel treatments targeting specific areas of PV pathogenesis, the signaling targets provide the most specific approach, thereby sparing patients complete immunosuppression. These signaling pathways are undoubtedly important in many other cellular functions and early studies will likely focus on safety profiles. However, the advancement in treatments truly gives credence to the idea of translational research.
5. Conclusion
In summary, the pemphigus phenotypes represent a complex spectrum of autoimmune disease with multiple genetic and environmental factors playing a role in disease pathogenesis. The pemphigus research community has collectively made great strides over the past several years identifying important concepts in pathogenesis as well as identifying novel targets for directed therapy. The endemic form of pemphigus foliaceus, Fogo Selvagem, provides a unique model in which to study each step that may play a role in the pathogenesis of pemphigus, and autoimmune disease in general. The strong epidemiological data collected through the years in FS and the recent immunologic findings may some day allow investigators to uncover the etiology of this fascinating disease. If this is accomplished, it would represent a major step forward in understanding the pathogenesis of human autoimmune diseases mediated by self reactive autoantibodies.
Acknowledgments
Funding support is provided by NIH grants R01-AR30281, RO1-AR32599, T32 AR07369 (LAD), and K01-AR052109 and R03-AR053313 (NL). Ye Qian is a recipient of the 2008 Dermatology Foundation Research Award.
Abbreviations
- Dsg
desmoglein
- PF
pemphigus foliaceus
- FS
Fogo Selvagem
- PV
pemphigus vulgaris
- MG
myasthenia gravis
- AchR
acetylcholine receptor
- IVIg
intravenous immunoglobulin
- IA
immunoadsorption
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
References
- 1.Anhalt GJ, Labib RS, Voorhees JJ, Beals TF, Diaz LA. Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med. 1982;306:1189–1196. doi: 10.1056/NEJM198205203062001. [DOI] [PubMed] [Google Scholar]
- 2.Roscoe JT, Diaz L, Sampaio SA, Castro RM, Labib RS, Takahashi Y, Patel H, Anhalt GJ. Brazilian pemphigus foliaceus autoantibodies are pathogenic to BALB/c mice by passive transfer. J Invest Dermatol. 1985;85:538–541. doi: 10.1111/1523-1747.ep12277362. [DOI] [PubMed] [Google Scholar]
- 3.Udey MC, Stanley JR. Pemphigus--diseases of antidesmosomal autoimmunity. Jama. 1999;282:572–576. doi: 10.1001/jama.282.6.572. [DOI] [PubMed] [Google Scholar]
- 4.Emery DJ, Diaz LA, Fairley JA, Lopez A, Taylor AF, Giudice GJ. Pemphigus foliaceus and pemphigus vulgaris autoantibodies react with the extracellular domain of desmoglein-1. J Invest Dermatol. 1995;104:323–328. doi: 10.1111/1523-1747.ep12665364. [DOI] [PubMed] [Google Scholar]
- 5.Futamura S, Martins C, Rivitti EA, Labib RS, Diaz LA, Anhalt GJ. Ultrastructural studies of acantholysis induced in vivo by passive transfer of IgG from endemic pemphigus foliaceus (Fogo Selvagem) J Invest Dermatol. 1989;93:480–485. doi: 10.1111/1523-1747.ep12284041. [DOI] [PubMed] [Google Scholar]
- 6.Hans-Filho G, dos Santos V, Katayama JH, Aoki V, Rivitti EA, Sampaio SA, Friedman H, Moraes JR, Moraes ME, Eaton DP, Lopez AL, Hoffman RG, Fairley JA, Giudice GJ, Diaz LA. An active focus of high prevalence of fogo selvagem on an Amerindian reservation in Brazil. J Invest Dermatol. 1996;107:68–75. doi: 10.1111/1523-1747.ep12298213. [DOI] [PubMed] [Google Scholar]
- 7.Aranha Campos J, editor. Penfigo foliaceo (Fogo Selvagem): Aspectos clinicos e epidemiologicos. Comp Melhoramentos; Sao Paulo, Brazil: 1942. [Google Scholar]
- 8.Auad A. Penfigo foliaceo Sul-Americano no Estado de Goias, Brazil. Revista Patologia Tropical. 1972;1:293–346. [Google Scholar]
- 9.Vieira JP. Empresa Grafica da “Revista dos Tribunais”. Sao Paulo, Brazil: 1940. Novas Contribuicoes ao Estudo do Penfigo Foliaceo (Fogo Selvagem) no Estado de Sao Paulo. [Google Scholar]
- 10.Diaz LA, Sampaio SA, Rivitti EA, Martins CR, Cunha PR, Lombardi C, Almeida FA, Castro RM, Macca ML, Lavrado C, et al. Endemic pemphigus foliaceus (fogo selvagem). I. Clinical features and immunopathology. J Am Acad Dermatol. 1989;20:657–669. doi: 10.1016/s0190-9622(89)70079-7. [DOI] [PubMed] [Google Scholar]
- 11.Diaz LA, Sampaio SA, Rivitti EA, Martins CR, Cunha PR, Lombardi C, Almeida FA, Castro RM, Macca ML, Lavrado C, et al. Endemic pemphigus foliaceus (Fogo Selvagem): II. Current and historic epidemiologic studies. J Invest Dermatol. 1989;92:4–12. doi: 10.1111/1523-1747.ep13070394. [DOI] [PubMed] [Google Scholar]
- 12.Minelli L. Geografia Medica do Penfigo Foliaceo Sul Americano no Estado do Parana. An Bras Dermatol Sifilol. 1976;51:173–181. [Google Scholar]
- 13.Dias JCP, Schofield CJ. The evolution of Chagas disease (American Trypanosomiasis) control after 90 years since Carlos Chagas discovery. Mem Inst Oswaldo Cruz, Rio de Janeiro. 1999;94:103–121. doi: 10.1590/S0074-02761999000700011. [DOI] [PubMed] [Google Scholar]
- 14.Kirchhoff LV. American trypanosomiasis (Chagas’ disease)--a tropical disease now in the United States. N Engl J Med. 1993;329:639–644. doi: 10.1056/NEJM199308263290909. [DOI] [PubMed] [Google Scholar]
- 15.Williams-Blangero S, Vanderberg JL, Teixeira ARL. Attitudes towards Chagas’ disease in a Brazilian community. Cad Saude Publ (Rio) 1999;15:7–13. doi: 10.1590/s0102-311x1999000100002. [DOI] [PubMed] [Google Scholar]
