

Abstract
Vasoactive peptides, such as endothelin-1 and angiotensin II are recognized by specific receptor proteins located in the cell membrane of target cells. Following receptor recognition, the specificity of the cellular response is achieved by G-protein coupling of ligand binding to the regulation of intracellular effectors. These intracellular effectors will be the subject of this brief review on contractile activity initiated by endothelin-1 and angiotensin II.
Activation of receptors by endothelin-1 and angiotensin II in smooth muscle cells results in phopholipase C (PLC) activation leading to the generation of the second messengers insitol trisphosphate (IP3) and diacylglycerol (DAG). IP3 stimulates intracellular Ca2+ release from the sarcoplasmic reticulum and DAG causes protein kinase C (PKC) activation. Additionally, different Ca2+ entry channels, such as voltage-operated (VOC), receptor-operated (ROC), and store-operated (SOC) Ca2+ channels, as well as Ca2+-permeable nonselective cation channels (NSCC), are involved in the elevation of intracellular Ca2+ concentration. The elevation in intracellular Ca2+ is transient and initiates contractile activity by a Ca2+-calmodulin interaction, stimulating myosin light chain (MLC) phosphorylation. When the Ca2+ concentration begins to decline, Ca2+-sensitization of the contractile proteins is signaled by the RhoA/Rho-kinase pathway to inhibit the dephosphorylation of MLC phosphatase (MLCP) thereby maintaining force generation. Removal of Ca2+ from the cytosol and stimulation of MLCP initiates the process of smooth muscle relaxation. In pathological conditions such as hypertension, alterations in these cellular signaling components can lead to an over stimulated state causing maintained vasoconstriction and blood pressure elevation.
Keywords: calcium channel, Rho-kinase, vasoactive peptides, phosphoinositides
In the vasculature the small arteries and arterioles regulate the majority of blood flow resistance in the circulation. Circulating neurotransmitters, hormones, endothelium-derived factors and even shear stress itself plays a role in modulating smooth muscle tone, and consequently lumen diameter (1-3). The final product is the control of blood pressure and organ blood flow.
Arteries are composed of three layers; the tunica adventitia, tunica media and tunica intima. While the outer and inner layers are composed mainly of connective tissue and endothelial cells, respectively, the tunica media is composed of smooth muscle cells (3). All smooth muscle cells, regardless of the stimulus, produce force or contraction through cross-bridge cycling between actin and myosin filaments. Although vascular smooth muscle cells are capable of dynamic changes in gene expression to either contractile or differentiation proteins, the contractile phenotype in vascular smooth muscle predominates (4). The contractile response of vascular smooth muscle is the product of myosin light chain kinase (MLCK) and myosin light chain phosphatase (MLCP) activation. In smooth muscle, this process is initiated by a calcium (Ca2+) mediated change in the thick (myosin) filaments (2).
In this review, we will discuss the molecular mechanisms by which two common vascoactive peptides, angiotensin II and endothelin-1, produce smooth muscle contraction. Since intracellular Ca2+ is fundamental to the contractile process, we will also provide a brief review of the cellular mechanisms that regulate this cation.
Angiotensin II Signaling
Angiotensin II, which is the predominate bioactive peptide in the renin-angiotensin system, promotes the maintenance of systemic pressure through various mechanisms in the cardiovascular and renal systems (5). Although angiotensin II is crucial to salt and water homeostasis, this peptide is also implicated in several cardiovascular conditions, such as hypertension, atherosclerosis and heart failure, and more specifically in vascular smooth muscle contraction; vascular remodeling including the induction of hypertrophy and hyperplasia. Canonical angiotensin II signaling occurs through a membrane bound heterotrimeric G-protein coupled receptor (GPCR). To date, there are two GPCRs known to mediate angiotensin II function, the AT1 and AT2 receptors (5, 6).
The AT1 receptor is expressed in a variety of tissues, including the kidney, heart, adrenal gland, brain, lung and adipose; however, the focus will be on AT1 receptor activation in arterial smooth muscle. The AT2 receptor is highly expressed in fetal tissues, then declining quickly after birth. In adults, the AT2 receptor has a varied tissue distribution as well, and is detectable in the kidney and adrenals, pancreas, ovary, brain, heart and vasculature. In vessels, the AT2 receptor is not localized on the smooth muscle, but is mainly expressed in the adventitia. After injury, levels of this receptor have been observed to increase, which may explain current dogma suggesting that the AT2 receptor is involved in cellular growth. In contrast to the vasoconstrictive effects of the AT1, the AT2 receptor has been shown to modulate vasodilation (7). Less is known about AT2 receptor mechanisms than the; however, it is known that it couples to Gi and activates tyrosine and serine/threonine phosphatases. This receptor isoform has also been documented to trigger the kinin-NO-cGMP pathway (8). However, it is through the AT1 receptor with which the majority of physiological and pathological actions of angiotensin II is mediated, and will be the focus of the signaling in this review.
The angiotensin II signaling cascade via the AT1 receptor is a consequence of the particular G-protein and/or signaling cascade which is activated: Gq/11, Gi, G12, and G13 (5, 6). The AT1 receptor signals through three main pathways: classical phospholipase C (PLC) signaling leading to inositol trisphosphate (IP3) and diacylglycerol (DAG) cleavage, phospholipase D (PLD), also culminating in DAG generation; and phosphatidylcholine, phosphatidic acid (PA) and phospholipase A2 (PLA2), which is responsible for arachidonic acid (AA) and prostaglandin (PG) production. Tyrosine phosphorylation, ie, mitogen activated protein kinase (MAPK), which is distinctive for its role in growth factor and cytokine activation, has been associated with increased levels of AT1 receptor activation. Non-receptor tyrosine kinase activity has also been associated with angiotensin II, including but not limited to: the Src family of kinases, proline-rich tyrosine kinases (Pyks), extracellular signal-regulated kinases (ERKs), paxillin, focal adhesion kinase (FAK), phosphoinositide 3 kinase (PI3K) and an inflammatory cytokine pathway, janus kinases/signal transducers and activators of transcription (JAK/STAT) (7). Angiotensin II is a pleiotropic signaling molecule, whose varied pathways is complex and specific, but yet also converges to bring forth multiple responses. This review will focus upon the PLC pathway culminating in vascular smooth muscle contraction.
Recent literature suggests that the AT1 receptor couples to Gq to mediate vasoconstriction, which is the pathway leading to smooth muscle contraction via PLC (9). In addition, angiotensin II was found to elicit a contractile response via activation of G12/G13 G-proteins and the RGS-RhoGEF signaling pathway. Recently, insights into this mechanism were discovered and will be discussed in a later section (10, 11). Following Gq/G11 GPCR activation, second messengers are generated, beginning with PLC activation. PLC is a membrane bound enzyme responsible for cleaving membrane lipid phosphoinositide 4, 5- bisphosphate (PIP2), resulting in the generation of IP3 and DAG (2, 12). IP3 and DAG regulate two distinct, but parallel pathways leading to increased intracellular Ca2+ and culminating in MLC phosphorylation (2, 13, 14).
IP3 regulates calcium release into the cytosol from the sarcoplasmic reticulum, mediated via the ryanodine receptor (RyR). The RyR, which has been shown to be localized centrally as well as peripherally on the sarcoplasmic reticulum (SR), is thought to facilitate optimal binding of IP3 (15, 16). Activation of the RyR opens Ca2+ channels, causing a rapid increase in Ca2+ concentration. After cytosolic Ca2+ concentrations peak and then decline, there is a sustained elevation of Ca2+ above that of basal levels (2). This sustained increase in Ca2+ is due to receptor-operated Ca2+ channels on plasma membrane, facilitating increased cytosolic Ca2+ levels from the extracellular space (2, 14). The EF hand Ca2+ -binding protein, calmodulin, is the primary target for elevated intracellular Ca2+. The Ca2+ -calmodulin complex is then able to activate MLCK (2). It is also of importance to note that the Ca2+-tension relationship changes for stimulation type and during the time of the contraction. In general, studies show that the receptor-mediated stimulations produce a greater tension for a given Ca2+ concentration. Moreover, during the sustained phase of contraction, lower levels of Ca2+ are needed, which is referred to as Ca2+ sensitization. These are two differential points of regulation for vascular smooth muscle contraction (17).
