

Abstract
Aims
Long QT syndrome (LQTS) is an inherited arrhythmia disorder with an estimated prevalence of 0.01%–0.05%. In Finland, four founder mutations constitute up to 70% of the known genetic spectrum of LQTS. In the present survey, we sought to estimate the actual prevalence of the founder mutations and to determine their effect sizes in the general Finnish population.
Methods and results
We genotyped 6334 subjects aged≥30 years from a population cohort (Health 2000 study) for the four Finnish founder mutations using Sequenom MALDI-TOF mass spectrometry. The electrocardiogram (ECG) parameters were measured from digital 12-lead ECGs, and QT intervals were adjusted for age, sex, and heart rate using linear regression. A total of 27 individuals carried one of the founder mutations resulting in their collective prevalence estimate of 0.4% (95% CI 0.3%–0.6%). The KCNQ1 G589D mutation (n=8) was associated with a 50 ms (SE 7.0) prolongation of the adjusted QT interval (P=9.0×10-13). The KCNH2 R176W variant (n=16) resulted in a 22 ms (SE 4.7) longer adjusted QT interval (P=2.1×10-6).
Conclusion
In Finland 1 individual out of 250 carries a LQTS founder mutation, which is the highest documented prevalence of LQTS mutations that lead to a marked QT prolongation.
Keywords: Epidemiology, ion channels, KCNH2, KCNQ1, long QT syndrome, QT interval
Introduction
Long QT syndrome (LQTS) is an inherited arrhythmic disorder characterized by prolonged QT interval on the electrocardiogram (ECG) and a risk for ventricular tachyarrhythmias and sudden death (1-3). A total of ten genes and several hundreds of mutations have been identified to underlie the disorder (2), most of them affecting the function of cardiac ion channels. In addition to the strong effects of disease-causing mutations, several common variants in the LQTS genes have been shown to prolong QT interval duration and predispose to the clinical manifestations of the disorder (3).
Key messages.
This is the first attempt to estimate the prevalence of LQTS at the population level using molecular methods.
The reported prevalence estimate of 0.4% is considerably more than the generally estimated prevalence of LQTS (0.01%–0.05%) in other populations.
The four Finnish LQTS founder mutations lead to a substantial 22–50 ms QT prolongation also at the population level and could thus mark individuals at risk for arrhythmias.
LQTS is assumed to be a rare disorder with a recent prevalence estimate of 1/2000 (4). Approximately 75% of genotyped LQTS patients carry a mutation in either KCNQ1, KCNH2 (HERG), or SCN5A gene that are responsible for LQTS subtypes 1–3 (5). In Finland, four previously described mutations, including G589D and IVS7-2A>G in the KCNQ1 gene and KCNH2 L552S and KCNH2 R176W, account for up to 70% of the known genetic spectrum of long QT syndrome (6). The aim of this survey was to estimate the prevalence of the LQTS founder mutations in Finland using an unselected population-based Health 2000 sample and to determine whether these mutations are associated with QT interval prolongation at the population level.
Methods
Study population
The study population consisted of a cross-sectional cohort of 8028 individuals drawn from the Finnish Population Information System (http://www.vaestorekisterikeskus.fi/vrk/home.nsf/www/populationinformationsystem) for the Health 2000 survey. The two-stage stratified cluster sample was collected between September 2000 and June 2001 and is representative of the entire Finnish population of 30 years and more (www.ktl.fi/health2000) (7). DNA samples were collected from 6334 individuals, and digital standard 12-lead ECGs were available from 6295 study participants (78% of the original sample). The prevalence estimates with 95% confidence intervals were calculated from the weighted genotyped study population (n=6334) as described earlier (7).