- 16.Goihman-Yahr M. American mucocutaneous leishmaniasis. Dermatol Clin. 1994;12:703–712. [PubMed] [Google Scholar]
- 17.Klaus SK, Frankemburg S. Cutaneous Leishmaniasis in the Middle East. Clin Dermatol. 1999;17:137–141. doi: 10.1016/s0738-081x(99)00006-1. [DOI] [PubMed] [Google Scholar]
- 18.Rapel-Medeiros AC, Roselina AMF. Leishmaniose tegumentaria Americana: do historico aos dias de hoje. An Bras Dermatol. 1999;74:329–336. [Google Scholar]
- 19.Molyneux DH, Morel C. Onchocerciasis and Chagas’ disease control: the evolution of control via applied research through changing development scenarios. Br Med Bull. 1998;54:327–339. doi: 10.1093/oxfordjournals.bmb.a011691. [DOI] [PubMed] [Google Scholar]
- 20.Routh HB, Bhowmik KR. Filariasis. Dermatol Clin. 1994;12:719–727. [PubMed] [Google Scholar]
- 21.Paes Leme C. Contribuicao ao estudo do Tokelau. Tese Facultade de Medicina do Rio de Janeiro. Rio de Janeiro; Brazil: 1903. [Google Scholar]
- 22.Cunha PR. Sero Epidemiologia do Fogo Selvagem em um Foco Endemico Remanescente do Estado de Sao Paulo. Facultade de Medicina da Universidade de Sao Paulo; Sao Paulo, Brazil: 1987. [Google Scholar]
- 23.Da Silva LJ, editor. Capitulo 3: A Evolucion da Doenca de Chagas no Estado de Sao Paulo. Funcraf-Hucite C; 1999. A Desarticulacao do Espaco. [Google Scholar]
- 24.Empinotti JC, Aoki V, Filgueira A, Sampaio SA, Rivitti EA, Sanches JA, Jr, Li N, Hilario-Vargas J, Diaz LA. Clinical and serological follow-up studies of endemic pemphigus foliaceus (fogo selvagem) in Western Parana, Brazil (2001–2002) Br J Dermatol. 2006;155:446–450. doi: 10.1111/j.1365-2133.2006.07302.x. [DOI] [PubMed] [Google Scholar]
- 25.Empinotti JC, Diaz LA, Martins CR, Rivitti EA, Sampaio SA, Lombardi C, Sanches JA. Endemic pemphigus foliaceus in western Parana, Brazil (1976–1988). Cooperative Group for Fogo Selvagem Research. Br J Dermatol. 1990;123:431–437. doi: 10.1111/j.1365-2133.1990.tb01446.x. [DOI] [PubMed] [Google Scholar]
- 26.Proenca NG. Declinio do Penfigo Foliaceo no Estado de Sao Paulo, Brasil. Rev Paulista Med. 1977;89:97–100. [PubMed] [Google Scholar]
- 27.Silveira AC, Vinhaes MC. Elimination of vector-borne transmission of Chagas disease. Mem Inst Oswaldo Cruz, Rio de Janeiro. 1999;94:405–411. doi: 10.1590/S0074-02761999000700080. [DOI] [PubMed] [Google Scholar]
- 28.Vinhaes MC, Pinto-Diaz JC. Doenca de Chagas no Brasil. Cad Saude Publ (Rio) 2000;16:7–12. [PubMed] [Google Scholar]
- 29.Da Silva LJ, editor. Anexo 3: A Evolucion da Doenca de Chagas no Estado de Sao Paulo. Funcraf-Hucite; 1999. A Agricultura Paulista a Partir de 1950. [Google Scholar]
- 30.Warren SJ, Lin MS, Giudice GJ, Hoffmann RG, Hans-Filho G, Aoki V, Rivitti EA, Santos V, Diaz LA. The prevalence of antibodies against desmoglein 1 in endemic pemphigus foliaceus in Brazil. Cooperative Group on Fogo Selvagem Research. N Engl J Med. 2000;343:23–30. doi: 10.1056/NEJM200007063430104. [DOI] [PubMed] [Google Scholar]
- 31.Eaton DP, Diaz LA, Hans-Filho G, Santos VD, Aoki V, Friedman H, Rivitti EA, Sampaio SA, Gottlieb MS, Giudice GJ, Lopez A, Cupp EW. Comparison of black fly species (Diptera: Simuliidae) on an Amerindian reservation with a high prevalence of fogo selvagem to neighboring disease-free sites in the State of Mato Grosso do Sul, Brazil. The Cooperative Group on Fogo Selvagem Research. J Med Entomol. 1998;35:120–131. doi: 10.1093/jmedent/35.2.120. [DOI] [PubMed] [Google Scholar]
- 32.Aoki V, Millikan RC, Rivitti EA, Hans-Filho G, Eaton DP, Warren SJ, Li N, Hilario-Vargas J, Hoffmann RG, Diaz LA. Environmental risk factors in endemic pemphigus foliaceus (fogo selvagem) J Investig Dermatol Symp Proc. 2004;9:34–40. doi: 10.1111/j.1087-0024.2004.00833.x. [DOI] [PubMed] [Google Scholar]
- 33.Lombardi C, Borges PC, Chaul A, Sampaio SA, Rivitti EA, Friedman H, Martins CR, Sanches Junior JA, Cunha PR, Hoffmann RG, et al. Environmental risk factors in endemic pemphigus foliaceus (Fogo selvagem). “The Cooperative Group on Fogo Selvagem Research”. J Invest Dermatol. 1992;98:847–850. doi: 10.1111/1523-1747.ep12456932. [DOI] [PubMed] [Google Scholar]
- 34.Hilario-Vargas J, Dasher DA, Li N, Aoki V, Hans-Filho G, dos Santos V, Qaqish BF, Rivitti EA, Diaz LA. Prevalence of anti-desmoglein-3 antibodies in endemic regions of Fogo selvagem in Brazil. J Invest Dermatol. 2006;126:2044–2048. doi: 10.1038/sj.jid.5700388. [DOI] [PubMed] [Google Scholar]
- 35.Li N, Aoki V, Hans-Filho G, Rivitti EA, Diaz LA. The role of intramolecular epitope spreading in the pathogenesis of endemic pemphigus foliaceus (fogo selvagem) J Exp Med. 2003;197:1501–1510. doi: 10.1084/jem.20022031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qaqish BF, Prisayanh P, Qian Y, Andraca E, Li N, Aoli V, Hans-Filho G, dos Santos V, Rivitti EA, Diaz LA. Development of an IgG4-based Classifier/Predictor of Endemic Pemphigus Foliaceus (Fogo Selvagem) J Invest Dermatol. 2008 doi: 10.1038/jid.2008.189. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abreu-Velez AM, Hashimoto T, Bollag WB, Tobon Arroyave S, Abreu-Velez CE, Londono ML, Montoya F, Beutner EH. A unique form of endemic pemphigus in northern Colombia. J Am Acad Dermatol. 2003;49:599–608. doi: 10.1067/s0190-9622(03)00851-x. [DOI] [PubMed] [Google Scholar]