DAG mediates vascular smooth muscle contraction via activation of protein kinase C (PKC). PKC is a serine/threonine kinase known to exist in several isoforms, all which contribute to various physiological activities and are activated differentially. Of the three known vascular smooth muscle isoforms, PKCα and PKCβ are dependent upon DAG activation and intracellular Ca2+ concentrations, while PKCε is DAG dependent only. PKC modulates vascular smooth muscle contraction by directly phosphorylating MLCK, which subsequently phosphorylates MLC. Furthermore, PKC activation leads to the activation of several other target proteins promoting smooth muscle contraction. PKC phosphorylates, and activates, ERK1/2, Rho-kinase (p160ROCK), and calmodulin-dependent protein kinase II. In addition, intracellular Ca2+ elevation was suggested to regulate angiotensin II-induced epidermal growth factor receptor (EGFR) and ERK activation (11). Various ion channels and ion transporters have been demonstrated as downstream targets of PKC. This phenomenon has been illustrated with the use of the specific PKC agonists (i.e. phorbol esters) which induce smooth muscle contraction (2).
All the earlier events described culminating in MLCK activation, resulting in contraction via the phosphorylation of Ser19 of the 20 kDa regulatory protein of MLC. The phosphorylation of MLC allows for the interaction between actin and myosin filaments. Intrinsic ATPase activity of myosin is crucial for this process, as the hydrolysis and subsequent release of ATP results in the cycling of the myosin cross bridges with actin, causing force generation (3, 18-21).
Smooth muscle cells also contain Ca2+-independent mechanisms to regulate contractility. The process of force generation is mediated by MLCK activation, and subsequent actin-myosin cross-bridging, but the process of force maintenance is mediated in a Ca2+-independent manner (2, 18, 22). MLCP is the contender, regulating the inactivation of MLC by removing the high-energy phosphate group, promoting smooth muscle relaxation. MLCP is composed of two subunits; one is the catalytic 38 kDa subunit of type 1 protein phosphatase (PP1c, δ isoform) and two other non-catalytic subunits (12, 17). This mechanism generates force maintenance by means of Ca2+-sensitization of the contractile proteins. This is signaled by the RhoA/Rho kinase pathway which inhibits the dephosphorylation of MLC by MLCP. RhoA is a small, monomeric G-protein, which is regulated by the binding of GTP. This process is facilitated by nucleotide exchange factors, RhoGEFs, which enable the exchange of GDP for GTP, activating RhoA. Upon RhoA activation, the RhoA-GTP complex is able to activate Rho kinase, a serine/threonine kinase. Rho kinase can then phosphorylate the myosin-binding subunit of MLCP, inhibiting it, and thus preventing the dephosphorylation of MLC, causing maintenance of contraction (2, 23, 24). In addition to the regulatory effects of Rho kinase, CPI-17 is another protein which regulates MYPT phosphorylation status by inhibiting PP1c, thus inactivating MLCP. CPI-17 phosphorylation occurs in a Ca2+-dependent and independent mechanism. Ca2+ dependent PKC was suggested to regulate the rapid phosphorylation, whereas phosphorylation by PKC and Rho kinase occurs independently of Ca2+ in the later phase. This has been demonstrated with the use of Rho kinase and PKC inhibitors; however, the effects were dependent upon the type of agonist and tissue used. Its activity and expression is a factor in the contractile state of vascular smooth muscle and has also been shown to be of importance in Ca2+ sensitization (12, 17). Much needs to be explored regarding MLCP regulation, but regardless, Rho kinase and CPI-17 are acknowledged to be of importance in vascular smooth muscle contraction and force maintenance.
Endothelin-1 Signaling
Endothelin-1, which is a 21-amino acid peptide, is known as one of the most potent endogenous vasoconstrictors as described by Yanagisawa, et al. in 1988 (25-27). Since its discovery, the endothelin-1 system has become increasingly complex; several endothelin-1 isoforms and receptors have been identified in a variety of tissues including neuronal, renal and vascular tissues. Interestingly, although some data show a correlation between endothelin-1 levels and hypertension, these data are not conclusive (4). Overall, endothelin-1 signaling in the vasculature is essentially the same as for angiotensin II signaling. A brief overview will be discussed, but the reader is directed to angiotensin II-GPCR coupled signaling pathways for a more in depth explanation. Differing pathways regarding to endothelin-1 signaling itself will be discussed further.
Endothelin-1 is the main isoform secreted by the endothelium, and has been shown to act in a paracrine or autocrine manner on its receptors in vascular smooth muscle. There are three known endothelin-1 receptors with have an assorted tissue distribution and functional role; ETA and ETB are present in mammals and a putative ETC in non-mammals. The typical receptor found on vascular smooth muscle cells is the ETA receptor, which mediates the vasoconstrictor effects of endothelin-1 (4, 25, 28). Endothelin-1 receptor activation can lead to diverse responses in the cell through interaction with pathways that are both pertussis toxin-sensitive and insensitive, leading to the conclusion that endothelin-1 acts through several GPCRs. Generally ETA receptors have been associated with vasoconstriction and cell growth, whilst ETB receptors are involved in the clearance of endothelin-1, inhibition of endothelial cell apoptosis, the release of NO and PGs leading to vasorelaxation, and inhibition of the expression of endothelin-1 converting enzyme; however, both receptors have been found to elicit vasoconstriction (29-31).
ETA receptors have been shown to be functionally coupled to the Gq/11 protein leading to PLC-β activation; Gs linked leading to increased cAMP and also to Gi thus inhibiting adenylate cyclase (30, 32-34). Activation of Gq/11, ending in IP3 and DAG cleavage, can stimulate Ca2+ release intra- and extra-cellularly, as described for angiotensin II signaling. In addition to second messenger generation, endothelin-1 has also been demonstrated to activate Ca2+ channels on the plasma membrane and stimulate Ca2+ flux from the extracellular space (4, 35). There is speculation that ETA receptor activation can activate the Na+/H+ exchanger, and activate the enzymes PLD generating DAG and PLA2 releasing AA (4). ETB receptors in rat carotid were shown to mediate vasorelaxation via a cGMP-NO pathway, the production of vasodilator cyclooxgenase (COX) products and the activation of voltage-activated K+ channels. Recently, endothelin-1 was shown to inhibit NADPH oxidase activity and superoxide generation via ETB1 receptors (36). The details of these mechanisms are still poorly understood (31).
Both agonists, angiotensin II and endothelin-1, mediate vasoconstriction via an increase in cytosolic Ca2+ although mechanisms of this process are different between the two agonists. Endothelin-1 increases intracellular Ca2+ by stimulating influx through Ca2+ channels and is known to produce a much more sustained contraction by the vascular smooth muscle cells, whereas angiotensin II elicits a potent biphasic response that is generated primarily by mobilization of Ca2+ from an intracellular store. Consequently, the actions of angiotensin II actions are relatively rapid in comparison (37-41).
Given the fact that both of these agonists mediate downstream signaling pathways via the same second messengers, it is easy to appreciate how crosstalk occurs between them. Both endothelin-1 and angiotensin II signal through distinct Gq coupled GPCR's leading to phosphoinositide production and MAPK activation. However, the mechanism by which this occurs varies at some points. It has been demonstrated that endothelin-1 and angiotensin II both stimulate this pathway via Ras-Raf; yet, angiotensin II produces phosphorylation of ERK1/2, SAPK/JNK, and p38MAPK via c-Src-dependent pathways. In contrast, endothelin-1 was shown to induce MAPK phosphorylation through c-Src independent pathways (42).
Calcium Mobilization
Given that Ca2+ is the trigger for smooth muscle contraction, it is necessary to understand the mechanisms by which intracellular Ca2+ levels increase. Hence, this is the focus of much research, and the subject of the remainder of this review. In vivo, arterial smooth muscle cells exist with an average intracellular Ca2+ concentration which is several orders of magnitude lower than that in the extracellular fluid. Intracellular Ca2+ concentration levels do not exist homogenously throughout the cell, but exhibit dynamic changes in the cell, temporally and spatially, due to Ca2+ flux. The changes seen in Ca2+ flux are a component of several different events, which are all dependent upon intracellular ultra structure, as well as the spatial relationship of ion pumps and channels in the plasma membrane (4).