When the QT-prolonging effect of each mutant allele was specifically investigated, subjects with complete left or right bundle branch block (n=143), QRS≥120 ms (n=205), atrial fibrillation or flutter (n=94), pacemaker (n=12), or using a potentially QT-altering medication (n=1064), including digoxin, were excluded from the calculations. A drug was considered to potentially prolong the QT interval if it was listed in any of the four categories at www.qtdrugs.org (accessed November 2006). In addition, we excluded the following agents that might also affect ventricular repolarization: carbamazepine, flupentixol, levomepromazine, me-floquine, olanzapine, oxcarbazepine, periciazine, sertindole, and trazodone. This study population used for QT interval analyses included 5068 individuals free of exclusions. Clinical characteristics, including medications, prior heart failure, history of myocardial infarction, prevalent diabetes, and smoking status, were determined as described previously (7). The occurrence of juvenile (<40 years) deaths in the first-degree relatives of each study participant was assessed using an investigator-administered questionnaire (7). The study was performed according to the declaration of Helsinki and was approved by the Ethical Committees of the Hospital District of Helsinki and Uusimaa and the National Public Health Institute. Written informed consent was obtained from the participants.
QT interval measurements
Digital standard 12-lead ECGs were recorded using Marquette MAC 5000 (GE Marquette Medical Systems, Milwaukee, WI, USA), and a digital median QRS-T complex was used for analyses. We used QT Guard software (GE Marquette Medical Systems, Milwaukee, WI, USA) to measure heart rate, and custom-made software for QT interval measurements. These measurements were based on a previously described and validated algorithm (8). The software calculates QT interval from QRS onset to T wave offset in each lead. A single observer (KP) reviewed all measurements in a blinded fashion, and the mean QT interval of all 12 leads was used for final analyses. The intraobserver coefficient of variation was 0.7% for the mean QT interval in repeated ECG measurements in our recent study (9), and the same measurement methods were used also in the present survey. In addition, the QT intervals of the founder mutation carriers were measured manually by a single cardiologist (HS) in order to allow comparison with the same measurement technique used by HS among clinical LQTS samples. The measured QT intervals were corrected for heart rate using Bazett’s formula (10). Left ventricular hypertrophy was assessed as a dichotomous variable based on the Sokolow-Lyon voltage >35 mm (11) and/or gender-adjusted Cornell voltage duration product >2440 mmms (12).
Genetic analyses
Genotyping for the four Finnish founder mutations was performed using Sequenom MALDI-TOF mass spectrometry (MassArray Compact Analyzer, Sequenom Inc., San Diego, USA). Sequences were evaluated in the ProxSNP and PreXTEND software (www.realsnp.com), and the reactions were designed utilizing Sequenom Assay Designer 3.1 software. Genotyping was performed with the iPLEX method for KCNQ1 (NM_000218) G589D G/A and IVS7-2A>G and KCNH2 (NM_000238) L552S T/C, and with homogeneous MassEXTEND method for KCNH2 R176W C/T according to standard protocols. The polymerase chain reaction (PCR) primer and extension primer sequences are available by contacting the authors. Reactions were tested with 32 DNAs prior to genotyping. Previously identified heterozygous DNA samples from an earlier study (6) served as positive controls in the genotyping process. The genotyping calls were made using Spectro-Analyzer 3.4 software. The heterozygous samples were confirmed by direct sequencing in forward and reverse directions using an ABI 3730 Automatic DNA sequencer (Applied Biosystems, Foster City, MA, USA) and the previously described primer sequences (6,13-15).
Statistical analyses
Histograms of all the variables were inspected for normality. We assessed correlations of crude values of sex-pooled and sex-specific age, heart rate (RR interval), and QT intervals. Using stepwise linear regression model, we adjusted the QT intervals for age, sex, and RR-interval (16,17) and determined the beta coefficient, two-tailed P-value, and partial R2 of the covariates in the model. In addition, we tested additional possible covariates, i.e. left ventricular hypertrophy, history of myocardial infarction, heart failure, prevalent diabetes, smoking status, diuretic usage, and geographical area, for a possible additional explanatory role in the model. As these covariates accounted for less than 1% of the variation of QT interval in the study population, did not reach strong statistical level of significance (P>0.001), and in order to use a measure comparable to other studies, we derived the QT residuals from the age-, sex-, and heart rate-adjusted model. The histograms of output residuals were reviewed for normality (skewness 0.37, SE 0.03, kurtosis 1.21, SE 0.07). Statistical analyses were performed using SPSS 13.0/15.0 (SPSS Inc., Chicago, Illinois, USA). The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
The characteristics of the initial study population have been described elsewhere (7). The mean age was 51±14 years. Women (53%) featured longer mean QTc (402±20 ms, 95% CI 401.2–402.8 ms) than men (389±22 ms, 95% CI 387.8–389.6 ms), P<0.001. A total of 120 (2.4%) subjects had a QTc interval greater than 440 ms (18), and 9 of them (0.2%) featured a QTc longer than 470 ms (18). The subjects on QT-prolonging drugs had a 4.0 ms longer QT interval compared to those study participants without such medication.