- 38.Morini JP, Jomaa B, Gorgi Y, Saguem MH, Nouira R, Roujeau JC, Revuz J. Pemphigus foliaceus in young women. An endemic focus in the Sousse area of Tunisia. Arch Dermatol. 1993;129:69–73. doi: 10.1001/archderm.129.1.69. [DOI] [PubMed] [Google Scholar]
- 39.Moraes ME, Fernandez-Vina M, Lazaro A, Diaz LA, Filho GH, Friedman H, Rivitti E, Aoki V, Stastny P, Moraes JR. An epitope in the third hypervariable region of the DRB1 gene is involved in the susceptibility to endemic pemphigus foliaceus (fogo selvagem) in three different Brazilian populations. Tissue Antigens. 1997;49:35–40. doi: 10.1111/j.1399-0039.1997.tb02707.x. [DOI] [PubMed] [Google Scholar]
- 40.Calvanico NJ, Swartz SJ, Diaz LA. Affinity immunoblotting studies on the restriction of autoantibodies from endemic pemphigus foliaceus patients. J Autoimmun. 1993;6:145–157. doi: 10.1006/jaut.1993.1012. [DOI] [PubMed] [Google Scholar]
- 41.Qian Y, Clarke SH, Aoki V, Hans-Filhio G, Rivitti EA, Diaz LA. Antigen selection of the anti-Dsg1 response in endemic pemphigus foliaceus (Fogo Selvagem) begins before the onset of clinical disease. 2008 Submitted for publication. [Google Scholar]
- 42.Dos Santos S. Perfil evolutivo das subclasses de imunoglobulinas gama em pacientes de penfigo foliaceo endimico. Universidada Federal do Rio de Janeiro; Rio de Janeiro: 1996. [Google Scholar]
- 43.Rock B, Martins CR, Theofilopoulos AN, Balderas RS, Anhalt GJ, Labib RS, Futamura S, Rivitti EA, Diaz LA. The pathogenic effect of IgG4 autoantibodies in endemic pemphigus foliaceus (fogo selvagem) N Engl J Med. 1989;320:1463–1469. doi: 10.1056/NEJM198906013202206. [DOI] [PubMed] [Google Scholar]
- 44.Warren SJ, Arteaga LA, Rivitti EA, Aoki V, Hans-Filho G, Qaqish BF, Lin MS, Giudice GJ, Diaz LA. The role of subclass switching in the pathogenesis of endemic pemphigus foliaceus. J Invest Dermatol. 2003;120:104–108. doi: 10.1046/j.1523-1747.2003.12017.x. [DOI] [PubMed] [Google Scholar]
- 45.Diaz LA, Prisayanh PS, Dasher DA, Li N, Evangelista F, Aoki V, Hans-Filho G, dos Santos V, Qaqish BF, Rivitti EA. The IgM anti-desmoglein 1 response distinguishes Brazilian pemphigus foliaceus (fogo selvagem) from other forms of pemphigus. J Invest Dermatol. 2008;128:667–675. doi: 10.1038/sj.jid.5701121. [DOI] [PubMed] [Google Scholar]
- 46.Kurniawan A, Yazdanbakhsh M, van Ree R, Aalberse R, Selkirk ME, Partono F, Maizels RM. Differential expression of IgE and IgG4 specific antibody responses in asymptomatic and chronic human filariasis. J Immunol. 1993;150:3941–3950. [PubMed] [Google Scholar]
- 47.Larche M, Akdis C, Valenta R. Immunological mechanisms of allergen-specific immunotherapy. Nat Rev Immunol. 2006;6:761–771. doi: 10.1038/nri1934. [DOI] [PubMed] [Google Scholar]
- 48.Rossi R, Monasterolo G, Coco G, Silvestro L, Operti D. Evaluation of serum IgG4 antibodies specific to grass pollen allergen components in the follow up of allergic patients undergoing subcutaneous and sublingual immunotherapy. Vaccine. 2007;25:957–964. doi: 10.1016/j.vaccine.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 49.Aalberse RC, van der Gaag R, van Leeuwen J. Serologic aspects of IgG4 antibodies. I. Prolonged immunization results in an IgG4-restricted response. J Immunol. 1983;130:722–726. [PubMed] [Google Scholar]
- 50.Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, Fukushima M, Nikaido T, Nakayama K, Usuda N, Kiyosawa K. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732–738. doi: 10.1056/NEJM200103083441005. [DOI] [PubMed] [Google Scholar]
- 51.Bhol K, Natarajan K, Nagarwalla N, Mohimen A, Aoki V, Ahmed AR. Correlation of peptide specificity and IgG subclass with pathogenic and nonpathogenic autoantibodies in pemphigus vulgaris: a model for autoimmunity. Proc Natl Acad Sci U S A. 1995;92:5239–5243. doi: 10.1073/pnas.92.11.5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hertl M, Riechers R. Analysis of the T cells that are potentially involved in autoantibody production in pemphigus vulgaris. J Dermatol. 1999;26:748–752. doi: 10.1111/j.1346-8138.1999.tb02086.x. [DOI] [PubMed] [Google Scholar]
- 53.Jones CC, Hamilton RG, Jordon RE. Subclass distribution of human IgG autoantibodies in pemphigus. J Clin Immunol. 1988;8:43–49. doi: 10.1007/BF00915155. [DOI] [PubMed] [Google Scholar]
- 54.Kricheli D, David M, Frusic-Zlotkin M, Goldsmith D, Rabinov M, Sulkes J, Milner Y. The distribution of pemphigus vulgaris-IgG subclasses and their reactivity with desmoglein 3 and 1 in pemphigus patients and their first-degree relatives. Br J Dermatol. 2000;143:337–342. doi: 10.1046/j.1365-2133.2000.03659.x. [DOI] [PubMed] [Google Scholar]
- 55.Sitaru C, Mihai S, Zillikens D. The relevance of the IgG subclass of autoantibodies for blister induction in autoimmune bullous skin diseases. Arch Dermatol Res. 2007;299:1–8. doi: 10.1007/s00403-007-0734-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manson JM, Mauri C, Ehrenstein MR. Natural serum IgM maintains immunological homeostasis and prevents autoimmunity. Spriger Semin Immunol. 2005;26:425–432. doi: 10.1007/s00281-004-0187-x. [DOI] [PubMed] [Google Scholar]
- 57.Coutinho A, Kazatchkine MD, Avrameas S. Natural autoantibodies. Curr Opin Immunol. 1995;7:812–818. doi: 10.1016/0952-7915(95)80053-0. [DOI] [PubMed] [Google Scholar]
- 58.Mouthon L, Lacroix-Desmazes S, Nobrega A, Barreau C, Coutinho A, Kazatchkine MD. The self-reactive antibody repertoire of normal human serum IgM is acquired in early childhood and remains conserved throughout life. Scand J Immunol. 1996;44:243–251. doi: 10.1046/j.1365-3083.1996.d01-306.x. [DOI] [PubMed] [Google Scholar]