Ca2+ entry through channels in the plasma membrane and release from the SR are the major sources of intracellular Ca2+ (1, 4, 15). Of the different Ca2+ entry channels the voltage-operated Ca2+ (VOC), receptor-operated Ca2+ (ROC), store-operated Ca2+ (SOC) channels and a Ca2+-permeable nonselective cation channels (NSCC) modulate most Ca2+ mobilization within the cell. We will discuss these particular vascular smooth muscle Ca2+ entry channels further below.
Voltage-Operated Ca2+ Channels
VOC channels represent a major route by which Ca2+ enters vascular smooth cells (43). VOC channel function is regulated by membrane potential such that hyper polarization closes them and depolarization opens them; the latter leading to vasoconstriction (1).
The majority of agonist-induced Ca2+ influx probably occurs through L-type VOC channels. Dyhydropyridine-sensitive (DHPR), L-type VOC channels are regulated by vasoconstrictors that are known to activate the PKC pathway. Vasodilators have also been shown to inactivate these channels; yet through cAMP production (1, 44). These gated channels also depolarize in response to membrane stretch, lending support to the hypothesis that these channels are important for the myogenic response and vascular tone. Inhibition of VOCs can occur primarily due to increasing intracellular Ca2+ concentration and activation of cGMP-dependent protein kinase (1, 2, 44).
Endothelin-1 has been shown to activate the VOC channel in smooth muscle cells from porcine coronary arteries, and has also been shown to augment Ca2+ channel currents in the smooth muscle cell membrane of guinea pig portal vein. In a model of cultured thoracic aorta vascular smooth muscle cells, Kawanabe and colleagues demonstrated that endothelin-1 produces a sustained increase in intracellular Ca2+ levels from VOC channels, as well as others (41, 45).
Although endothelin-1 increases intracellular Ca2+ by stimulating influx through Ca2+ channels, the role of VOC channels is limited, and current data suggest that other channels are more important; namely the NSCC, as evidenced by experiments using nifedipine (41, 46). Even though described over sixty years ago, the exact mechanisms of action of this peptide are not completely understood.
Angiotensin II-GPCR stimulation leading to PLC is an accepted mechanism acknowledged to enhance the L-type Ca2+ channel. Interestingly, intracellular angiotensin II has been demonstrated to stimulate VOC channels similarly, but yet independently of extracellular ligand binding of angiotensin II to the AT1 receptor, possibly through the actions of PLC, PKC and/or tyrosine kinase (47, 48).
As shown in the smooth muscle cells of the anococygeous, there are two very distinct mechanisms for inward Ca2+ currents and Ca2+ store depletion (49). The increase in Ca2+ concentration after the activation of VOC channels can also lead to a phenomenon known as Ca2+ induced Ca2+ release (CICR), where the increase in Ca2+ concentration leads to Ca2+ release from intracellular stores. The RyR on the SR mediates this process, as they are activated by Ca2+ (50). This produces a membrane depolarization that promotes further VOC channel opening and Ca2+ entry.
Receptor-Operated Ca2+ Channels
Only twenty years ago the exact mechanism of Ca2+ channel activation by extracellular ligands, such as hormones and neurotransmitters, was largely unknown. In addition, there was still uncertainty concerning the mechanism by which Ca2+ entry was mediated until a model, proposed by Putney and colleagues, detailing the mechanism by which activation of surface membrane receptors causes a sustained Ca2+ entry into cells from the extracellular space (51). Furthermore, Benham and Tsien suggested that agonists might activate Ca2+ influx directly, by receptor interaction with Ca2+-permeable, NSCC. (52) Currently, ROC channels are defined by the following: channels where molecules are separate from the ligand-binding protein, are capable of activating a range of GPCRs via circulating agonists, and are neither VOC nor SOC channels (28). After ligand binding, ROC channels are generally thought to be activated by GPCRs which are coupled to PLC, leading to IP3 and DAG generation. (53, 54)
Several members of the transient receptor potential channel (TRPC) family are accepted to form ROC channels, including TRPC3, TRPC6 and TRPC7. Angiotensin II has been proposed to activate TRPC6 in rabbit mesenteric arteries, while endothelin-1 activates TRPC3 and TRPC7 (55-57).
Store-Operated Ca2+ Channels
SOC channels represent yet another mechanism of excitation-contraction coupling in smooth muscle. SOC channels are activated by means of intracellular Ca2+ level depletion, and as a result, are inhibited when Ca2+ stores are filled. This process was called capacitative Ca2+ entry, before store-operated Ca2+ entry (SOCE) was coined. It should be noted that SOCE does not denote any one mechanism of Ca2+ entry, nor does it refer to any particular channel (1, 58-62). These channels are highly selective for Ca2+, and have been found to be bradykinin and ATP sensitive (60). RyR or IP3 sensitive stores can induce Ca2+ release sequentially, possibly amplifying the rise in intracellular Ca2+ and further stimulating Ca2+ channel dependent Cl- channels. SOC channels were described in 1986, when Putney, et al. demonstrated that the same Ca2+ entry mechanism normally activated as a result of Ca2+-mobilizing agonists can be triggered equally as well by depleting the intracellular Ca2+ pool, even in the absence of receptor activation or elevated cellular levels of inositol polyphosphates (63-65). SOC channel entry has been proposed to increase cytoplasmic Ca2+ levels not only for contraction, but also cell proliferation and apoptosis (4). SERCA pumps, which are found on the membrane of the sarcoplasmic reticulum, function by pumping the Ca2+ ions back into the SR after a contractile event replenishing Ca2+ concentrations (14, 15). SERCA pumps can be inhibited by cyclopiazonic acid or thapsigargin, which deplete intracellular Ca2+ stores and thereby activate SOC channels (66, 67). Ca2+ entering through the SOC channels can then be pumped into the stores, replenishing them. According to functional and pharmacological experiments, SOC channel entry elicited by specific inhibitors of SERCA pumps induced a sustained elevation of intracellular Ca2+, which is also dependent upon extracellular Ca2+ entry and is attributed to SOCE (68). Thus, Ca2+ signals generated in response to receptors involve two coupled components: a transient release of Ca2+ stored in the SR, followed by a slowly developing extracellular Ca2+ entry (14, 15, 69, 70). With depleted Ca2+ stores, Ca2+ influx factor (CIF) is generated and diffuses to the plasma membrane where, through a series of reactions CIF activates Ca2+-independent phospholipase A2 (iPLA2). This generation of lysophospholipids, in turn, activates the SOC channels (62).
The family of TRPC is well known for their role in SOCE, although the mechanism allowing for TRPC activation from store depletion is largely unknown. There is evidence for and against TRPCs actually being a SOC channel, including TRPC1 (56, 58, 62). Also, interesting data from recent high throughput RNAi screens of thapsigargin-activated Ca2+ entry revealed a stromal-interacting molecule (STIM) in Drosophila S2 cells and in mammalian HeLa cells, which are now thought to play an essential role in SOCE and conductance through Ca2+-release activated Ca2+ (CRAC) channels (71-73). This single membrane spanning protein is now thought to activate the SOC channels by actually sensing Ca2+ within the stores. Pharmacological experiments showed that the actual contribution of SOCE to excitation/contraction coupling seems to depend upon the smooth muscle type, and may be more important in tonic smooth muscle (62). A canonical TRP channel, TRPC7, which is activated by AT1 coupled GPCR activation and DAG, was recently hypothesized to mediate angiotensin II-induced myocardial apoptosis. Moreover, angiotensin II has been postulated to activate TRPC1, as evidenced by data showing that angiotensin II stimulation increased SOCE together with TRPC1 expression. These data were obtained using a cell culture model, but given the heterogeneity of TRPC channels in vascular smooth muscle, there is no reason to believe that this occurrence does not exist in vivo (74). In a recent study in vascular smooth muscle cells, endothelin-1 activation of TRPC1 was found to not only be involved in a SOCE, but also in a ROC channel entry (ROCE), which requires IP3 receptor activation. A possible explanation for this exciting phenomenon is the report that TRPCs may exist in heterotrimeric complexes, possibly accounting for the varying pharmacological properties seen (56).