Successful genotyping calls were obtained for 97.6%–99.9% of the DNA samples, and 6263 study participants were successfully genotyped for all four founder mutations. No homozygous nor compound heterozygous carriers were identified in the study population. A total of 27 individuals were heterozygous carriers of one of the four Finnish founder mutations, KCNQ1 G589D (n=8), KCNQ1 IVS7-2A>G (n=1), KCNH2 L552S (n=2), or KCNH2 R176W (n=16). The sex distribution was similar in the four groups of founder mutation carriers. Collectively, these four founder mutations have a prevalence estimate of 0.4% (95% CI 0.3%–0.6%) in the Finnish population.
Table I summarizes the clinical characteristics of the founder mutation carriers and non-carriers in the Health 2000 survey, after exclusion of one carrier of KCNQ1 G589D (QTc 438 ms) and one carrier of KCNQ1 IVS7-2A>G (QTc 409 ms) who were treated with a potentially QT-prolonging drug (salbutamol and terbutaline, respectively). None of the 27 individuals possessed other exclusion criteria. The differences in the QTc intervals between founder mutation carriers and non-carriers are illustrated in Figure 1.
Table I.
The clinical characteristics of the founder mutation carriers (n=25) and non-carriers (n=4758) after exclusion of cases with potentially QTc-altering traits (bundle branch block, QRS≥120 ms, atrial fibrillation or flutter, pacemaker) or medications.
|
KCNQ1 G589D n=7 |
KCNH2 L552S n=2 |
KCNH2 R176W n=16 |
Non-carriers n=4758 |
|
|---|---|---|---|---|
| Females/males | 4/3 | 1/1 | 8/8 | 2524/2234 |
| Age, years | 43±7 (34–57) | 38±2 (36–39) | 53±11 (30–70) | 51±14 (30-97) |
| Heart rate, bpm | 55±5 (48–63) | 60±16 (49–71) | 61±7 (50–72) | 63±11 (34–120) |
| QT interval, ms | 451±33 (406–507) | 442±32 (420–465) | 414±33 (357–480) | 388±30 (279–536) a |
| QTc interval, ms | 435±40 (403–520) | 438±26 (420–457) | 430±20 (396–459) | 396±22 (323–514) b |
Data are presented as a mean±SD (range) unless otherwise indicated.
P 2.17 × 10-10
P 1.26 × 10-9 in comparison of QT parameters among founder mutation carriers and non-carriers. Because of failures in genotyping for the individual mutations, the genotype numbers do not add up to the total of 5068 individuals.
bpm=beats per minute; QTc=QT interval corrected for heart rate using Bazett’s formula.
Figure 1.
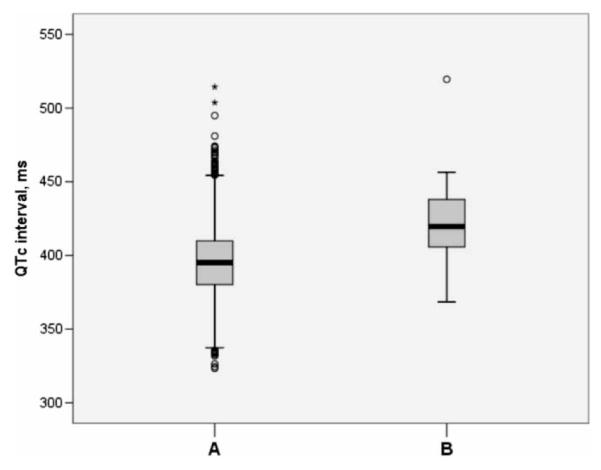
The QTc intervals in founder mutation non-carriers (A) (n=4758) and carriers (B) (n=25) (P=1.26×10-9), after exclusion of cases with potentially QTc-altering traits or medications. The boxes represent the median values (bolded horizontal lines) and the 25th to 75th percentiles of the mean QTc intervals. The fine lines refer to the highest and lowest values that are not outliers. Circles represent values >1.5-fold the interquartile range, and asterisks refer to values >3-fold the range indicated by the box.