- 59.Hamkar R, Jalilvand S, Mokhtari-Azad T, Nouri Jelyani K, Dahi-Far H, Soleimanjahi H, Nategh R. Assessment of IgM enzyme immunoassay and IgG avidity assay for distinguishing between primary and secondary immune response to rubella vaccine. J Virol Methods. 2005;130:59–65. doi: 10.1016/j.jviromet.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 60.Obel N, Hoier-Madsen M, Kangro H. Serological and clinical findings in patients with serological evidence of reactivated Epstein-Barr virus infection. APMIS. 1996;104:424–428. doi: 10.1111/j.1699-0463.1996.tb00737.x. [DOI] [PubMed] [Google Scholar]
- 61.Chu CM. Immunoglobulin class M anti-hepatitis B core antigen for serodiagnosis of acute hepatitis: pitfalls and recommendations. J Gastroenterol Hepatol. 2006;21:789–791. doi: 10.1111/j.1440-1746.2006.04337.x. [DOI] [PubMed] [Google Scholar]
- 62.Cermakova Z, Ryskova O, Honegr K, Cermakova E, Hanovcova I. Diagnosis of Lyme borreliosis using enzyme immunoanalysis. Med Sci Monit. 2005;11:BR121–125. [PubMed] [Google Scholar]
- 63.Montoya JG. Laboratory diagnosis of Toxoplasma gondii infection and toxoplasmosis. J Infect Dis. 2002;185 (Suppl 1):S73–82. doi: 10.1086/338827. [DOI] [PubMed] [Google Scholar]
- 64.Reis MM, Tessaro MM, D’Azevedo PA. Toxoplasma-IgM and IgG-avidity in single samples from areas with a high infection rate can determine the risk of mother-to-child transmission. Rev Inst Med Trop Sao Paulo. 2006;48:93–98. doi: 10.1590/s0036-46652006000200007. [DOI] [PubMed] [Google Scholar]
- 65.Ferreira R, Barreto M, Santos E, Pereira C, Martins B, Andreia R, Crespo F, Viana JF, Vasconcelos C, Ferreira C, Vicente AM, Fesel C. Heritable factors shape natural human IgM reactivity to Ro60/SS-A and may predispose for SLE-associated IgG anti-Ro and anti-La autoantibody production. J Autoimmun. 2005;25:155–163. doi: 10.1016/j.jaut.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 66.Forger F, Matthias T, Oppermann M, Becker H, Helmke K. Clinical significance of anti-dsDNA antibody isotypes: IgG/IgM ratio of anti-dsDNA antibodies as a prognostic marker for lupus nephritis. Lupus. 2004;13:36–44. doi: 10.1191/0961203304lu485oa. [DOI] [PubMed] [Google Scholar]
- 67.Li N, Zhao M, Hilario-Vargas J, Prisayanh P, Warren S, Diaz LA, Roopenian DC, Liu Z. Complete FcRn dependence for intravenous Ig therapy in autoimmune skin blistering diseases. J Clin Invest. 2005;115:3440–3450. doi: 10.1172/JCI24394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li QZ, Xie C, Wu T, Mackay M, Aranow C, Putterman C, Mohan C. Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J Clin Invest. 2005;115:3428–3439. doi: 10.1172/JCI23587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stahl D, Sibrowski W. Warm autoimmune hemolytic anemia is an IgM-IgG immune complex disease. J Autoimmun. 2005;25:272–282. doi: 10.1016/j.jaut.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 70.Stahl D, Hoemberg M, Cassens U, Pachmann U, Sibrowski W. Evidence that human autoimmune thrombocytopenia mediated by both immunoglobulin isotypes IgM and IgG is an independent disease entity. Eur J Haematol. 2005;75:318–327. doi: 10.1111/j.1600-0609.2005.00516.x. [DOI] [PubMed] [Google Scholar]
- 71.Werwitzke S, Trick D, Kamino K, Matthias T, Kniesch K, Schlegelberger B, Schmidt RE, Witte T. Inhibition of lupus disease by anti-double-stranded DNA antibodies of the IgM isotype in the (NZB × NZW)F1 mouse. Arthritis Rheum. 2005;52:3629–3638. doi: 10.1002/art.21379. [DOI] [PubMed] [Google Scholar]
- 72.Mamula MJ. Epitope spreading: the role of self peptides and autoantigen processing by B lymphocytes. Immunol Rev. 1998;164:231–239. doi: 10.1111/j.1600-065x.1998.tb01223.x. [DOI] [PubMed] [Google Scholar]
- 73.Vanderlugt CJ, Miller SD. Epitope spreading. Curr Opin Immunol. 1996;8:831–836. doi: 10.1016/S0952-7915(96)80012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Diaz LA, Arteaga LA, Hilario-Vargas J, Valenzuela JG, Li N, Warren S, Aoki V, Hans-Filho G, Eaton D, dos Santos V, Nutman TB, de Mayolo AA, Qaqish BF, Sampaio SA, Rivitti EA. Anti-desmoglein-1 antibodies in onchocerciasis, leishmaniasis and Chagas disease suggest a possible etiological link to Fogo selvagem. J Invest Dermatol. 2004;123:1045–1051. doi: 10.1111/j.0022-202X.2004.23438.x. [DOI] [PubMed] [Google Scholar]
- 75.Hertl M, Amagai M, Sundaram H, Stanley J, Ishii K, Katz SI. Recognition of desmoglein 3 by autoreactive T cells in pemphigus vulgaris patients and normals. J Invest Dermatol. 1998;110:62–66. doi: 10.1046/j.1523-1747.1998.00086.x. [DOI] [PubMed] [Google Scholar]
- 76.Lin MS, Swartz SJ, Lopez A, Ding X, Fernandez-Vina MA, Stastny P, Fairley JA, Diaz LA. Development and characterization of desmoglein-3 specific T cells from patients with pemphigus vulgaris. J Clin Invest. 1997;99:31–40. doi: 10.1172/JCI119130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wucherpfennig KW, Yu B, Bhol K, Monos DS, Argyris E, Karr RW, Ahmed AR, Strominger JL. Structural basis for major histocompatibility complex (MHC)-linked susceptibility to autoimmunity: charged residues of a single MHC binding pocket confer selective presentation of self-peptides in pemphigus vulgaris. Proc Natl Acad Sci U S A. 1995;92:11935–11939. doi: 10.1073/pnas.92.25.11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin MS, Swartz SJ, Lopez A, Ding X, Fairley JA, Diaz LA. T lymphocytes from a subset of patients with pemphigus vulgaris respond to both desmoglein-3 and desmoglein-1. J Invest Dermatol. 1997;109:734–737. doi: 10.1111/1523-1747.ep12340738. [DOI] [PubMed] [Google Scholar]