Non-Selective Cation Channels
The NSCC, so called because it is ‘nonselective’, being equally permeable to monovalent cations, such as Na+ and K+ in the extra- and intracellular compartments (25, 28). This channel has been revealed to be activated by stimulation of native ETA receptors in vascular smooth muscle, and the majority of endothelin-1 response being mediated by NSCCs; namely, NSCC-1 and NSCC-2 (28, 41, 46, 75). It was previously shown, pharmacologically and with electrophysiology, that the response to endothelin-1 is dose-dependent. More specifically, with lower concentrations of endothelin-1, Ca2+ entry via IP3 formation from intracellular stores occurs; however, with higher endothelin- 1 concentrations both the former in addition to extracellular Ca2+ entry occurs as well (25, 28, 38, 76, 77).
The molecular mechanisms of NSCC activation are still not entirely clear. Recent evidence in rabbit internal carotid artery shows that endothelin-1 may induce an intracellular response after GPCR activation via Pyk2 and the ETA receptor. Data from the same laboratory indicate that PI3K also regulates the activation of Ca2+ entry after endothelin-1 GPCR activation and stimulation of NSCC-2 (46, 78).
Hot Topics in Vascular Smooth Muscle Signaling
Within just the past year, exciting new insights into vascular smooth muscle signaling have been discovered. We will discuss several recent advancements in the field which have been especially stimulating.
As described earlier, GPCR signaling can elicit stimulatory or inhibitory signals in the cell. With regards to vascular smooth muscle, the Gq/G11 G-proteins have been accepted as producing contraction via the Ca2+-dependent PLC pathway; however, Ca2+-independent mechanisms, not only for force maintenance, but for contraction have emerged. By means of a novel murine knock-out model for Gq/G11 and G12/G13 G-proteins in smooth muscle cells, Wirth and colleagues elaborated upon the knowledge of how angiotensin II and endothelin-1 mediate contraction via G-proteins. Using aortic segments they demonstrated that in G12/G13 knock-out mice, angiotensin II and endothelin-1 were able to elicit a contraction; however, both potency and efficacy were affected. In the Gq/G11 knock-out mice, although endothelin-1 still produced a severely reduced contraction, angiotensin II was not capable of eliciting any contraction. It was also demonstrated that G12/G13 coupled G-proteins activate the RhoA/Rho kinase signaling pathway via interaction of RhoGEF's; in particular LARG. Using a LARG murine knock-out model, they showed that LARG is necessary for full contraction with angiotensin II and endothelin-1, as demonstrated by the severely restriction constriction elicited in aortic segments from LARG knock-out mice (10, 79).
As described earlier, Pyk2 is a Ca2+ sensitive non-receptor protein tyrosine kinase that is known to associate with focal adhesion sites and is a downstream effector of angiotensin II and endothelin-1 GPCR stimulation (7, 15, 80, 81). Pyk2 phosphorylation and activation has been postulated in several different pathways, including ERK1/2 signaling in cardiomyocytes, p38MAPK in mesangial cells and recently in RhoA/Rho kinase activation via PDZ-RhoGEF vascular smooth muscle cell migration (15, 80, 82, 83). It has even been suggested that tyrosine kinase pathways such as these may even be important in angiotensin II signaling leading to vasoconstriction (84). These alternative signaling pathways in angiotensin II signaling should be a new directive for investigation into increased vascular smooth muscle reactivity in conditions such as hypertension. This was a focus of a recent paper from Giachini and colleagues, in press. It is known that increased vascular reactivity to contractile stimuli is present in vessels from DOCA-salt hypertensive mice. Using this model with a pharmacological approach, mechanistic studies were performed to determine the role that Pyk2 plays in the hyper-reactivity exhibited in DOCA-salt hypertension. In mesenteric arteries and aorta, Pyk2 inhibition attenuated the increased vascular constriction to phenylephrine and western blot data showed increased Pyk2 and phospho-Pyk2 in vessels from DOCA-salt treated mice vs. sham, thus confirming a role for Pyk2 in hypertension (80, 85).
A recent discovery of a transcription factor named the repressor element 1-silencing transcriptional factor, or REST, was shown to have a consensus sequence, KCNN4 which is known to encode for the intermediate conductance Ca2+-activated K+ channels (IKCa). These channels are of the utmost importance in small resistance vessels where a primary vasodilating agent is endothelium-derived hyperpolarizing factor (EDHF). EDHF not only remains elusive as a factor, but the mechanism by which it produces vasodilation is only speculation. One such hypothesis is that K+ efflux from endothelial cells via these IKCa channels in coordination with small-conductance Ca2+-activated K+ channels (SKCa), activates inward rectifier K+ channels (KIR). This would lead to vascular smooth muscle relaxation. Unpublished results from the laboratory of R.C. Webb and R.C. Tostes show for the first time that REST expression is closely associated with IKCa expression in arteries from hypertensive animals. Giachini and colleagues demonstrated that REST expression was down-regulated, whereas IKCa channels were over expressed in arteries obtained from spontaneously hypertensive stroke-prone rats (SHRSP), when compared to their Wistar-Kyoto controls. Protein levels of SKCa and IKCa were also assessed, and alterations were observed in mesenteric arteries from hypertensive animals. It seems as though REST may be a negatively modulatory mechanism, which controls the levels of IKCa in the SHRSP vasculature.
As mentioned previously, the STIM molecule was shown to play an essential role for SOCE and conductance through CRAC channels (71). The recently discovered subunit of the CRAC channel pore, termed Orai1, is essential for CRAC channel activation (62, 72, 86, 87). However, the exact mechanism for STIM/Orai1 signaling is not yet known. STIM can undergo phosphorylation at specific serine/threonine sites, as well as undergo N-linked glycosylation (88). Sobolff et al. determined that while STIM1 is expressed at the cell surface and within the endoplasmic reticulum (ER), the STIM2 protein is expressed only intracellularly, which likely reflects an ER-retention signal that is present in STIM2 but not STIM1 (86, 87, 89, 90). It was apparent that suppressed STIM1 expression, but not STIM2, was able to prevent SOCE and eliminate the store-dependent activation of CRAC channels (71, 73). Thus, the function of STIM1 was postulated to act as a Ca2+ sensor in the ER (72).
On a final note, the question of ‘blame’ for hypertension is argued between two schools of thought, cardiovascular and renal physiologists; the former believing that hypertension is due to increased vascular resistance and an overall hyper-reactivity of vascular tissue, the latter believing that the kidney has the final say in blood pressure control. Until recently, renal physiologists everywhere cited papers by Guyton, who was the first to clearly demonstrate the phenomenon of ‘pressure natriuresis’ and argue for the central role for the kidney in blood pressure control and Coffman, who uses a model of AT1A receptor knock-out to illustrate the necessity of the renin-angiotensin system in hypertension (91, 92). These elegant studies used the AT1 receptor knock-out mice and kidney cross-transplantation to show that the AT1A receptor is crucial to basal blood pressure regulation, as well as hypertension and that AT1A receptors in the kidney are paramount in hypertension and the renal response to hypertension. However, these studies also demonstrated that blood pressure regulation by AT1A receptors in non-renal tissues, i.e., vasculature, were also a major contributor to systemic blood pressure (93, 94). A paper recently published in PNAS by Michael et al., complements these studies, using a knock-in mutant of the cGMP-dependent protein kinase, PKGIα. PKGI is expressed in vascular and smooth muscle cells and has been shown to regulate vascular relaxation via endothelial-derived NO and other nitrovasodilators. The most remarkable data from this study show that the LZM-PKGIα mutant mice exhibit elevated blood pressures, even in the presence of normal renal function and normal renal salt handling, suggesting an important mechanism for vascular smooth muscle in the normal and pathophysiological control of blood pressure (95). Overall, these studies reiterate our awareness of the complexity of hypertension, and the necessity for systemic integration.
Concluding Remarks
Arterial vascular smooth muscle cells constitute the majority of the arterial wall, playing a foremost role in vascular resistance and blood flow. Angiotensin II and endothelin-1 are potent agonists inducing contraction of vascular smooth muscle. The contractile apparatus of vascular smooth muscle, actin and myosin, can be activated in a Ca2+-dependent and Ca2+-independent manner. Via ligand binding to plasma membrane GPCRs, second messengers are generated and induce the release of Ca2+through channels located on the plasma membrane or on the SR producing a rapid and transient increase in intracellular Ca2+. Channels discussed in this review are activated through ligand binding, store depletion and membrane depolarization. In the form of a Ca2+-calmodulin complex, subsequent activation of MLCK occurs, inducing contraction via actin-myosin cross bridges. Contraction also occurs, Ca2+ independently, through the activation of the RhoA/Rho kinase pathway leading to MLCP inactivation, and the maintenance of contraction.