The mutant allele A of KCNQ1 G589D was identifiable in eight individuals in heterozygous form and was found to be associated with a 50.5 ms (SE 7.1, 2.7 SD, P=8.99×10-13) increase of the QT interval when compared to mutation non-carriers. A total of 16 study participants carried the minor KCNH2 R176W allele and showed a 22.2 ms (SE 4.7, 1.2 SD, P=2.15×10-6) longer QT interval compared to mutation non-carriers. No systematic ascertainment of personal or family history of arrhythmia symptoms was undertaken as part of the Health 2000 population-based survey. One mutation carrier, a person with KCNQ1 G589D and a QTc of 520 ms, had previously been symptomatic. None of the parents of the 27 mutation carriers had died before 40 years of age. Ten (37%) founder mutation carriers had siblings who had suffered from juvenile (<40 years) death, but the difference to the corresponding frequency in mutation non-carriers (n=1541, 25%) was not statistically significant (P=0.14).
It was pertinent to exclude the possibility that the high prevalence estimate would have resulted because of enrolment of a given geographic area showing an exceptional clustering of a specific founder mutation. Therefore, we compared the distribution of founder mutation carriers, first, in the Health 2000 material, and second, in the updated material of clinical LQTS indexes (described in reference (7), and supplemented by additional cases by H. Swan). The geographical distribution of the four founder mutations in these two groups are shown in Supplementary Figure 1 (http://www.informaworld.com/annmed). The grey areas represent municipalities enrolled for the Health 2000 study. These maps indicate that the four founder mutations are widely distributed in Finland, and no preferential area enrichment affects the Health 2000 study in this respect. The only exception is the KCNQ1 IVS7-2A>G mutation that appears to be clustered to the southern Finland, as based on the clinical index material. As only one Health 2000 study participant carried the KCNQ1 IVS7-2A>G, its contribution to the prevalence calculations is negligible.
Discussion
Using a large, epidemiological study cohort of more than 6000 individuals, we report a 0.4% prevalence of disease-causing LQTS founder mutations in Finland. This is considerably more than the generally estimated prevalence of 0.01% (19) to 0.05% (20) in other populations and may reflect the special genetic composition of the Finnish population. Increasing evidence suggests that these founder mutations may be enriched in the isolated Finnish population as they were found to be relatively frequent in a subset of acquired long QT syndrome (21). Because genotyping was targeted only to these four founder mutations and the Health 2000 Study did not include subjects younger than 30 years old, the actual prevalence of disease-causing LQTS mutations in Finland is likely to be even higher.
Previously, we have reported that the KCNQ1 G589D is associated with a mean QTc of 462±38 ms (n=508), KCNQ1 IVS7-2A>G with a QTc of 470±31 ms (n=55), KCNH2 L552S with a QTc of 463±45 ms (n=90), and KCNH2 R176W with a mean QTc of 459±40 ms (n=30) in clinical LQTS families (6). In addition to the prolongation of the QTc interval, these four mutations have been associated with symptoms, i.e. syncopal spells or sudden death, in 23%–38% of the mutation carriers among previous clinical samples (6) thus being sufficient to cause a life-threatening form of LQTS. In the present epidemiological data set, the mean QTc intervals are slightly shorter than in our previous report on the clinical sample and may reflect an unavoidable bias in clinical settings. For comparison, we also made manual QTc measurements in the founder mutation carriers of the present study; the QTc intervals in the founder mutation carriers were 444±10 ms for KCNQ1 G589D, 456±12 ms for KCNH2 L552S, and 433±27 ms for KCNH2 R176W. The patient population ascertained on the presence of overt syncope or arrhythmia may possess other uncharacterized genetic modifier factors that contribute to the QT prolongation and thus to the clinical manifestations of the disorder.