- 79.Ahmed AR, Wagner R, Khatri K, Notani G, Awdeh Z, Alper CA, Yunis EJ. Major histocompatibility complex haplotypes and class II genes in non-Jewish patients with pemphigus vulgaris. Proc Natl Acad Sci U S A. 1991;88:5056–5060. doi: 10.1073/pnas.88.11.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ahmed AR, Yunis EJ, Khatri K, Wagner R, Notani G, Awdeh Z, Alper CA. Major histocompatibility complex haplotype studies in Ashkenazi Jewish patients with pemphigus vulgaris. Proc Natl Acad Sci U S A. 1990;87:7658–7662. doi: 10.1073/pnas.87.19.7658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hertl M, Karr RW, Amagai M, Katz SI. Heterogeneous MHC II restriction pattern of autoreactive desmoglein 3 specific T cell responses in pemphigus vulgaris patients and normals. J Invest Dermatol. 1998;110:388–392. doi: 10.1046/j.1523-1747.1998.00156.x. [DOI] [PubMed] [Google Scholar]
- 82.Riechers R, Grotzinger J, Hertl M. HLA class II restriction of autoreactive T cell responses in pemphigus vulgaris: review of the literature and potential applications for the development of a specific immunotherapy. Autoimmunity. 1999;30:183–196. doi: 10.3109/08916939908993852. [DOI] [PubMed] [Google Scholar]
- 83.Sinha AA, Brautbar C, Szafer F, Friedmann A, Tzfoni E, Todd JA, Steinman L, McDevitt HO. A newly characterized HLA DQ beta allele associated with pemphigus vulgaris. Science. 1988;239:1026–1029. doi: 10.1126/science.2894075. [DOI] [PubMed] [Google Scholar]
- 84.Veldman C, Stauber A, Wassmuth R, Uter W, Schuler G, Hertl M. Dichotomy of autoreactive Th1 and Th2 cell responses to desmoglein 3 in patients with pemphigus vulgaris (PV) and healthy carriers of PV-associated HLA class II alleles. J Immunol. 2003;170:635–642. doi: 10.4049/jimmunol.170.1.635. [DOI] [PubMed] [Google Scholar]
- 85.Veldman CM, Gebhard KL, Uter W, Wassmuth R, Grotzinger J, Schultz E, Hertl M. T cell recognition of desmoglein 3 peptides in patients with pemphigus vulgaris and healthy individuals. J Immunol. 2004;172:3883–3892. doi: 10.4049/jimmunol.172.6.3883. [DOI] [PubMed] [Google Scholar]
- 86.Hacker-Foegen MK, Fairley JA, Lin MS. T cell receptor gene usage in desmoglein-3-specific T lymphocytes from patients with pemphigus vulgaris. J Invest Dermatol. 2003;121:1365–1372. doi: 10.1111/j.1523-1747.2003.12601.x. [DOI] [PubMed] [Google Scholar]
- 87.Moesta AK, Lin MS, Diaz LA, Sinha AA. T cell receptor Beta chain gene usage in endemic pemphigus foliaceus (fogo selvagem) J Invest Dermatol. 2002;119:377–383. doi: 10.1046/j.1523-1747.2002.01815.x. [DOI] [PubMed] [Google Scholar]
- 88.Nishifuji K, Amagai M, Kuwana M, Iwasaki T, Nishikawa T. Detection of antigen-specific B cells in patients with pemphigus vulgaris by enzyme-linked immunospot assay: requirement of T cell collaboration for autoantibody production. J Invest Dermatol. 2000;114:88–94. doi: 10.1046/j.1523-1747.2000.00840.x. [DOI] [PubMed] [Google Scholar]
- 89.Amagai M, Tsunoda K, Suzuki H, Nishifuji K, Koyasu S, Nishikawa T. Use of autoantigen-knockout mice in developing an active autoimmune disease model for pemphigus. J Clin Invest. 2000;105:625–631. doi: 10.1172/JCI8748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tsunoda K, Ota T, Suzuki H, Ohyama M, Nagai T, Nishikawa T, Amagai M, Koyasu S. Pathogenic autoantibody production requires loss of tolerance against desmoglein 3 in both T and B cells in experimental pemphigus vulgaris. Eur J Immunol. 2002;32:627–633. doi: 10.1002/1521-4141(200203)32:3<627::AID-IMMU627>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 91.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 92.Sugiyama H, Matsue H, Nagasaka A, Nakamura Y, Tsukamoto K, Shibagaki N, Kawamura T, Kitamura R, Ando N, Shimada S. CD4+CD25high regulatory T cells are markedly decreased in blood of patients with pemphigus vulgaris. Dermatology. 2007;214:210–220. doi: 10.1159/000099585. [DOI] [PubMed] [Google Scholar]
- 93.Veldman C, Hohne A, Dieckmann D, Schuler G, Hertl M. Type I regulatory T cells specific for desmoglein 3 are more frequently detected in healthy individuals than in patients with pemphigus vulgaris. J Immunol. 2004;172:6468–6475. doi: 10.4049/jimmunol.172.10.6468. [DOI] [PubMed] [Google Scholar]
- 94.Veldman C, Pahl A, Beissert S, Hansen W, Buer J, Dieckmann D, Schuler G, Hertl M. Inhibition of the transcription factor Foxp3 converts desmoglein 3-specific type 1 regulatory T cells into Th2-like cells. J Immunol. 2006;176:3215–3222. doi: 10.4049/jimmunol.176.5.3215. [DOI] [PubMed] [Google Scholar]
- 95.Evangelista F, Dasher DA, Diaz LA, Prisayanh PS, Li N. E-cadherin is an additional immunological target for pemphigus autoantibodies. J Invest Dermatol. 2008;128:1710–1718. doi: 10.1038/sj.jid.5701260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ding X, Aoki V, Mascaro JM, Jr, Lopez-Swiderski A, Diaz LA, Fairley JA. Mucosal and mucocutaneous (generalized) pemphigus vulgaris show distinct autoantibody profiles. J Invest Dermatol. 1997;109:592–596. doi: 10.1111/1523-1747.ep12337524. [DOI] [PubMed] [Google Scholar]