Given the varied mechanisms for smooth muscle contraction, one can imagine the wide-range of areas where deregulation can occur, leading to increased blood pressure, or hypertension. On a molecular level, alterations in various cellular signaling components, can lead to over stimulation, causing an increased and maintained vasoconstriction, decreased relaxation and consequently, elevation of systemic blood pressure.
Figure 1. Overview of major contributors to vascular smooth muscle contraction.
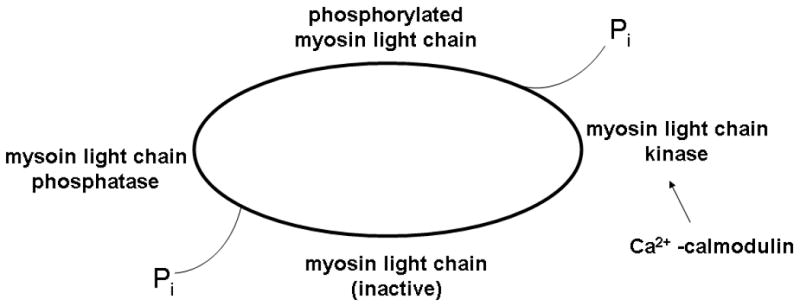
In smooth muscle, contraction is initiated by a Ca2+ mediated change in the thick filaments, or myosin. With myosin light chain phosphorylation, the actin and myosin filaments are capable of interacting. ATP hydrolysis is the source for force generation in smooth muscle; with contraction, inorganic phosphate leaves the myosin head. Relaxation occurs via the action of myosin light chain phosphatase, which de-phosphorylates myosin light chain, inactivating it.
Figure 2. Molecular mechanisms of smooth muscle contraction.
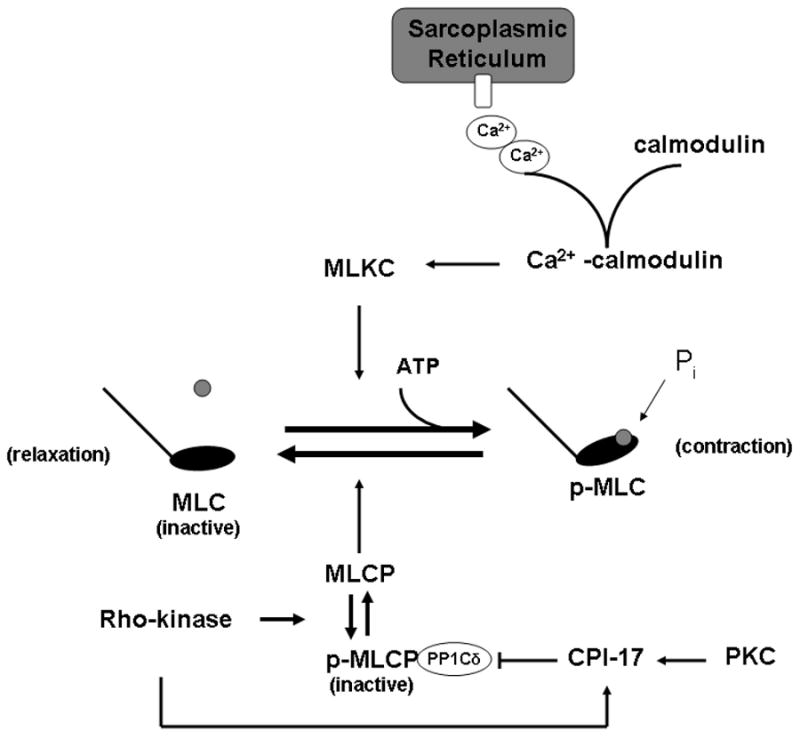
Vascular smooth muscle contraction is the summation of myosin light chain kinase (MLCK) and myosin light chain phosphatase (MLCP) activity. With receptor binding, an increase in intracellular Ca2+ occurs, both via channels located in the membrane and intracellular stores in the sarcoplasmic reticulum. The Ca2+ interacts with calmodulin, forming a Ca2+-calmodulin complex which activates MLCK. MLCK can then phosphorylate myosin light chain (MLC-P), allowing for the close interaction of the actin and myosin filaments for force generation. Relaxation occurs with MLCP dephosphorylating MLC. In some instances, force generation can be tonic; this is mediated in a Ca2+-independent manner. Rho-kinase becomes activated via the small, activated RhoA protein, which subsequently phosphorylates MLCP, rendering the enzyme inactive and incapable of de-phosphorylating MLC. In addition, PKC and Rho kinase work in concert to activate CPI-17, which inhibits MLCP. Thus, the vascular smooth muscle cannot relax and a tonix contraction occurs.
Figure 3. The concept of receptor-operated Ca2+ (ROC) channels, voltage-operated Ca2+ (VOC) channels and store-operated Ca2+ (SOC) channels.

ROC channels are activated via receptor stimulation of G-protein coupled receptors (GPCRs) directly or through the production of second messengers such as IP3 or DAG. IP3 can then bind to and activate the IP3 receptor on the sarcoplasmic reticulum membrane, causing discharge and release of the stored Ca2+. The release of Ca2+ from the sarcoplasmic reticulum induces Cl- efflux or the influx of Na+ and Ca+ from the ROC channels, which can then activate VOC channels. The second messenger, IP3 activates the IP3 receptor on the sarcoplasmic reticulum membrane, causing discharge and release of the stored Ca2+. After Ca2+ depletion from the sarcoplasmic reticulum, the SOC channels are activated. A, agonist; DAG, diacylglycerol; G, G-protein; IP3, inositol 1,4,5-trisphosphate; IP3R, IP3 receptor; PIP2, phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; R, receptor; TK, tyrosine kinase.
Figure 4. New pathways in vascular smooth muscle signaling.

Orai1 and STIM work together to function as Ca2+ sensors within the cell. Orai1 makes up the pore of the CRAC channel, allowing for Ca2+ entry while STIM1 senses Ca2+ levels within the SR. Their interaction facilitates this process. The IKCa channels are intermediate conductance Ca2+-activated K+ channels which are of importance in small resistance vessels. G12/13 signaling through LARG was recently found to be a mechanism for RhoA/Rho kinase activation, leading to MLCP inactivation.
Acknowledgments
This study was supported by grants from the National Institutes of Health (NIH -HL71138 and HL74167).