The Finnish type of KCNQ1 founder mutations has not to our knowledge been described in other patient populations. KCNH2 L552S has been identified in a single Caucasian family (22) as a disease-causing LQTS mutation. In contrast, the KCNH2 R176W has been reported elsewhere as an innocent polymorphism with a minor allele frequency of 0.3%–0.5% in the healthy white population (19,23). In Finland, the variant was previously detectable in 0.9% (3/317) of randomly collected blood donors, but it was significantly enriched in LQTS patients with an 8% genotype frequency among LQTS probands (6). At present, we have identified the KCNH2 R176W mutation in 112 LQTS patients, of which 18 (16%) are symptomatic (Swan et al., unpublished observations). Up to 40% of male mutation carriers (16 of the 40 men free of QT-affecting medication) have QTc over 440 ms. Similarly, 22% (12/55) of female R176W carriers show a QT prolongation over 460 ms (Swan et al., unpublished observations). Furthermore, the KCNH2 R176W was identified in six KCNQ1 founder mutation carriers from four LQTS1 families (24). Five patients with compound KCNQ1 G589D and KCNH2 R176W mutations had a mean QTc of 473±30 ms as opposed to the QTc of 457±33 ms in KCNQ1 G589D carriers (n=371) alone (24). In addition, four (67%) of these compound heterozygous patients were symptomatic (24). However, until now the actual effect size of this variant in the general population has remained uncertain.
Recently, it was shown that up to one-third of the variation in the QT interval is heritable (25,26). The growing interest in understanding genetic modulation of cardiac repolarization has led to the identification of gene variants that affect QT interval duration in unselected population-based samples (27-31). However, the effects of common gene variants on QT interval duration have remained relatively modest in these epidemiological surveys (28-30). In the present study, the KCNH2 R176W is shown to be associated with a 22 ms prolongation of the age-, sex-, and heart rate-adjusted QT interval. This is considerably stronger than the previously presented effect sizes among common gene variants (28-30) and, along with the data on clinical KCNH2 R176W samples, strongly supports the role of the KCNH2 R176W in predisposing to repolarization-related arrhythmogenesis, at least in the Finnish population.
Previous in vitro experiments of mutant channels provide further evidence of the pathogenicity of the Finnish LQTS founder mutations. KCNQ1 G589D channels, once coexpressed with minK in vitro, feature smaller currents and result in a pronounced rightward shift in the voltage threshold of activation (13). In addition, the intronic KCNQ1 IVS7-2A>G mutation that alters the splicing acceptor site and results in insertion of several amino acids to the C-terminal region of the protein, has been shown to lead to a complete loss-of-function defect (6). We have also demonstrated that the KCNH2 R176W results in a reduced current density and slight acceleration in deactivation kinetics in vitro (24).
Even after excluding the 16 carriers of the KCNH2 R176W mutation with moderate QT-prolonging effect, the prevalence estimate is 0.2% (95% CI 0.1%–0.3%). The prevalence estimate of 1/250 to 1/500 is striking and suggests that a considerable proportion of the Finnish population may be genetically predisposed to severe ventricular arrhythmias due to the four LQTS founder mutations. Despite the potency of these mutations to cause a disease phenotype, their penetrance is variable ranging from 21% to 34% (6,14) among clinical LQTS families. Moreover, the information on risk stratification is derived from LQTS patients and their relatives only, which precludes any definitive statements regarding the risk of life-threatening ventricular arrhythmias among the mutation carriers in the general population. Nevertheless, as the founder mutation carriers in the present survey have substantially longer QT intervals than non-carriers, it is evident that these individuals are at increased risk for arrhythmias if the repolarization reserve (32) is further challenged due to exposure to a QT-prolonging medication, hypokalaemia, or ischaemic or structural heart disease (33).