- 97.Getsios S, Huen AC, Green KJ. Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol. 2004;5:271–281. doi: 10.1038/nrm1356. [DOI] [PubMed] [Google Scholar]
- 98.Wheelock MJ, Johnson KR. Cadherins as modulators of cellular phenotype. Annu Rev Cell Dev Biol. 2003;19:207–235. doi: 10.1146/annurev.cellbio.19.011102.111135. [DOI] [PubMed] [Google Scholar]
- 99.Mahoney M, Wang Z, Rothenberger K, Koch P, Amagai M, Stanley J. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 1999;103:461–468. doi: 10.1172/JCI5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wu H, Wang ZH, Yan A, Lyle S, Fakharzadeh S, Wahl JK, Wheelock MJ, Ishikawa H, Uitto J, Amagai M, Stanley JR. Protection against pemphigus foliaceus by desmoglein 3 in neonates. N Engl J Med. 2000;343:31–35. doi: 10.1056/NEJM200007063430105. [DOI] [PubMed] [Google Scholar]
- 101.Rocha-Alvarez R, Ortega-Loayza AG, Friedman H, Campbell I, Aoki V, Rivitti EA, Dasher D, Li N, Diaz LA. Endemic pemphigus vulgaris. Arch Dermatol. 2007;143:895–899. doi: 10.1001/archderm.143.7.895. [DOI] [PubMed] [Google Scholar]
- 102.Diaz LA, Marcelo CL. Pemphigoid and pemphigus antigens in cultured epidermal cells. Br J Dermatol. 1978;98:631–637. doi: 10.1111/j.1365-2133.1978.tb03581.x. [DOI] [PubMed] [Google Scholar]
- 103.Stanley JR, Amagai M. Pemphigus, bullous impetigo, and the staphylococcal scalded-skin syndrome. N Engl J Med. 2006;355:1800–1810. doi: 10.1056/NEJMra061111. [DOI] [PubMed] [Google Scholar]
- 104.Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, Murphy GF, Whitaker-Menezes D, Stanley JR. Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol. 1997;137:1091–1102. doi: 10.1083/jcb.137.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Espana A, Diaz LA, Mascaro JM, Jr, Giudice GJ, Fairley JA, Till GO, Liu Z. Mechanisms of acantholysis in pemphigus foliaceus. Clin Immunol Immunopathol. 1997;85:83–89. doi: 10.1006/clin.1997.4407. [DOI] [PubMed] [Google Scholar]
- 106.Mascaro JM, Jr, Espana A, Liu Z, Ding X, Swartz SJ, Fairley JA, Diaz LA. Mechanisms of acantholysis in pemphigus vulgaris: role of IgG valence. Clin Immunol Immunopathol. 1997;85:90–96. doi: 10.1006/clin.1997.4408. [DOI] [PubMed] [Google Scholar]
- 107.Rock B, Labib RS, Diaz LA. Monovalent Fab’ immunoglobulin fragments from endemic pemphigus foliaceus autoantibodies reproduce the human disease in neonatal Balb/c mice. J Clin Invest. 1990;85:296–299. doi: 10.1172/JCI114426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Waschke J, Bruggeman P, Baumgartner W, Zillikens D, Drenckhahn D. Pemphigus foliaceus IgG causes dissociation of desmoglein 1-containing junctions without blocking desmoglein 1 transinteraction. J Clin Invest. 2005;115:3157–3165. doi: 10.1172/JCI23475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Patel HP, Diaz LA, Anhalt GJ, Labib RS, Takahashi Y. Demonstration of pemphigus antibodies on the cell surface of murine epidermal cell monolayers and their internalization. J Invest Dermatol. 1984;83:409–415. doi: 10.1111/1523-1747.ep12273480. [DOI] [PubMed] [Google Scholar]
- 110.Delva E, Jennings JM, Calkins CC, Kottke MD, Faundez V, Kowalczyk AP. Pemphigus Vulgaris IgG-induced Desmoglein-3 Endocytosis and Desmosomal Disassembly Are Mediated by a Clathrin- and Dynamin-independent Mechanism. J Biol Chem. 2008;283:18303–18313. doi: 10.1074/jbc.M710046200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Aoyama Y, Kitajima Y. Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the Triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J Invest Dermatol. 1999;112:67–71. doi: 10.1046/j.1523-1747.1999.00463.x. [DOI] [PubMed] [Google Scholar]
- 112.Cirillo N, Femiano F, Gombos F, Lanza A. Serum from pemphigus vulgaris reduces desmoglein 3 half-life and perturbs its de novo assembly to desmosomal sites in cultured keratinocytes. FEBS Lett. 2006;580:3276–3281. doi: 10.1016/j.febslet.2006.04.089. [DOI] [PubMed] [Google Scholar]
- 113.Seishima M, Esaki C, Osada K, Mori S, Hashimoto T, Kitajima Y. Pemphigus IgG, but not bullous pemphigoid IgG, causes a transient increase in intracellular calcium and inositol 1,4,5-triphosphate in DJM-1 cells, a squamous cell carcinoma line. J Invest Dermatol. 1995;104:33–37. doi: 10.1111/1523-1747.ep12613469. [DOI] [PubMed] [Google Scholar]
- 114.Aoyama Y, Owada MK, Kitajima Y. A pathogenic autoantibody, pemphigus vulgaris-IgG, induces phosphorylation of desmoglein 3, and its dissociation from plakoglobin in cultured keratinocytes. Eur J Immunol. 1999;29:2233–2240. doi: 10.1002/(SICI)1521-4141(199907)29:07<2233::AID-IMMU2233>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 115.Christofori G. Changing neighbours, changing behaviour: cell adhesion molecule-mediated signalling during tumour progression. EMBO J. 2003;22:2318–2323. doi: 10.1093/emboj/cdg228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Stappert J, Kemler R. A short core region of E-cadherin is essential for catenin binding and is highly phosphorylated. Cell Adhes Commun. 1994;2:319–327. doi: 10.3109/15419069409014207. [DOI] [PubMed] [Google Scholar]