Footnotes
No disclosures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jackson WF. Ion channels and vascular tone. Hypertension. 2000 Jan;35(1 Pt 2):173–8. doi: 10.1161/01.hyp.35.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hilgers RH, Webb RC. Molecular aspects of arterial smooth muscle contraction: focus on Rho. Exp Biol Med (Maywood) 2005 Dec;230(11):829–35. doi: 10.1177/153537020523001107. [DOI] [PubMed] [Google Scholar]
- 3.Woodrum DA, Brophy CM. The paradox of smooth muscle physiology. Mol Cell Endocrinol. 2001 May 25;177(12):135–43. doi: 10.1016/s0303-7207(01)00407-5. [DOI] [PubMed] [Google Scholar]
- 4.Wamhoff BR, Bowles DK, Owens GK. Excitation-transcription coupling in arterial smooth muscle. Circ Res. 2006 Apr 14;98(7):868–78. doi: 10.1161/01.RES.0000216596.73005.3c. [DOI] [PubMed] [Google Scholar]
- 5.Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S. Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin Sci (Lond) 2007 Apr;112(8):417–28. doi: 10.1042/CS20060342. [DOI] [PubMed] [Google Scholar]
- 6.Ohtsu H, Suzuki H, Nakashima H, Dhobale S, Frank GD, Motley ED, et al. Angiotensin II signal transduction through small GTP-binding proteins: mechanism and significance in vascular smooth muscle cells. Hypertension. 2006 Oct;48(4):534–40. doi: 10.1161/01.HYP.0000237975.90870.eb. [DOI] [PubMed] [Google Scholar]
- 7.Touyz RM, Berry C. Recent advances in angiotensin II signaling. Braz J Med Biol Res. 2002 Sep;35(9):1001–15. doi: 10.1590/s0100-879x2002000900001. [DOI] [PubMed] [Google Scholar]
- 8.Matsubara H. Pathophysiological role of angiotensin II type 2 receptor in cardiovascular and renal diseases. Circ Res. 1998 Dec 14-28;83(12):1182–91. doi: 10.1161/01.res.83.12.1182. [DOI] [PubMed] [Google Scholar]
- 9.Harris DM, Cohn HI, Pesant S, Zhou RH, Eckhart AD. Vascular smooth muscle G(q) signaling is involved in high blood pressure in both induced renal and genetic vascular smooth muscle-derived models of hypertension. Am J Physiol Heart Circ Physiol. 2007 Nov;293(5):H3072–9. doi: 10.1152/ajpheart.00880.2007. [DOI] [PubMed] [Google Scholar]
- 10.Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008 Jan;14(1):64–8. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 11.Ohtsu H, Higuchi S, Shirai H, Eguchi K, Suzuki H, Hinoki A, et al. Central role of Gq in the hypertrophic signal transduction of angiotensin II in vascular smooth muscle cells. Endocrinology. 2008 Jul;149(7):3569–75. doi: 10.1210/en.2007-1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woodsome TP, Polzin A, Kitazawa K, Eto M, Kitazawa T. Agonist- and depolarization-induced signals for myosin light chain phosphorylation and force generation of cultured vascular smooth muscle cells. J Cell Sci. 2006 May 1;119(Pt 9):1769–80. doi: 10.1242/jcs.02805. [DOI] [PubMed] [Google Scholar]
- 13.Del Valle-Rodriguez A, Calderon E, Ruiz M, Ordonez A, Lopez-Barneo J, Urena J. Metabotropic Ca(2+) channel-induced Ca(2+) release and ATP-dependent facilitation of arterial myocyte contraction. Proc Natl Acad Sci U S A. 2006 Mar 14;103(11):4316–21. doi: 10.1073/pnas.0508781103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Urena J, del Valle-Rodriguez A, Lopez-Barneo J. Metabotropic Ca2+ channel-induced calcium release in vascular smooth muscle. Cell Calcium. 2007 Oct-Nov;42(45):513–20. doi: 10.1016/j.ceca.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 15.Bouallegue A, Daou GB, Srivastava AK. Nitric oxide attenuates endothelin-1-induced activation of ERK1/2, PKB, and Pyk2 in vascular smooth muscle cells by a cGMP-dependent pathway. Am J Physiol Heart Circ Physiol. 2007 Oct;293(4):H2072–9. doi: 10.1152/ajpheart.01097.2006. [DOI] [PubMed] [Google Scholar]
- 16.Lesh RE, Nixon GF, Fleischer S, Airey JA, Somlyo AP, Somlyo AV. Localization of ryanodine receptors in smooth muscle. Circ Res. 1998 Feb 9;82(2):175–85. doi: 10.1161/01.res.82.2.175. [DOI] [PubMed] [Google Scholar]
- 17.Hirano K. Current topics in the regulatory mechanism underlying the Ca2+ sensitization of the contractile apparatus in vascular smooth muscle. J Pharmacol Sci. 2007 Jun;104(2):109–15. doi: 10.1254/jphs.cp0070027. [DOI] [PubMed] [Google Scholar]
- 18.Hilgers RH, Todd J, Jr, Webb RC. Increased PDZ-RhoGEF/RhoA/Rho kinase signaling in small mesenteric arteries of angiotensin II-induced hypertensive rats. J Hypertens. 2007 Aug;25(8):1687–97. doi: 10.1097/HJH.0b013e32816f778d. [DOI] [PubMed] [Google Scholar]
- 19.Kamm KE, Murphy RA. Velocity and myosin phosphorylation transients in arterial smooth muscle: effects of agonist diffusion. Experientia. 1985 Aug 15;41(8):1010–7. doi: 10.1007/BF01952123. [DOI] [PubMed] [Google Scholar]
- 20.Kamm KE, Stull JT. The function of myosin and myosin light chain kinase phosphorylation in smooth muscle. Annu Rev Pharmacol Toxicol. 1985;25:593–620. doi: 10.1146/annurev.pa.25.040185.003113. [DOI] [PubMed] [Google Scholar]
- 21.Gallagher PJ, Herring BP, Stull JT. Myosin light chain kinases. J Muscle Res Cell Motil. 1997 Feb;18(1):1–16. doi: 10.1023/a:1018616814417. [DOI] [PubMed] [Google Scholar]
- 22.Chitaley K, Weber D, Webb RC. RhoA/Rho-kinase, vascular changes, and hypertension. Curr Hypertens Rep. 2001 Apr;3(2):139–44. doi: 10.1007/s11906-001-0028-4. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002 Jul 1;16(13):1587–609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- 24.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997 Oct 30;389(6654):990–4. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 25.Miwa S, Iwamuro Y, Zhang XF, Inoki T, Okamoto Y, Okazawa M, et al. Ca2+ entry channels in rat thoracic aortic smooth muscle cells activated by endothelin-1. Jpn J Pharmacol. 1999 Aug;80(4):281–8. doi: 10.1254/jjp.80.281. [DOI] [PubMed] [Google Scholar]
- 26.Yanagisawa M, Inoue A, Ishikawa T, Kasuya Y, Kimura S, Kumagaye S, et al. Primary structure, synthesis, and biological activity of rat endothelin, an endothelium-derived vasoconstrictor peptide. Proc Natl Acad Sci U S A. 1988 Sep;85(18):6964–7. doi: 10.1073/pnas.85.18.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yanagisawa M, Kurihara H, Kimura S, Goto K, Masaki T. A novel peptide vasoconstrictor, endothelin, is produced by vascular endothelium and modulates smooth muscle Ca2+ channels. J Hypertens Suppl. 1988 Dec;6(4):S188–91. doi: 10.1097/00004872-198812040-00056. [DOI] [PubMed] [Google Scholar]
- 28.Miwa S, Kawanabe Y, Okamoto Y, Masaki T. Ca2+ entry channels involved in endothelin-1-induced contractions of vascular smooth muscle cells. J Smooth Muscle Res. 2005 Apr;41(2):61–75. doi: 10.1540/jsmr.41.61. [DOI] [PubMed] [Google Scholar]
- 29.Mohacsi A, Magyar J, Tamas B, Nanasi PP. Effects of endothelins on cardiac and vascular cells: new therapeutic target for the future? Curr Vasc Pharmacol. 2004 Jan;2(1):53–63. doi: 10.2174/1570161043476528. [DOI] [PubMed] [Google Scholar]
- 30.Neylon CB. Vascular biology of endothelin signal transduction. Clin Exp Pharmacol Physiol. 1999 Feb;26(2):149–53. doi: 10.1046/j.1440-1681.1999.03013.x. [DOI] [PubMed] [Google Scholar]
- 31.Tirapelli CR, Casolari DA, Yogi A, Montezano AC, Tostes RC, Legros E, et al. Functional characterization and expression of endothelin receptors in rat carotid artery: involvement of nitric oxide, a vasodilator prostanoid and the opening of K+ channels in ETB-induced relaxation. Br J Pharmacol. 2005 Nov;146(6):903–12. doi: 10.1038/sj.bjp.0706388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robin P, Boulven I, Desmyter C, Harbon S, Leiber D. ET-1 stimulates ERK signaling pathway through sequential activation of PKC and Src in rat myometrial cells. Am J Physiol Cell Physiol. 2002 Jul;283(1):C251–60. doi: 10.1152/ajpcell.00601.2001. [DOI] [PubMed] [Google Scholar]