The findings and implications of the present study are not directly applicable to other populations where LQTS founder mutations do not exist or have not been detected until now. However, we feel that the present study stands as a forerunner to demonstrate that relatively prevalent genetic causes of markedly prolonged ventricular repolarization may indeed exist in the population. With the advent of constantly improving DNA sequencing techniques, this may also soon be explored in other populations. Early, presymptomatic identification of risk for ventricular arrhythmias, particularly upon exposure to certain medications, is of high priority since preventive measures can effectively avert sudden death (34). Even though in 75% of the cases the DNA analysis of the five most common LQTS genes confirms the LQTS diagnosis (35,36), prophylactic screening at the population level has not been practical due to the costs and rarity of the disorder. In Finland, targeted pre-prescription screening could become cost-effective in the future along with the rapidly evolving genetic screening measures. Especially in population isolates like Finland showing enrichment of a limited number of variants, such screening could be exceptionally beneficial for the health care system.
We conclude that four LQTS mutations, KCNQ1 G589D, KCNQ1 IVS7-2A>G, KCNH2 L552S, and KCNH2 R176W, are surprisingly common in the Finnish population and seemingly represent founder mutations which have become enriched in this isolated population. These mutations lead to a considerable QT prolongation at the population level and could mark individuals at risk for arrhythmias even if clinical LQTS is not apparent.
Supplementary Material
Acknowledgements
Susanna Tverin, Pirkko Alha, Harri Rissanen, Anu Yliperttula, and Sirkka Ekström are acknowledged for skilful technical assistance. The study was financially supported by grants from the Sigrid Juselius Foundation and the Finnish Academy to Dr Kontula and by the Center of Excellence of Common Disease Genetics of the Academy of Finland. Dr Marjamaa has received funding from the Finska Läkaresällskapet Foundation and the Biomedicum Helsinki Foundation. Dr Newton-Cheh is supported by the National Institutes of Health (HL080025), a Doris Duke Charitable Foundation Clinical Scientist Development Award, and a Burroughs Wellcome Fund Career Award for Medical Scientists. Dr Porthan has received funding from the Aarne Koskelo Foundation, the Finnish Foundation for Cardiovascular Research, the Ida Montin Foundation, the Paavo and Eila Salonen Foundation. Dr Salomaa was also supported by the Sigrid Juselius Foundation.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Viskin S. Long QT syndromes and torsade de pointes. Lancet. 1999;354:1625–33. doi: 10.1016/S0140-6736(99)02107-8. [DOI] [PubMed] [Google Scholar]
- 2.Priori SG, Napolitano C, Schwartz PJ. Genetics of cardiac arrhythmias. In: Libby P, Bonow RO, Mann DL, Zipes DP, editors. Braunwald’s Heart Disease. 8th ed Saunders Elsevier; Philadelphia, PA: 2008. [Google Scholar]
- 3.Schwartz PJ. The congenital long QT syndromes from genotype to phenotype: clinical implications. J Intern Med. 2006;259:39–47. doi: 10.1111/j.1365-2796.2005.01583.x. [DOI] [PubMed] [Google Scholar]
- 4.Stramba-Badiale M, Crotti L, Goulene K, Pedrazzini M, Mannarino S, Salice P, et al. Electrocardiographic and genetic screening for long QT syndrome: results from a prospective study on 44,596 neonates. Circulation. 2007;116 Supplement:II_377. [Google Scholar]
- 5.Wilde AA, Bezzina CR. Genetics of cardiac arrhythmias. Heart. 2005;91:1352–8. doi: 10.1136/hrt.2004.046334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fodstad H, Swan H, Laitinen P, Piippo K, Paavonen K, Viitasalo M, et al. Four potassium channel mutations account for 73% of the genetic spectrum underlying long-QT syndrome (LQTS) and provide evidence for a strong founder effect in Finland. Ann Med. 2004;36:53–63. doi: 10.1080/17431380410032689. [DOI] [PubMed] [Google Scholar]
- 7.Aromaa A, Koskinen S. Health and functional capacity in Finland. Baseline Results of the Health 2000 Health Examination Survey. Publications of the National Public Health Institute B12/2004. Helsinki 2004. [accessed 1 August, 2007]. Available at www.terveys2000.fi/julkaisut/baseline.pdf.