- 117.Caldelari R, de Bruin A, Baumann D, Suter MM, Bierkamp C, Balmer V, Muller E. A central role for the armadillo protein plakoglobin in the autoimmune disease pemphigus vulgaris. J Cell Biol. 2001;153:823–834. doi: 10.1083/jcb.153.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Williamson L, Raess N, Caldelari R, Zakher A, de Bruin A, Posthaus H, Bolli R, Hunziker T, Suter M, Müller E. Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. EMBO J. 2006;25:3298–3309. doi: 10.1038/sj.emboj.7601224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, Chua MP, Rubenstein DS. Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem. 2005;280:23778–23784. doi: 10.1074/jbc.M501365200. [DOI] [PubMed] [Google Scholar]
- 120.Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci U S A. 2006;103:12855–12860. doi: 10.1073/pnas.0602973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Berkowitz P, Diaz LA, Hall RP, Rubenstein DS. Induction of p38MAPK and HSP27 phosphorylation in pemphigus patient skin. J Invest Dermatol. 2008;128:738–740. doi: 10.1038/sj.jid.5701080. [DOI] [PubMed] [Google Scholar]
- 122.Arredondo J, Chernyavsky AI, Karaouni A, Grando SA. Novel mechanisms of target cell death and survival and of therapeutic action of IVIg in Pemphigus. Am J Pathol. 2005;167:1531–1544. doi: 10.1016/S0002-9440(10)61239-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Baroni A, Buommino E, Paoletti I, Orlando M, Ruocco E, Ruocco V. Pemphigus serum and captopril induce heat shock protein 70 and inducible nitric oxide synthase overexpression, triggering apoptosis in human keratinocytes. Br J Dermatol. 2004;150:1070–1080. doi: 10.1111/j.1365-2133.2004.05919.x. [DOI] [PubMed] [Google Scholar]
- 124.Pelacho B, Natal C, Espana A, Sanchez-Carpintero I, Iraburu MJ, Lopez-Zabalza MJ. Pemphigus vulgaris autoantibodies induce apoptosis in HaCaT keratinocytes. FEBS Lett. 2004;566:6–10. doi: 10.1016/j.febslet.2004.03.107. [DOI] [PubMed] [Google Scholar]
- 125.Puviani M, Marconi A, Cozzani E, Pincelli C. Fas ligand in pemphigus sera induces keratinocyte apoptosis through the activation of caspase-8. J Invest Dermatol. 2003;120:164–167. doi: 10.1046/j.1523-1747.2003.12014.x. [DOI] [PubMed] [Google Scholar]
- 126.Wang X, Bregegere F, Frusic-Zlotkin M, Feinmesser M, Michel B, Milner Y. Possible apoptotic mechanism in epidermal cell acantholysis induced by pemphigus vulgaris autoimmunoglobulins. Apoptosis. 2004;9:131–143. doi: 10.1023/B:APPT.0000018795.05766.1f. [DOI] [PubMed] [Google Scholar]
- 127.Gniadecki R, Jemec GB, Thomsen BM, Hansen M. Relationship between keratinocyte adhesion and death: anoikis in acantholytic diseases. Arch Dermatol Res. 1998;290:528–532. doi: 10.1007/s004030050347. [DOI] [PubMed] [Google Scholar]
- 128.Li N, Zhao ML, Wang J, Liu Z, Diaz LA. Pathogenic Role of the Apoptotic Mechanism in Pemphigus Foliaceus Autoimmune Skin Blistering. 2008 doi: 10.4049/jimmunol.182.1.711. Submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fischer U, Janicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 2003;10:76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dusek RL, Getsios S, Chen F, Park JK, Amargo EV, Cryns VL, Green KJ. The differentiation-dependent desmosomal cadherin desmoglein 1 is a novel caspase-3 target that regulates apoptosis in keratinocytes. J Biol Chem. 2006;281:3614–3624. doi: 10.1074/jbc.M508258200. [DOI] [PubMed] [Google Scholar]
- 131.Lanza A, Cirillo N, Rossiello R, Rienzo M, Cutillo L, Casamassimi A, de Nigris F, Schiano C, Rossiello L, Femiano F, Gombos F, Napoli C. Evidence of key role of Cdk2 overexpression in pemphigus vulgaris. J Biol Chem. 2008;283:8736–8745. doi: 10.1074/jbc.M702186200. [DOI] [PubMed] [Google Scholar]
- 132.Aho S. Plakin proteins are coordinately cleaved during apoptosis but preferentially through the action of different caspases. Exp Dermatol. 2004;13:700–707. doi: 10.1111/j.0906-6705.2004.00217.x. [DOI] [PubMed] [Google Scholar]
- 133.Kalinin AE, Aho M, Uitto J, Aho S. Breaking the connection: caspase 6 disconnects intermediate filament-binding domain of periplakin from its actin-binding N-terminal region. J Invest Dermatol. 2005;124:46–55. doi: 10.1111/j.0022-202X.2004.23507.x. [DOI] [PubMed] [Google Scholar]
- 134.Weiske J, Schoneberg T, Schroder W, Hatzfeld M, Tauber R, Huber O. The fate of desmosomal proteins in apoptotic cells. J Biol Chem. 2001;276:41175–41181. doi: 10.1074/jbc.M105769200. [DOI] [PubMed] [Google Scholar]
- 135.Schmeiser K, Grand RJ. The fate of E- and P-cadherin during the early stages of apoptosis. Cell Death Differ. 1999;6:377–386. doi: 10.1038/sj.cdd.4400504. [DOI] [PubMed] [Google Scholar]
- 136.Steinhusen U, Weiske J, Badock V, Tauber R, Bommert K, Huber O. Cleavage and shedding of E-cadherin after induction of apoptosis. J Biol Chem. 2001;276:4972–4980. doi: 10.1074/jbc.M006102200. [DOI] [PubMed] [Google Scholar]
- 137.Steinhusen U, Badock V, Bauer A, Behrens J, Wittman-Liebold B, Dorken B, Bommert K. Apoptosis-induced cleavage of beta-catenin by caspase-3 results in proteolytic fragments with reduced transactivation potential. J Biol Chem. 2000;275:16345–16353. doi: 10.1074/jbc.M001458200. [DOI] [PubMed] [Google Scholar]
- 138.Lever W. Pemphigus. Medicine (Baltimore) 1953;32:1–123. doi: 10.1097/00005792-195302000-00001. [DOI] [PubMed] [Google Scholar]
- 139.Herbst A, Bystryn JC. Patterns of remission in pemphigus vulgaris. J Am Acad Dermatol. 2000;42:422–427. doi: 10.1016/s0190-9622(00)90213-5. [DOI] [PubMed] [Google Scholar]