- 33.Hilal-Dandan R, Ramirez MT, Villegas S, Gonzalez A, Endo-Mochizuki Y, Brown JH, et al. Endothelin ETA receptor regulates signaling and ANF gene expression via multiple G protein-linked pathways. Am J Physiol. 1997 Jan;272(1 Pt 2):H130–7. doi: 10.1152/ajpheart.1997.272.1.H130. [DOI] [PubMed] [Google Scholar]
- 34.Aramori I, Nakanishi S. Coupling of two endothelin receptor subtypes to differing signal transduction in transfected Chinese hamster ovary cells. J Biol Chem. 1992 Jun 25;267(18):12468–74. [PubMed] [Google Scholar]
- 35.Smith L, Payne JA, Sedeek MH, Granger JP, Khalil RA. Endothelin-induced increases in Ca2+ entry mechanisms of vascular contraction are enhanced during high-salt diet. Hypertension. 2003 Mar;41(3 Pt 2):787–93. doi: 10.1161/01.HYP.0000051643.05700.56. [DOI] [PubMed] [Google Scholar]
- 36.Dammanahalli JK, Sun Z. Endothelin-1 Inhibits NADPH Oxidase Activity in Human Abdominal Aortic Endothelial Cells: A Novel Function of ETB1 Receptors. Endocrinology. 2008 Jun 5; doi: 10.1210/en.2008-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Douglas JT, Curiel DT. Strategies to accomplish targeted gene delivery to muscle cells employing tropism-modified adenoviral vectors. Neuromuscul Disord. 1997 Jul;7(5):284–98. doi: 10.1016/s0960-8966(97)00053-9. [DOI] [PubMed] [Google Scholar]
- 38.Rubanyi GM, Polokoff MA. Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacol Rev. 1994 Sep;46(3):325–415. [PubMed] [Google Scholar]
- 39.Komuro T, Miwa S, Zhang XF, Minowa T, Enoki T, Kobayashi S, et al. Physiological role of Ca2+-permeable nonselective cation channel in endothelin-1-induced contraction of rabbit aorta. J Cardiovasc Pharmacol. 1997 Oct;30(4):504–9. doi: 10.1097/00005344-199710000-00015. [DOI] [PubMed] [Google Scholar]
- 40.Dostal DE, Murahashi T, Peach MJ. Regulation of cytosolic calcium by angiotensins in vascular smooth muscle. Hypertension. 1990 Jun;15(6 Pt 2):815–22. doi: 10.1161/01.hyp.15.6.815. [DOI] [PubMed] [Google Scholar]
- 41.Kawanabe Y, Hashimoto N, Masaki T. Characterization of Ca2+ channels involved in endothelin-1-induced contraction of rabbit basilar artery. J Cardiovasc Pharmacol. 2002 Sep;40(3):438–47. doi: 10.1097/00005344-200209000-00013. [DOI] [PubMed] [Google Scholar]
- 42.Yogi A, Callera GE, Montezano AC, Aranha AB, Tostes RC, Schiffrin EL, et al. Endothelin-1, but not Ang II, activates MAP kinases through c-Src independent Ras-Raf dependent pathways in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2007 Sep;27(9):1960–7. doi: 10.1161/ATVBAHA.107.146746. [DOI] [PubMed] [Google Scholar]
- 43.Bolton TB. Mechanisms of action of transmitters and other substances on smooth muscle. Physiol Rev. 1979 Jul;59(3):606–718. doi: 10.1152/physrev.1979.59.3.606. [DOI] [PubMed] [Google Scholar]
- 44.Hughes AD. Calcium channels in vascular smooth muscle cells. J Vasc Res. 1995 Nov-Dec;32(6):353–70. doi: 10.1159/000159111. [DOI] [PubMed] [Google Scholar]
- 45.Goto K, Kasuya Y, Matsuki N, Takuwa Y, Kurihara H, Ishikawa T, et al. Endothelin activates the dihydropyridine-sensitive, voltage-dependent Ca2+ channel in vascular smooth muscle. Proc Natl Acad Sci U S A. 1989 May;86(10):3915–8. doi: 10.1073/pnas.86.10.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawanabe Y, Hashimoto N, Masaki T. Involvements of voltage-independent Ca2+ channels and phosphoinositide 3-kinase in endothelin-1-induced PYK2 tyrosine phosphorylation. Mol Pharmacol. 2003 Apr;63(4):808–13. doi: 10.1124/mol.63.4.808. [DOI] [PubMed] [Google Scholar]
- 47.Eto K, Ohya Y, Nakamura Y, Abe I, Iida M. Intracellular angiotensin II stimulates voltage-operated Ca(2+) channels in arterial myocytes. Hypertension. 2002 Feb;39(2 Pt 2):474–8. doi: 10.1161/hy02t2.102961. [DOI] [PubMed] [Google Scholar]
- 48.Ohya Y, Sperelakis N. Involvement of a GTP-binding protein in stimulating action of angiotensin II on calcium channels in vascular smooth muscle cells. Circ Res. 1991 Mar;68(3):763–71. doi: 10.1161/01.res.68.3.763. [DOI] [PubMed] [Google Scholar]
- 49.Wayman CP, McFadzean I, Gibson A, Tucker JF. Two distinct membrane currents activated by cyclopiazonic acid-induced calcium store depletion in single smooth muscle cells of the mouse anococcygeus. Br J Pharmacol. 1996 Feb;117(3):566–72. doi: 10.1111/j.1476-5381.1996.tb15228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang AY, Li PL. Vascular physiology of a Ca2+ mobilizing second messenger - cyclic ADP-ribose. J Cell Mol Med. 2006 Apr-Jun;10(2):407–22. doi: 10.1111/j.1582-4934.2006.tb00408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986 Feb;7(1):1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 52.Benham CD, Hess P, Tsien RW. Two types of calcium channels in single smooth muscle cells from rabbit ear artery studied with whole-cell and single-channel recordings. Circ Res. 1987 Oct;61(4 Pt 2):I10–6. [PubMed] [Google Scholar]
- 53.Morris AP, Gallacher DV, Irvine RF, Petersen OH. Synergism of inositol trisphosphate and tetrakisphosphate in activating Ca2+-dependent K+ channels. Nature. 1987 Dec 17-23;330(6149):653–5. doi: 10.1038/330653a0. [DOI] [PubMed] [Google Scholar]
- 54.Kuno M, Gardner P. Ion channels activated by inositol 1,4,5-trisphosphate in plasma membrane of human T-lymphocytes. Nature. 1987 Mar 19-25;326(6110):301–4. doi: 10.1038/326301a0. [DOI] [PubMed] [Google Scholar]
- 55.Saleh SN, Albert AP, Peppiatt CM, Large WA. Angiotensin II activates two cation conductances with distinct TRPC1 and TRPC6 channel properties in rabbit mesenteric artery myocytes. J Physiol. 2006 Dec 1;577(Pt 2):479–95. doi: 10.1113/jphysiol.2006.119305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tai K, Hamaide MC, Debaix H, Gailly P, Wibo M, Morel N. Agonist-evoked calcium entry in vascular smooth muscle cells requires IP3 receptor-mediated activation of TRPC1. Eur J Pharmacol. 2008 Mar 31;583(1):135–47. doi: 10.1016/j.ejphar.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 57.Peppiatt-Wildman CM, Albert AP, Saleh SN, Large WA. Endothelin-1 activates a Ca2+-permeable cation channel with TRPC3 and TRPC7 properties in rabbit coronary artery myocytes. J Physiol. 2007 May 1;580(Pt3):755–64. doi: 10.1113/jphysiol.2006.126656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smyth JT, Dehaven WI, Jones BF, Mercer JC, Trebak M, Vazquez G, et al. Emerging perspectives in store-operated Ca2+ entry: roles of Orai, Stim and TRP. Biochim Biophys Acta. 2006 Nov;1763(11):1147–60. doi: 10.1016/j.bbamcr.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 59.Putney JW, Jr, McKay RR. Capacitative calcium entry channels. Bioessays. 1999 Jan;21(1):38–46. doi: 10.1002/(SICI)1521-1878(199901)21:1<38::AID-BIES5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 60.Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiological reviews. 2001 Oct;81(4):1415–59. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- 61.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiological reviews. 2005 Apr;85(2):757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 62.Leung FP, Yung LM, Yao X, Laher I, Huang Y. Store-operated calcium entry in vascular smooth muscle. Br J Pharmacol. 2008 Mar;153(5):846–57. doi: 10.1038/sj.bjp.0707455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Putney JW., Jr Capacitative calcium entry revisited. Cell Calcium. 1990 Nov-Dec;11(10):611–24. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- 64.Putney JW., Jr Receptor-regulated calcium entry. Pharmacol Ther. 1990;48(3):427–34. doi: 10.1016/0163-7258(90)90059-b. [DOI] [PubMed] [Google Scholar]