- 8.Oikarinen L, Paavola M, Montonen J, Viitasalo M, Makijarvi M, Toivonen L, et al. Magnetocardiographic QT interval dispersion in postmyocardial infarction patients with sustained ventricular tachycardia: validation of automated QT measurements. Pacing Clin Electrophysiol. 1998;21:1934–42. doi: 10.1111/j.1540-8159.1998.tb00013.x. [DOI] [PubMed] [Google Scholar]
- 9.Porthan K, Virolainen J, Hiltunen TP, Viitasalo M, Vaananen H, Dabek J, et al. Relationship of electrocardiographic repolarization measures to echocardiographic left ventricular mass in men with hypertension. J Hypertens. 2007;25:1951–7. doi: 10.1097/HJH.0b013e328263088b. [DOI] [PubMed] [Google Scholar]
- 10.Bazett H. An analysis of time relations of the electrocardiogram. Heart. 1920;7:353–70. [Google Scholar]
- 11.Sokolow M, Freidlander RD. The normal unipolar precordial and limb lead electrocardiogram. Am Heart J. 1949;38:665–87. doi: 10.1016/0002-8703(49)90525-6. [DOI] [PubMed] [Google Scholar]
- 12.Molloy TJ, Okin PM, Devereux RB, Kligfield P. Electrocardiographic detection of left ventricular hypertrophy by the simple QRS voltage-duration product. J Am Coll Cardiol. 1992;20:1180–6. doi: 10.1016/0735-1097(92)90376-x. [DOI] [PubMed] [Google Scholar]
- 13.Piippo K, Swan H, Pasternack M, Chapman H, Paavonen K, Viitasalo M, et al. A founder mutation of the potassium channel KCNQ1 in long QT syndrome: implications for estimation of disease prevalence and molecular diagnostics. J Am Coll Cardiol. 2001;37:562–8. doi: 10.1016/s0735-1097(00)01124-4. [DOI] [PubMed] [Google Scholar]
- 14.Laitinen P, Fodstad H, Piippo K, Swan H, Toivonen L, Viitasalo M, et al. Survey of the coding region of the HERG gene in long QT syndrome reveals six novel mutations and an amino acid polymorphism with possible phenotypic effects. Hum Mutat. 2000;15:580–1. doi: 10.1002/1098-1004(200006)15:6<580::AID-HUMU16>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 15.Piippo K, Laitinen P, Swan H, Toivonen L, Viitasalo M, Pasternack M, et al. Homozygosity for a HERG potassium channel mutation causes a severe form of long QT syndrome: identification of an apparent founder mutation in the Finns. J Am Coll Cardiol. 2000;35:1919–25. doi: 10.1016/s0735-1097(00)00636-7. [DOI] [PubMed] [Google Scholar]
- 16.Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, et al. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome: findings from the International LQTS Registry. Circulation. 1998;97:2237–44. doi: 10.1161/01.cir.97.22.2237. [DOI] [PubMed] [Google Scholar]
- 17.Susmano A. Effect of heart rate and autonomic tone on the QT interval. Circulation. 1982;66:478. [PubMed] [Google Scholar]
- 18.Vincent GM, Timothy KW, Leppert M, Keating M. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N Engl J Med. 1992;327:846–52. doi: 10.1056/NEJM199209173271204. [DOI] [PubMed] [Google Scholar]
- 19.Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin Proc. 2003;78:1479–87. doi: 10.4065/78.12.1479. [DOI] [PubMed] [Google Scholar]
- 20.Hofman N, Wilde AA, Kaab S, van Langen IM, Tanck MW, Mannens MM, et al. Diagnostic criteria for congenital long QT syndrome in the era of molecular genetics: do we need a scoring system? Eur Heart J. 2007;28:575–80. doi: 10.1093/eurheartj/ehl355. [DOI] [PubMed] [Google Scholar]