- 140.Nagao K, Tanikawa A, Yamamoto N, Amagai M. Decline of anti-desmoglein 1 IgG ELISA scores by withdrawal of D-penicillamine in drug-induced pemphigus foliaceus. Clin Exp Dermatol. 2005;30:43–45. doi: 10.1111/j.1365-2230.2004.01655.x. [DOI] [PubMed] [Google Scholar]
- 141.Bialy-Golan A, Brenner S. Penicillamine-induced bullous dermatoses. J Am Acad Dermatol. 1996;35:732–742. doi: 10.1016/s0190-9622(96)90729-x. [DOI] [PubMed] [Google Scholar]
- 142.Kaplan RP, Potter TS, Fox JN. Drug-induced pemphigus related to angiotensin-converting enzyme inhibitors. J Am Acad Dermatol. 1992;26:364–366. doi: 10.1016/0190-9622(92)70057-m. [DOI] [PubMed] [Google Scholar]
- 143.Rizzo C, Fotino M, Zhang Y, Chow S, Spizuoco A, Sinha AA. Direct characterization of human T cells in pemphigus vulgaris reveals elevated autoantigen-specific Th2 activity in association with active disease. Clin Exp Dermatol. 2005;30:535–540. doi: 10.1111/j.1365-2230.2005.01836.x. [DOI] [PubMed] [Google Scholar]
- 144.Takahashi H, Amagai M, Tanikawa A, Suzuki S, Ikeda Y, Nishikawa T, Kawakami Y, Kuwana M. T helper type 2-biased natural killer cell phenotype in patients with pemphigus vulgaris. J Invest Dermatol. 2007;127:324–330. doi: 10.1038/sj.jid.5700527. [DOI] [PubMed] [Google Scholar]
- 145.Oshima M, Deitiker P, Atassi MZ. Targeting the antigen-binding site of HLA-restricting alleles in treatment of autoimmune disease. Crit Rev Immunol. 2007;27:271–288. doi: 10.1615/critrevimmunol.v27.i3.60. [DOI] [PubMed] [Google Scholar]
- 146.Anhalt GJ, Werth VP, Strober B. An open-label phase I clinical study to assess the safety of PI-0824 in patients with pemphigus vulgaris. J Invest Dermatol. 2005;125:1088. [Google Scholar]
- 147.Werth VP, Strober B, Connolly MK, et al. An open-label phase I clinical study to assess the safety of PI-0824 in patients with pemphigus vulgaris. J Invest Dermatol. 2005;125:A5 #930. [Google Scholar]
- 148.Peptimmune.com. 2008.
- 149.Hertl M, Eming R, Veldman C. T cell control in autoimmune bullous skin disorders. J Clin Invest. 2006;116:1159–1166. doi: 10.1172/JCI28547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Eming R, Nagel A, Wolff-Franke S, Podstawa E, Debus D, Hertl M. Rituximab Exerts a Dual Effect in Pemphigus Vulgaris. J Invest Dermatol. 2008 doi: 10.1038/jid.2008.172. [DOI] [PubMed] [Google Scholar]
- 151.Ahmed AR, Spigelman Z, Cavacini LA, Posner MR. Treatment of pemphigus vulgaris with rituximab and intravenous immune globulin. N Engl J Med. 2006;355:1772–1779. doi: 10.1056/NEJMoa062930. [DOI] [PubMed] [Google Scholar]
- 152.Joly P, Mouquet H, Roujeau J-C, D/Incan M, Gilbert D, Jacquot S, et al. The effect of the anti-CD20 monoclonal antibody rituximab on severe pemphigus. N Engl J Med. 2007;357:545–552. doi: 10.1056/NEJMoa067752. [DOI] [PubMed] [Google Scholar]
- 153.Cianchini G, Corona R, Frezzolini A, Ruffeli M, Didona B, Puddu P. Treatment of severe pemphigus with rituximab: Report of 12 cases and a review of the literature. Arch Dermatol. 2007;143:1033–1038. doi: 10.1001/archderm.143.8.1033. [DOI] [PubMed] [Google Scholar]
- 154.Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dorner T, Hiepe F. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. 2006;6:741–750. doi: 10.1038/nri1886. [DOI] [PubMed] [Google Scholar]
- 155.Caproni M, Antiga E, Torchia D, Volpi W, del Bianco E, Cappetti A, Feliciani C, Fabbri P. The CD40/CD40 ligand system is involved in the pathogenesis of pemphigus. Clin Immunol. 2007;124:22–25. doi: 10.1016/j.clim.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 156.Ahmed AR. Intravenous immunoglobulin therapy in the treatment of patients with pemphigus vulgaris unresponsive to conventional immunosuppressive treatment. J Am Acad Dermatol. 2001;45:679–690. doi: 10.1067/mjd.2001.116339. [DOI] [PubMed] [Google Scholar]
- 157.Bystryn JC, Jiao D, Natow S. Treatment of pemphigus with intravenous immunoglobulin. J Am Acad Dermatol. 2002;47:358–363. doi: 10.1067/mjd.2002.122735. [DOI] [PubMed] [Google Scholar]
- 158.Bystryn JC, Jiao D. IVIg selectively and rapidly decreases circulating pathogenic autoantibodies in pemphigus vulgaris. Autoimmunity. 2006;39:601–607. doi: 10.1080/08916930600972016. [DOI] [PubMed] [Google Scholar]
- 159.Czernik A, Beutner EH, Bystryn JC. Intravenous immunoglobulin selectively decreases circulating autoantibodies in pemphigus. J Am Acad Dermatol. 2008;58:796–801. doi: 10.1016/j.jaad.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 160.Eming R, Hertl M. Immunoadsorption in pemphigus. Autoimmunity. 2006;39:609–616. doi: 10.1080/08916930600972040. [DOI] [PubMed] [Google Scholar]
- 161.Alvarado-Flores E, Avalos-Diaz E, Diaz LA, Herrera-Esparza R. Anti-idiotype antibodies neutralize in vivo the blistering effect of Pemphigus foliaceus IgG. Scand J Immunol. 2001;53:254–258. doi: 10.1046/j.1365-3083.2001.00863.x. [DOI] [PubMed] [Google Scholar]
- 162.Payne AS, Siegel DL, Stanley JR. Targeting pemphigus autoantibodies through their heavy-chain variable region genes. J Invest Dermatol. 2007;127:1681–1691. doi: 10.1038/sj.jid.5700790. [DOI] [PubMed] [Google Scholar]
- 163.Li N, Liu Z, Wang J, Warren SJP, Diaz LA. Inhibition of experimental pemphigus by the p53 inhibitor pifithrin-alpha. J Invest Dermatol. 2007;127 Abstract 64. [Google Scholar]
- 164.Li N, Liu Z, Park M, Diaz LA. p53-knockout mice are resistant to the induction of skin blisters and acantholysis by pemphigus autoantibodies. Annual Meeting SID; Japan. 2008. [Google Scholar]