- 65.Putney JW., Jr The integration of receptor-regulated intracellular calcium release and calcium entry across the plasma membrane. Curr Top Cell Regul. 1990;31:111–27. doi: 10.1016/b978-0-12-152831-7.50004-4. [DOI] [PubMed] [Google Scholar]
- 66.Kasuya Y, Ishikawa T, Yanagisawa M, Kimura S, Goto K, Masaki T. Mechanism of contraction to endothelin in isolated porcine coronary artery. Am J Physiol. 1989 Dec;257(6 Pt 2):H1828–35. doi: 10.1152/ajpheart.1989.257.6.H1828. [DOI] [PubMed] [Google Scholar]
- 67.Takemura H, Thastrup O, Putney JW., Jr Calcium efflux across the plasma membrane of rat parotid acinar cells is unaffected by receptor activation or by the microsomal calcium ATPase inhibitor, thapsigargin. Cell Calcium. 1990 Jan;11(1):11–7. doi: 10.1016/0143-4160(90)90044-u. [DOI] [PubMed] [Google Scholar]
- 68.Putney JW, Jr, Broad LM, Braun FJ, Lievremont JP, Bird GS. Mechanisms of capacitative calcium entry. J Cell Sci. 2001 Jun;114(Pt 12):2223–9. doi: 10.1242/jcs.114.12.2223. [DOI] [PubMed] [Google Scholar]
- 69.Bouallegue A, Yamaguchi N. Nitric oxide inhibits the bradykinin B2 receptor-mediated adrenomedullary catecholamine release but has no effect on adrenal blood flow response in vivo. J Pharmacol Sci. 2005 Jun;98(2):151–60. doi: 10.1254/jphs.fpj04048x. [DOI] [PubMed] [Google Scholar]
- 70.Venkatachalam K, van Rossum DB, Patterson RL, Ma HT, Gill DL. The cellular and molecular basis of store-operated calcium entry. Nat Cell Biol. 2002 Nov;4(11):E263–72. doi: 10.1038/ncb1102-e263. [DOI] [PubMed] [Google Scholar]
- 71.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005 May 9;169(3):435–45. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005 Oct 6;437(7060):902–5. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005 Jul 12;15(13):1235–41. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takahashi Y, Watanabe H, Murakami M, Ohba T, Radovanovic M, Ono K, et al. Involvement of transient receptor potential canonical 1 (TRPC1) in angiotensin II-induced vascular smooth muscle cell hypertrophy. Atherosclerosis. 2007 Dec;195(2):287–96. doi: 10.1016/j.atherosclerosis.2006.12.033. [DOI] [PubMed] [Google Scholar]
- 75.Chen C, Wagoner PK. Endothelin induces a nonselective cation current in vascular smooth muscle cells. Circ Res. 1991 Aug;69(2):447–54. doi: 10.1161/01.res.69.2.447. [DOI] [PubMed] [Google Scholar]
- 76.Enoki T, Miwa S, Sakamoto A, Minowa T, Komuro T, Kobayashi S, et al. Long-lasting activation of cation current by low concentration of endothelin-1 in mouse fibroblasts and smooth muscle cells of rabbit aorta. Br J Pharmacol. 1995 Jun;115(3):479–85. doi: 10.1111/j.1476-5381.1995.tb16358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kasuya Y, Takuwa Y, Yanagisawa M, Kimura S, Goto K, Masaki T. Endothelin-1 induces vasoconstriction through two functionally distinct pathways in porcine coronary artery: contribution of phosphoinositide turnover. Biochem Biophys Res Commun. 1989 Jun 30;161(3):1049–55. doi: 10.1016/0006-291x(89)91349-1. [DOI] [PubMed] [Google Scholar]
- 78.Kawanabe Y, Hashimoto N, Masaki T. Effects of phosphoinositide 3-kinase on endothelin-1-induced activation of voltage-independent Ca2+ channels and vasoconstriction. Biochem Pharmacol. 2004 Jul 15;68(2):215–21. doi: 10.1016/j.bcp.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 79.Schoner W. Salt abuse: the path to hypertension. Nat Med. 2008 Jan;14(1):16–7. doi: 10.1038/nm0108-16. [DOI] [PubMed] [Google Scholar]
- 80.Fernanda RC, Giachini FSC, Lima Victor V, Carniero Zidonia N, Carvalho Maria Helena C, Fortes Zuleica B, Webb R Clinton, Tostes Rita C. Pyk2 mediates increased adrenergic contractile responses in arteries from DOCA-salt mice. Journal of the American Society of Hypertension. 2008 doi: 10.1016/j.jash.2008.05.001. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yin G, Yan C, Berk BC. Angiotensin II signaling pathways mediated by tyrosine kinases. Int J Biochem Cell Biol. 2003 Jun;35(6):780–3. doi: 10.1016/s1357-2725(02)00300-x. [DOI] [PubMed] [Google Scholar]
- 82.Bouallegue A, Daou GB, Srivastava AK. Endothelin-1-induced signaling pathways in vascular smooth muscle cells. Curr Vasc Pharmacol. 2007 Jan;5(1):45–52. doi: 10.2174/157016107779317161. [DOI] [PubMed] [Google Scholar]
- 83.Schaller MD. Calcium-dependent Pyk2 activation: a role for calmodulin? Biochem J. 2008 Mar 15;410(3):e3–4. doi: 10.1042/BJ20080133. [DOI] [PubMed] [Google Scholar]
- 84.Berk BC. Angiotensin II signal transduction in vascular smooth muscle: pathways activated by specific tyrosine kinases. J Am Soc Nephrol. 1999 Jan;10 11:S62–8. [PubMed] [Google Scholar]
- 85.Giachini FRCCF, Lima VV, Carneiro ZN, Carvalho MHC, Fortes ZB, Webb RC, Tostes RC. Pyk2 mediates increased adrenergic contractile responses in arteries from DOCA-salt mice. Journal of the American Society of Hypertension. 2008 doi: 10.1016/j.jash.2008.05.001. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006 Jul 28;281(30):20661–5. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 87.Spassova MA, Soboloff J, He LP, Xu W, Dziadek MA, Gill DL. STIM1 has a plasma membrane role in the activation of store-operated Ca(2+) channels. Proc Natl Acad Sci U S A. 2006 Mar 14;103(11):4040–5. doi: 10.1073/pnas.0510050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Manji SS, Parker NJ, Williams RT, van Stekelenburg L, Pearson RB, Dziadek M, et al. STIM1: a novel phosphoprotein located at the cell surface. Biochim Biophys Acta. 2000 Aug 31;1481(1):147–55. doi: 10.1016/s0167-4838(00)00105-9. [DOI] [PubMed] [Google Scholar]
- 89.Soboloff J, Spassova MA, Dziadek MA, Gill DL. Calcium signals mediated by STIM and Orai proteins--a new paradigm in inter-organelle communication. Biochim Biophys Acta. 2006 Nov;1763(11):1161–8. doi: 10.1016/j.bbamcr.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 90.Soboloff J, Spassova MA, Hewavitharana T, He LP, Xu W, Johnstone LS, et al. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry. Curr Biol. 2006 Jul 25;16(14):1465–70. doi: 10.1016/j.cub.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 91.Coffman TM, Crowley SD. Kidney in hypertension: guyton redux. Hypertension. 2008 Apr;51(4):811–6. doi: 10.1161/HYPERTENSIONAHA.105.063636. [DOI] [PubMed] [Google Scholar]
- 92.Guyton AC. Blood pressure control--special role of the kidneys and body fluids. Science. 1991 Jun 28;252(5014):1813–6. doi: 10.1126/science.2063193. [DOI] [PubMed] [Google Scholar]
- 93.Coffman TM, Himmelstein S, Best C, Klotman PE. Post-transplant hypertension in the rat: effects of captopril and native nephrectomy. Kidney Int. 1989 Jul;36(1):35–40. doi: 10.1038/ki.1989.157. [DOI] [PubMed] [Google Scholar]
- 94.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, et al. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006 Nov 21;103(47):17985–90. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Michael SK, Surks HK, Wang Y, Zhu Y, Blanton R, Jamnongjit M, et al. High blood pressure arising from a defect in vascular function. Proc Natl Acad Sci U S A. 2008 May 6;105(18):6702–7. doi: 10.1073/pnas.0802128105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res. 2002 Sep 6;91(5):406–13. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]