- 21.Lehtonen A, Fodstad H, Laitinen-Forsblom P, Toivonen L, Kontula K, Swan H. Further evidence of inherited long QT syndrome gene mutations in antiarrhythmic drug-associated torsades de pointes. Heart Rhythm. 2007;4:603–7. doi: 10.1016/j.hrthm.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 22.Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–85. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 23.Mank-Seymour AR, Richmond JL, Wood LS, Reynolds JM, Fan YT, Warnes GR, et al. Association of torsades de pointes with novel and known single nucleotide polymorphisms in long QT syndrome genes. Am Heart J. 2006;152:1116–22. doi: 10.1016/j.ahj.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 24.Fodstad H, Bendahhou S, Rougier JS, Laitinen-Forsblom PJ, Barhanin J, Abriel H, et al. Molecular characterization of two founder mutations causing long QT syndrome and identification of compound heterozygous patients. Ann Med. 2006;38:294–304. doi: 10.1080/07853890600756065. [DOI] [PubMed] [Google Scholar]
- 25.Newton-Cheh C, Larson MG, Corey DC, Benjamin EJ, Herbert AG, Levy D, et al. QT interval is a heritable quantitative trait with evidence of linkage to chromosome 3 in a genome-wide linkage analysis: The Framingham Heart Study. Heart Rhythm. 2005;2:277–84. doi: 10.1016/j.hrthm.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 26.Akylbekova EL, Crow RS, Johnson WD, Buxbaum SG, Njemanze S, Fox E, et al. Clinical correlates and heritability of QT interval duration in African Americans: the Jackson Heart Study. Circulation. 2007;116:II_781. doi: 10.1161/CIRCEP.109.858894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gouas L, Nicaud V, Berthet M, Forhan A, Tiret L, Balkau B, et al. Association of KCNQ1, KCNE1, KCNH2 and SCN5A polymorphisms with QTc interval length in a healthy population. Eur J Hum Genet. 2005;13:1213–22. doi: 10.1038/sj.ejhg.5201489. [DOI] [PubMed] [Google Scholar]
- 28.Newton-Cheh C, Guo CY, Larson MG, Musone SL, Surti A, Camargo AL, et al. Common genetic variation in KCNH2 is associated with QT interval duration: the Framingham Heart Study. Circulation. 2007;116:1128–36. doi: 10.1161/CIRCULATIONAHA.107.710780. [DOI] [PubMed] [Google Scholar]
- 29.Arking DE, Pfeufer A, Post W, Kao WH, Newton-Cheh C, Ikeda M, et al. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet. 2006;38:644–51. doi: 10.1038/ng1790. [DOI] [PubMed] [Google Scholar]
- 30.Aarnoudse AJ, Newton-Cheh C, de Bakker PI, Straus SM, Kors JA, Hofman A, et al. Common NOS1AP variants are associated with a prolonged QTc interval in the Rotterdam Study. Circulation. 2007;116:10–6. doi: 10.1161/CIRCULATIONAHA.106.676783. [DOI] [PubMed] [Google Scholar]
- 31.Gouas L, Nicaud V, Chaouch S, Berthet M, Forhan A, Tichet J, et al. Confirmation of associations between ion channel gene SNPs and QTc interval duration in healthy subjects. Eur J Hum Genet. 2007;15:974–9. doi: 10.1038/sj.ejhg.5201866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roden DM. Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med. 2006;259:59–69. doi: 10.1111/j.1365-2796.2005.01589.x. [DOI] [PubMed] [Google Scholar]
- 33.Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–22. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- 34.Shimizu W. The long QT syndrome: therapeutic implications of a genetic diagnosis. Cardiovasc Res. 2005;67:347–56. doi: 10.1016/j.cardiores.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 35.Hofman N, Wilde AA, Tan HL. Diagnostic criteria for congenital long QT syndrome in the era of molecular genetics: do we need a scoring system? Eur Heart J. 2007;28:1399. doi: 10.1093/eurheartj/ehm118. [DOI] [PubMed] [Google Scholar]
- 36.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–17. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.